
The Assessment of Anti-Melanoma Potential of Tigecycline—Cellular and Molecular Studies of Cell Proliferation, Apoptosis and Autophagy on Amelanotic and Melanotic Melanoma Cells
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Screening Cytotoxicity Assessment
2.4. Analysis of Cell Number and Cell Viability
2.5. Cell Cycle Analysis
2.6. Reduced Thiols Analysis
2.7. Mitochondrial Membrane Potential Assay
2.8. Annexin V Assay
2.9. Caspase 3/7 Activity Assay
2.10. Western Blotting Analysis
2.11. Confocal Microscopy Imaging
2.12. Statistical Analysis
3. Results
3.1. Screening Assessment of Cytotoxic Effect of Tigecycline on Melanoma Cells
3.2. Influence of Tigecycline on Proliferation, Viability, and Apoptosis of Melanoma Cells
3.3. Tigecycline Decreases the Level of Reduced Thiols in Melanoma Cells
3.4. Tigecycline Decreases Mitochondrial Membrane Potential (MMP) in Melanoma Cells
3.5. Tigecycline Disturbs Cell Cycle of Melanoma Cells
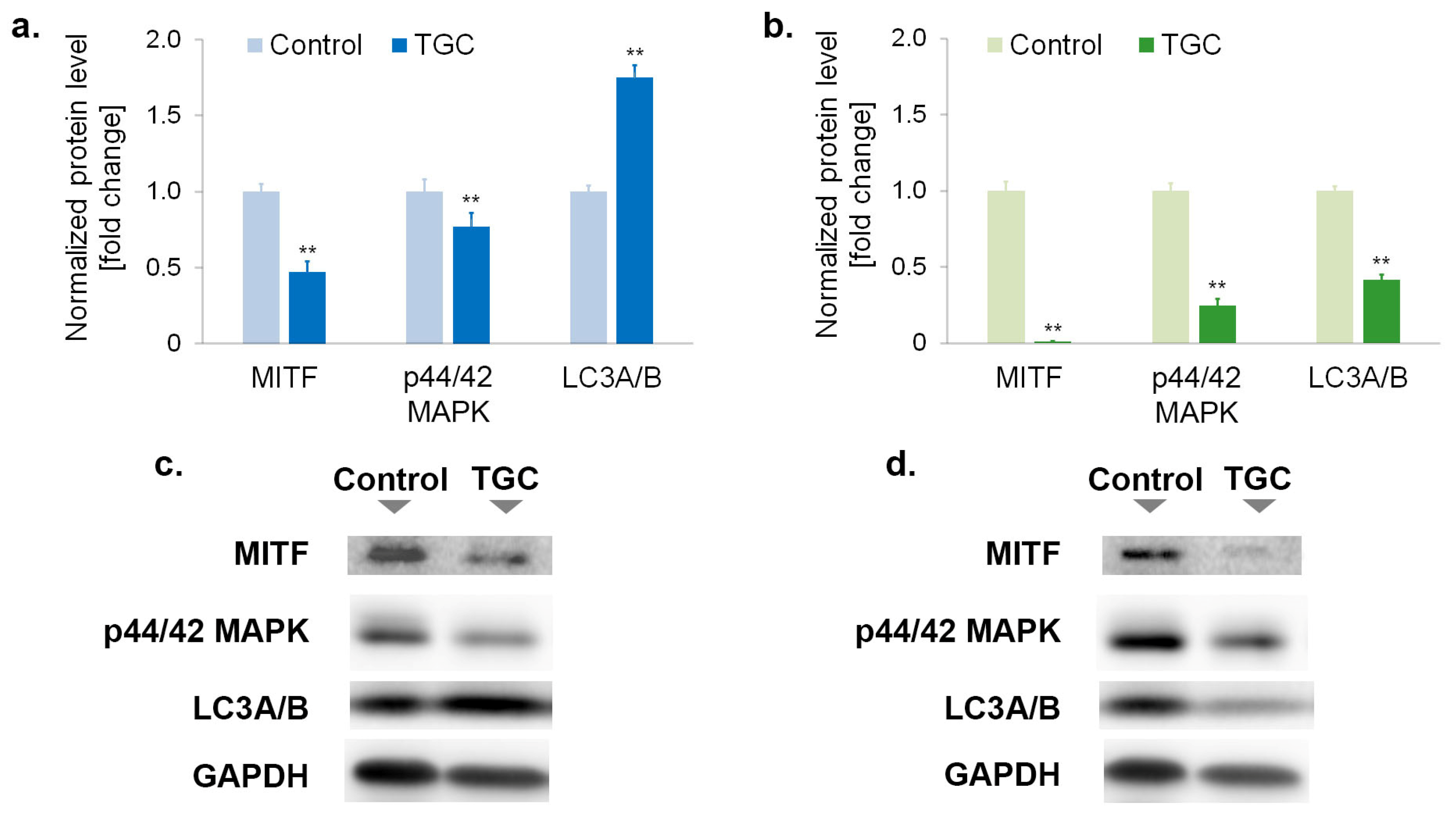
3.6. Tigecycline Changes the Level of Proteins Regulating Proliferation and Survival of Melanoma Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ahmed, B.; Qadir, M.I.; Ghafoor, S. Malignant Melanoma: Skin Cancer-Diagnosis, Prevention, and Treatment. Crit. Rev. Eukaryot. Gene Expr. 2020, 30, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef]
- Conforti, C.; Zalaudek, I. Epidemiology and Risk Factors of Melanoma: A Review. Dermatol. Pract. Concept. 2021, 11, e2021161S. [Google Scholar] [CrossRef] [PubMed]
- Moran, B.; Silva, R.; Perry, A.S.; Gallagher, W.M. Epigenetics of malignant melanoma. Semin. Cancer Biol. 2018, 51, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.G.; Miller, N.; McInerney, N.M. A Review of Epidemiology and Cancer Biology of Malignant Melanoma. Cureus 2021, 13, e15087. [Google Scholar] [CrossRef] [PubMed]
- Newton-Bishop, J.; Bishop, D.T.; Harland, M. Melanoma Genomics. Acta Derm. Venereol. 2020, 100, adv00138. [Google Scholar] [CrossRef]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [Green Version]
- Ziogas, D.C.; Theocharopoulos, C.; Koutouratsas, T.; Haanen, J.; Gogas, H. Mechanisms of resistance to immune checkpoint inhibitors in melanoma: What we have to overcome? Cancer Treat. Rev. 2023, 113, 102499. [Google Scholar] [CrossRef]
- Curti, B.D.; Faries, M.B. Recent Advances in the Treatment of Melanoma. N. Engl. J. Med. 2021, 384, 2229–2240. [Google Scholar] [CrossRef]
- Stachyra-Strawa, P.; Ciesielka, M.; Janiszewski, M.; Grzybowska-Szatkowska, L. The role of immunotherapy and molecular-targeted therapy in the treatment of melanoma (Review). Oncol. Rep. 2021, 46, 158. [Google Scholar] [CrossRef]
- Rok, J.; Karkoszka, M.; Rzepka, Z.; Respondek, M.; Banach, K.; Beberok, A.; Wrześniok, D. Cytotoxic and proapoptotic effect of doxycycline—An in vitro study on the human skin melanoma cells. Toxicol. Vitro 2020, 65, 104790. [Google Scholar] [CrossRef] [PubMed]
