
Mustard Gas Exposure Actuates SMAD2/3 Signaling to Promote Myofibroblast Generation in the Cornea
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. In Vivo Rabbit SM Studies
2.3. Animal Tissue Processing
2.4. In Vitro NM Studies
2.5. Real-Time PCR
2.6. Western Blot
2.7. Immunocytochemistry
2.8. Immunohistochemistry
2.9. Statistical Analysis
3. Results
3.1. SM Exposure Induces Myofibroblast Formation in Rabbit Cornea In Vivo
3.1.1. Time-Dependent Expression of Profibrotic Genes in Rabbit CORNEAS Exposed to SM In Vivo
3.1.2. Localization of αSMA in Rabbit Corneas Exposed to SM In Vivo
3.1.3. Time-Dependent Expression of SMAD Signaling Proteins in Rabbit Corneas Exposed to SM In Vivo
3.2. NM Exposure Induces Myofibroblast Formation in a Time-Dependent Expression In Vitro
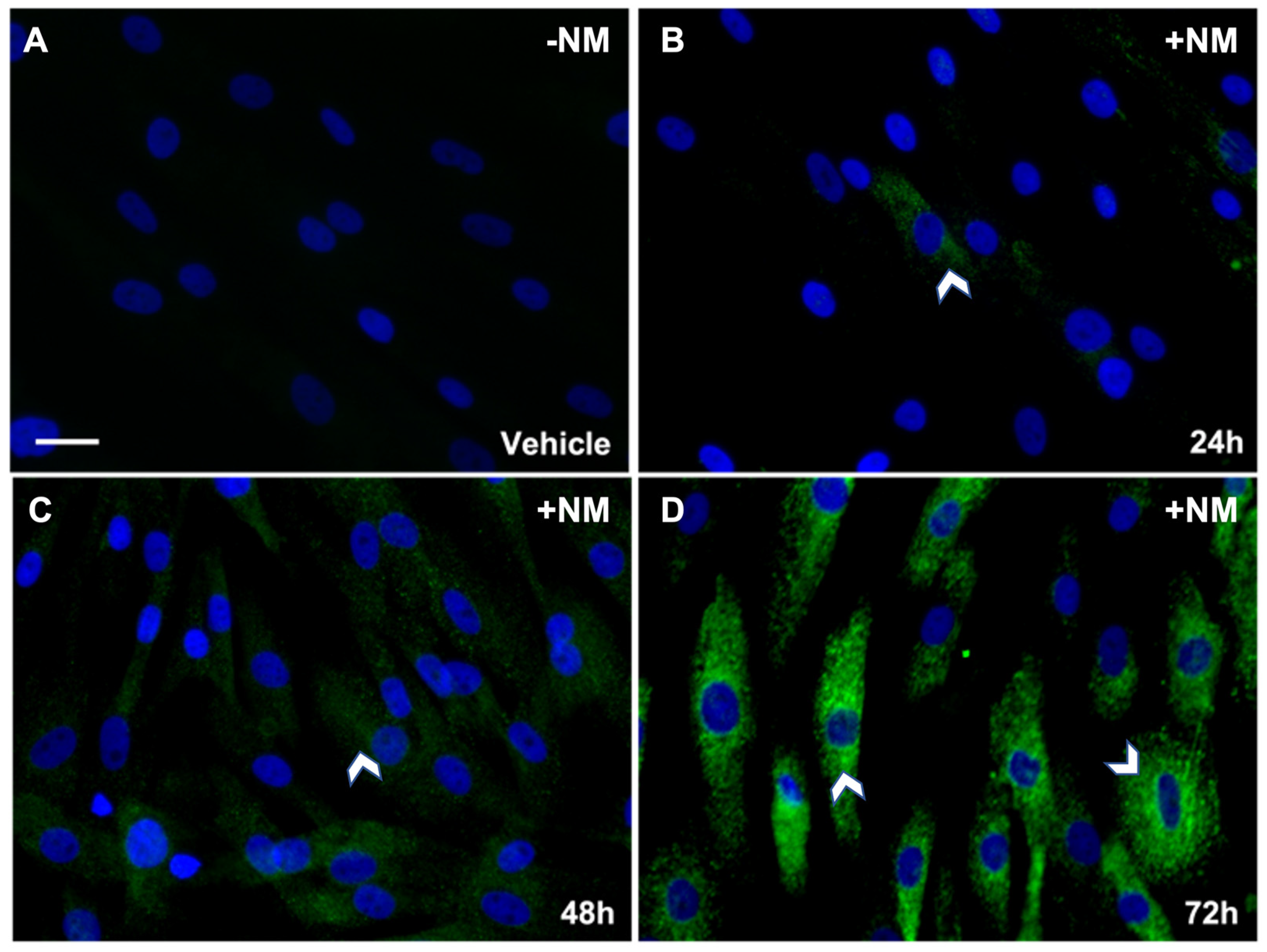
3.2.1. Time-Dependent Analysis of αSMA in hCSFs Treated with NM In Vitro
3.2.2. Profibrotic Protein Analysis of hCSFs Treated −/+ NM In Vitro
3.3. NM-Induced Myofibroblast Formation Involves Activation of SMAD Signaling Pathway
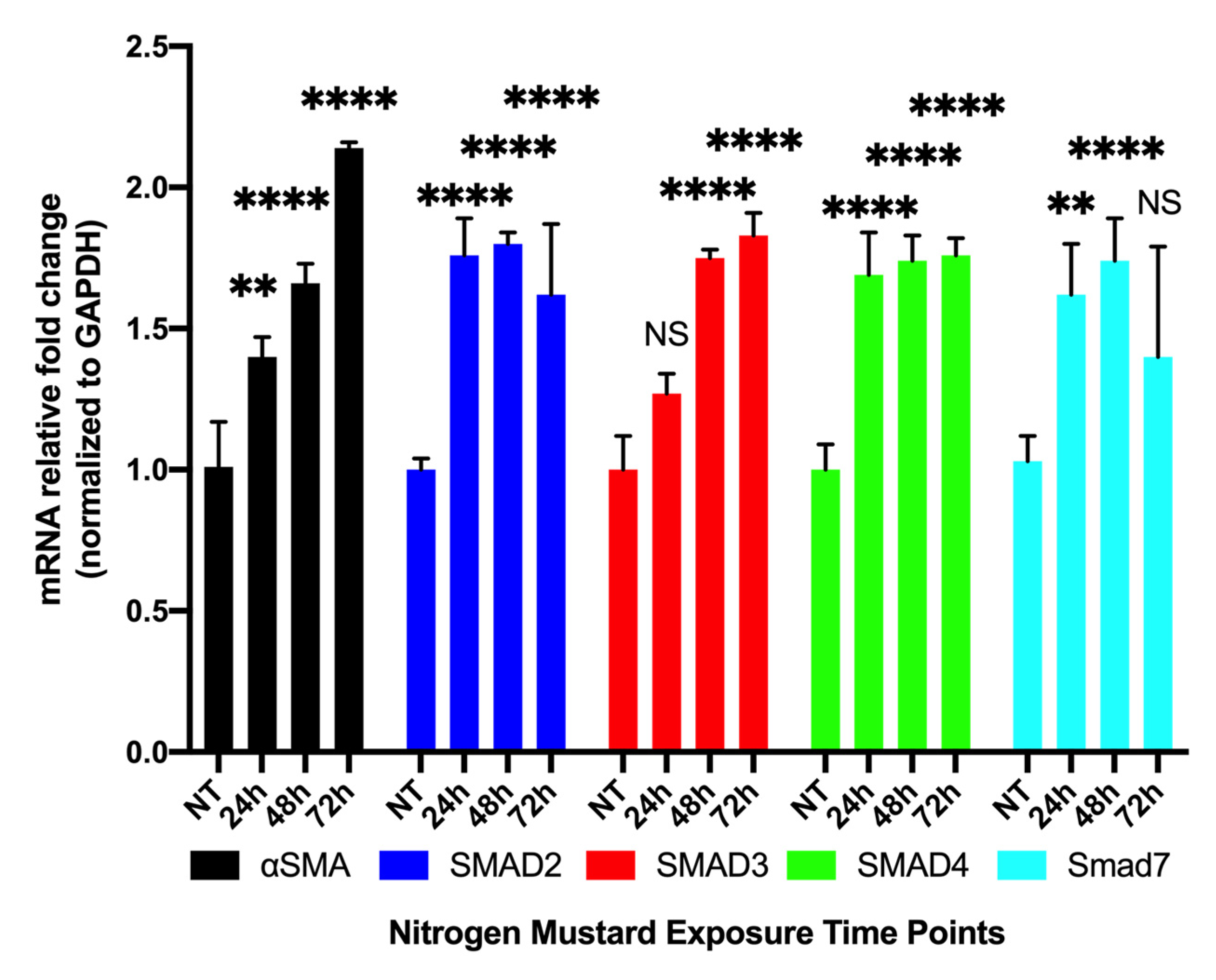
3.3.1. Time-Dependent Expression of Profibrotic Genes in hCSFs Treated with NM In Vitro
3.3.2. Time-Dependent Expression of Profibrotic Proteins in hCSFs Treated with NM In Vitro
3.4. Modulation of NM-Induced SMAD Expression by Specific Inhibitor In Vitro
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wattana, M.; Bey, T. Mustard Gas or Sulfur Mustard: An Old Chemical Agent as a New Terrorist Threat. Prehospital Disaster Med. 2009, 24, 19–29. [Google Scholar] [CrossRef] [PubMed]
- OPCW Technical Secretariat Report of the OPCW Fact-Finding Mission in Syria Regarding the Incident of 2 August 2016 as Reported in the Note Verbale of the Syrian Arab Republic Number 69 Dated 16 August 2016. 2016/12/21 2016, S14442016, 30. Available online: https://www.opcw.org/sites/default/files/documents/2018/11/s-1444-2016%28e%29.pdf (accessed on 24 August 2020).
- Smith, S.L. Toxic Legacy: Mustard Gas in the Sea Around Us. J. Law Med. Ethics 2011, 39, 34–40. [Google Scholar] [CrossRef]
- Safarinejad, M.R.; Moosavi, S.A.; Montazeri, B. Ocular Injuries Caused by Mustard Gas: Diagnosis, Treatment, and Medical Defense. Mil. Med. 2001, 166, 67–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNutt, P.; Lyman, M.; Swartz, A.; Tuznik, K.; Kniffin, D.; Whitten, K.; Milhorn, D.; Hamilton, T. Architectural and Biochemical Expressions of Mustard Gas Keratopathy: Preclinical Indicators and Pathogenic Mechanisms. PLoS ONE 2012, 7, e42837. [Google Scholar] [CrossRef] [Green Version]
- Kilic, E.; Ortatatli, M.; Sezigen, S.; Eyison, R.K.; Kenar, L. Acute Intensive Care Unit Management of Mustard Gas Victims: The Turkish Experience*. Cutan. Ocul. Toxicol. 2018, 37, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, H.; Ghazanfari, T.; Ghassemi-Broumand, M.; Javadi, M.A.; Babaei, M.; Soroush, M.R.; Yaraee, R.; Faghihzadeh, S.; Poorfarzam, S.; Owlia, P.; et al. Long-Term Ocular Consequences of Sulfur Mustard in Seriously Eye-Injured War Veterans. Cutan. Ocul. Toxicol. 2009, 28, 71–77. [Google Scholar] [CrossRef]
- Javadi, M.A.; Jafarinasab, M.R.; Feizi, S.; Karimian, F.; Negahban, K. Management of Mustard Gas-Induced Limbal Stem Cell Deficiency and Keratitis. Ophthalmology 2011, 118, 1272–1281. [Google Scholar] [CrossRef]
- Baradaran-Rafii, A.; Eslani, M.; Tseng, S.C.G. Sulfur Mustard-Induced Ocular Surface Disorders. Ocul. Surf. 2011, 9, 163–178. [Google Scholar] [CrossRef]