- Rok, J.; Rzepka, Z.; Beberok, A.; Pawlik, J.; Wrześniok, D. Cellular and Molecular Aspects of Anti-Melanoma Effect of Minocycline-A Study of Cytotoxicity and Apoptosis on Human Melanotic Melanoma Cells. Int. J. Mol. Sci. 2020, 21, 6917. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.; Kaysiewicz, J.; Markowska, J.; Huczyński, A. Doxycycline, salinomycin, monensin and ivermectin repositioned as cancer drugs. Bioorg. Med. Chem. Lett. 2019, 29, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Hadjimichael, A.C.; Foukas, A.F.; Savvidou, O.D.; Mavrogenis, A.F.; Psyrri, A.K.; Papagelopoulos, P.J. The anti-neoplastic effect of doxycycline in osteosarcoma as a metalloproteinase (MMP) inhibitor: A systematic review. Clin. Sarcoma Res. 2020, 10, 7. [Google Scholar] [CrossRef]
- Afshari, A.R.; Mollazadeh, H.; Sahebkar, A. Minocycline in Treating Glioblastoma Multiforme: Far beyond a Conventional Antibiotic. J. Oncol. 2020, 2020, 8659802. [Google Scholar] [CrossRef]
- Araújo, D.; Ribeiro, E.; Amorim, I.; Vale, N. Repurposed Drugs in Gastric Cancer. Molecules 2022, 28, 319. [Google Scholar] [CrossRef]
- Liu, H.; Tao, H.; Wang, H.; Yang, Y.; Yang, R.; Dai, X.; Ding, X.; Wu, H.; Chen, S.; Sun, T. Doxycycline Inhibits Cancer Stem Cell-Like Properties via PAR1/FAK/PI3K/AKT Pathway in Pancreatic Cancer. Front. Oncol. 2021, 10, 619317. [Google Scholar] [CrossRef]
- Ali, I.; Alfarouk, K.O.; Reshkin, S.J.; Ibrahim, M.E. Doxycycline as Potential Anti-cancer Agent. Anticancer Agents Med. Chem. 2017, 17, 1617–1623. [Google Scholar] [CrossRef]
- Ghasemi, K.; Ghasemi, K. A Brief look at antitumor effects of doxycycline in the treatment of colorectal cancer and combination therapies. Eur. J. Pharmacol. 2022, 916, 174593. [Google Scholar] [CrossRef]
- Jung, H.J.; Seo, I.; Jha, B.K.; Suh, S.I.; Suh, M.H.; Baek, W.K. Minocycline inhibits angiogenesis in vitro through the translational suppression of HIF-1α. Arch. Biochem. Biophys. 2014, 545, 74–82. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Karlowsky, J.A.; Rubinstein, E.; Hoban, D.J. Tigecycline: A novel glycylcycline antibiotic. Expert Rev. Anti Infect. Ther. 2006, 4, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Meagher, A.K.; Ambrose, P.G.; Grasela, T.H.; Ellis-Grosse, E.J. Pharmacokinetic/pharmacodynamic profile for tigecycline-a new glycylcycline antimicrobial agent. Diagn. Microbiol. Infect. Dis. 2005, 52, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubi, S.; Zekiy, A.O.; Krutova, M.; Gholami, M.; Kouhsari, E.; Sholeh, M.; Ghafouri, Z.; Maleki, F. Tigecycline antibacterial activity, clinical effectiveness, and mechanisms and epidemiology of resistance: Narrative review. Eur. J. Clin. Microbiol. Infect. Dis. 2022, 41, 1003–1022. [Google Scholar] [CrossRef] [PubMed]
- Rose, W.E.; Rybak, M.J. Tigecycline: First of a new class of antimicrobial agents. Pharmacotherapy 2006, 26, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Skrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Abbas, M.N.; Kausar, S.; Yang, J.; Li, L.; Tan, L.; Cui, H. Biological Functions and Molecular Mechanisms of Antibiotic Tigecycline in the Treatment of Cancers. Int. J. Mol. Sci. 2019, 20, 3577. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Yan, Y.; Li, Z.; Qian, L.; Gong, Z. The Antibiotic Drug Tigecycline: A Focus on its Promising Anticancer Properties. Front. Pharmacol. 2016, 7, 473. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Yang, L.; Jiang, X.; Xu, C.; Wang, M.; Wang, Q.; Zhou, Z.; Xiang, Z.; Cui, H. Antibiotic drug tigecycline inhibited cell proliferation and induced autophagy in gastric cancer cells. Biochem. Biophys. Res. Commun. 2014, 446, 105–112. [Google Scholar] [CrossRef]
- Hu, H.; Dong, Z.; Tan, P.; Zhang, Y.; Liu, L.; Yang, L.; Liu, Y.; Cui, H. Antibiotic drug tigecycline inhibits melanoma progression and metastasis in a p21CIP1/Waf1-dependent manner. Oncotarget 2016, 7, 3171–3185. [Google Scholar] [CrossRef] [Green Version]
- Sapadin, A.N.; Fleischmajer, R. Tetracyclines: Nonantibiotic properties and their clinical implications. J. Am. Acad. Dermatol. 2006, 54, 258–265. [Google Scholar] [CrossRef]
- Rok, J.; Rzepka, Z.; Respondek, M.; Beberok, A.; Wrześniok, D. Chlortetracycline and melanin biopolymer—The risk of accumulation and implications for phototoxicity: An in vitro study on normal human melanocytes. Chem. Biol. Interact. 2019, 303, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Banning, T.P.; Heard, C.M. Binding of doxycycline to keratin, melanin and human epidermal tissue. Int. J. Pharm. 2002, 235, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Rok, J.; Rzepka, Z.; Kowalska, J.; Banach, K.; Beberok, A.; Wrześniok, D. The Anticancer Potential of Doxycycline and Minocycline-A Comparative Study on Amelanotic Melanoma Cell Lines. Int. J. Mol. Sci. 2022, 23, 831. [Google Scholar] [CrossRef]
- Rok, J.; Rzepka, Z.; Banach, K.; Kowalska, J.; Wrześniok, D. The Assessment of the Phototoxic Action of Chlortetracycline and Doxycycline as a Potential Treatment of Melanotic Melanoma-Biochemical and Molecular Studies on COLO 829 and G-361 Cell Lines. Int. J. Mol. Sci. 2023, 24, 2353. [Google Scholar] [CrossRef] [PubMed]
- Slominski, R.M.; Sarna, T.; Płonka, P.M.; Raman, C.; Brożyna, A.A.; Slominski, A.T. Melanoma, Melanin, and Melanogenesis: The Yin and Yang Relationship. Front. Oncol. 2022, 12, 842496. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Wlodkowic, D.; Skommer, J.; Darzynkiewicz, Z. Cytometry of apoptosis. Historical perspective and new advances. Exp. Oncol. 2012, 34, 255–262. [Google Scholar]