- Safi, S.; Javadi, M.A.; Jafarinasab, M.R.; Feizi, S.; Moghadam, M.S.; Jadidi, K.; Babaei, M.; Shirvani, A.; Baradaran-Rafii, A.; Mohammad-Rabei, H.; et al. Clinical Practice Guidelines for Prevention, Diagnosis and Management of Early and Delayed-Onset Ocular Injuries Due to Mustard Gas Exposure. J. Ophthalmic Vis. Res. 2017, 12, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Solberg, Y.; Alcalay, M.; Belkin, M. Ocular Injury by Mustard Gas. Surv. Ophthalmol. 1997, 41, 461–466. [Google Scholar] [CrossRef]
- McNutt, P.; Hamilton, T.; Nelson, M.; Adkins, A.; Swartz, A.; Lawrence, R.; Milhorn, D. Pathogenesis of Acute and Delayed Corneal Lesions after Ocular Exposure to Sulfur Mustard Vapor. Cornea 2012, 31, 280–290. [Google Scholar] [CrossRef]
- Milhorn, D.M.; Hamilton, T.A.; Nelson, M.R.; McNutt, P.M. Progression of Ocular Sulfur Mustard Injury: Development of a Model System. Ann. N. Y. Acad. Sci. 2010, 1194, 72–80. [Google Scholar] [CrossRef]
- Rowell, M.; Kehe, K.; Balszuweit, F.; Thiermann, H. The Chronic Effects of Sulfur Mustard Exposure. Toxicology 2009, 263, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Jafarinasab, M.R.; Zarei-Ghanavati, S.; Kanavi, M.R.; Karimian, F.; Soroush, M.R.; Javadi, M.A. Confocal Microscopy in Chronic and Delayed Mustard Gas Keratopathy. Cornea 2010, 29, 889–894. [Google Scholar] [CrossRef] [PubMed]
- McNutt, P.; Tuznik, K.; Nelson, M.; Adkins, A.; Lyman, M.; Glotfelty, E.; Hughes, J.; Hamilton, T. Structural, Morphological, and Functional Correlates of Corneal Endothelial Toxicity Following Corneal Exposure to Sulfur Mustard Vapor. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6735–6744. [Google Scholar] [CrossRef] [Green Version]