- Darzynkiewicz, Z.; Zhao, H.; Halicka, D.H.; Pozarowski, P.; Lee, B. Fluorochrome-Labeled Inhibitors of Caspases: Expedient In Vitro and In Vivo Markers of Apoptotic Cells for Rapid Cytometric Analysis. Methods Mol. Biol. 2017, 1644, 61–73. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Van Loenhout, J.; Peeters, M.; Bogaerts, A.; Smits, E.; Deben, C. Oxidative Stress-Inducing Anticancer Therapies: Taking a Closer Look at Their Immunomodulating Effects. Antioxidants 2020, 9, 1188. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef] [PubMed]
- Azmanova, M.; Pitto-Barry, A. Oxidative Stress in Cancer Therapy: Friend or Enemy? Chembiochem 2022, 23, e202100641. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer. 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.L.; Ponneri Babuharisankar, A.; Lin, Y.C.; Lien, H.W.; Lo, Y.K.; Chou, H.Y.; Tangeda, V.; Cheng, L.C.; Cheng, A.N.; Lee, A.Y. Mitochondrial oxidative stress in the tumor microenvironment and cancer immunoescape: Foe or friend? J. Biomed. Sci. 2022, 29, 74. [Google Scholar] [CrossRef]
- Lopez, J.; Tait, S.W. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, K.; Jakob, U. The role of thiols in antioxidant systems. Free Radic. Biol. Med. 2019, 140, 14–27. [Google Scholar] [CrossRef]
- Niu, B.; Liao, K.; Zhou, Y.; Wen, T.; Quan, G.; Pan, X.; Wu, C. Application of glutathione depletion in cancer therapy: Enhanced ROS-based therapy, ferroptosis, and chemotherapy. Biomaterials 2021, 277, 121110. [Google Scholar] [CrossRef]
- Kennedy, L.; Sandhu, J.K.; Harper, M.E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. Pro-survival role of MITF in melanoma. J. Investig. Dermatol. 2015, 135, 352–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelmi, M.C.; Houtzagers, L.E.; Strub, T.; Krossa, I.; Jager, M.J. MITF in Normal Melanocytes, Cutaneous and Uveal Melanoma: A Delicate Balance. Int. J. Mol. Sci. 2022, 23, 6001. [Google Scholar] [CrossRef] [PubMed]
- Vlčková, K.; Vachtenheim, J.; Réda, J.; Horák, P.; Ondrušová, L. Inducibly decreased MITF levels do not affect proliferation and phenotype switching but reduce differentiation of melanoma cells. J. Cell. Mol. Med. 2018, 22, 2240–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, A.; Fisher, D.E. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab. Investig. 2017, 97, 649–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, M.L.; Czyz, M. MITF in melanoma: Mechanisms behind its expression and activity. Cell. Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vachtenheim, J.; Ondrušová, L. Microphthalmia-associated transcription factor expression levels in melanoma cells contribute to cell invasion and proliferation. Exp. Dermatol. 2015, 24, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Chambard, J.C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef]
- Mirmohammadsadegh, A.; Mota, R.; Gustrau, A.; Hassan, M.; Nambiar, S.; Marini, A.; Bojar, H.; Tannapfel, A.; Hengge, U.R. ERK1/2 is highly phosphorylated in melanoma metastases and protects melanoma cells from cisplatin-mediated apoptosis. J. Investig. Dermatol. 2007, 127, 2207–2215. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.M.; Mohamad Hanif, E.A.; Chin, S.F. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi-Dehlaghi, F.; Mohammadi, P.; Valipour, E.; Pournaghi, P.; Kiani, S.; Mansouri, K. Autophagy: A challengeable paradox in cancer treatment. Cancer Med. 2023, 00, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Wang, S.; Wang, B.; Hong, X.; Liu, X.; Li, M.; Shen, R.; Dong, Q. The role of interaction between autophagy and apoptosis in tumorigenesis (Review). Oncol. Rep. 2022, 48, 208. [Google Scholar] [CrossRef]
- Hou, W.; Han, J.; Lu, C.; Goldstein, L.A.; Rabinowich, H. Autophagic degradation of active caspase-8: A crosstalk mechanism between autophagy and apoptosis. Autophagy 2010, 6, 891–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Chen, M.; Chen, N.; Ma, A.; Zhu, C.; Zhao, R.; Jiang, M.; Zhou, J.; Ye, L.; Fu, H.; et al. Glycyrrhetinic acid induces G1-phase cell cycle arrest in human non-small cell lung cancer cells through endoplasmic reticulum stress pathway. Int. J. Oncol. 2015, 46, 981–988. [Google Scholar] [CrossRef] [Green Version]
- Ciołczyk-Wierzbicka, D.; Gil, D.; Laidler, P. Treatment of melanoma with selected inhibitors of signaling kinases effectively reduces proliferation and induces expression of cell cycle inhibitors. Med. Oncol. 2017, 35, 7. [Google Scholar] [CrossRef] [Green Version]
- Slominski, R.M.; Raman, C.; Chen, J.Y.; Slominski, A.T. How cancer hijacks the body’s homeostasis through the neuroendocrine system. Trends Neurosci. 2023, 46, 263–275. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rok, J.; Kowalska, J.; Rzepka, Z.; Stencel, D.; Skorek, A.; Banach, K.; Wrześniok, D. The Assessment of Anti-Melanoma Potential of Tigecycline—Cellular and Molecular Studies of Cell Proliferation, Apoptosis and Autophagy on Amelanotic and Melanotic Melanoma Cells. Cells 2023, 12, 1564. https://doi.org/10.3390/cells12121564
Rok J, Kowalska J, Rzepka Z, Stencel D, Skorek A, Banach K, Wrześniok D. The Assessment of Anti-Melanoma Potential of Tigecycline—Cellular and Molecular Studies of Cell Proliferation, Apoptosis and Autophagy on Amelanotic and Melanotic Melanoma Cells. Cells. 2023; 12(12):1564. https://doi.org/10.3390/cells12121564
Chicago/Turabian StyleRok, Jakub, Justyna Kowalska, Zuzanna Rzepka, Dominika Stencel, Anna Skorek, Klaudia Banach, and Dorota Wrześniok. 2023. "The Assessment of Anti-Melanoma Potential of Tigecycline—Cellular and Molecular Studies of Cell Proliferation, Apoptosis and Autophagy on Amelanotic and Melanotic Melanoma Cells" Cells 12, no. 12: 1564. https://doi.org/10.3390/cells12121564