- Kadar, T.; Horwitz, V.; Sahar, R.; Cohen, M.; Cohen, L.; Gez, R.; Tveria, L.; Gutman, H.; Buch, H.; Fishbine, E.; et al. Delayed Loss of Corneal Epithelial Stem Cells in a Chemical Injury Model Associated with Limbal Stem Cell Deficiency in Rabbits. Curr. Eye Res. 2011, 36, 1098–1107. [Google Scholar] [CrossRef]
- Murray, V.S.G.; Volans, G.N. Management of Injuries Due to Chemical Weapons. Br. Med. J. 1991, 302, 129–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNutt, P.M.; Mohan, R.R. The Need for Improved Therapeutic Approaches to Protect the Cornea against Chemotoxic Injuries. Transl. Vis. Sci. Technol. 2020, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Meek, K.M. Corneal Collagen-Its Role in Maintaining Corneal Shape and Transparency. Biophys. Rev. 2009, 1, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Michelacci, Y.M. Collagens and Proteoglycans of the Corneal Extracellular Matrix. Braz. J. Med. Biol. Res. 2003, 36, 1037–1046. [Google Scholar] [CrossRef] [Green Version]
- Hassell, J.R.; Birk, D.E. The Molecular Basis of Corneal Transparency. Exp. Eye Res. 2010, 91, 326–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balne, P.K.; Gupta, S.; Zhang, J.; Bristow, D.; Faubion, M.; Heil, S.D.; Sinha, P.R.; Green, S.L.; Iozzo, R.V.; Mohan, R.R. The Functional Role of Decorin in Corneal Neovascularization in Vivo. Exp. Eye Res. 2021, 207, 108610. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Martin, L.M.; Sinha, N.R.; Smith, K.E.; Sinha, P.R.; Emilee, M.; Hesemann, N.P.; Mohan, R.R. Role of Inhibitor of Differentiation 3 Gene in Cellular Differentiation of Human Corneal Stromal Fibroblasts. Mol. Vis. 2020, 26, 742–756. [Google Scholar] [PubMed]
- Kamil, S.; Mohan, R.R. Corneal Stromal Wound Healing: Major Regulators and Therapeutic Targets. Ocul. Surf. 2021, 19, 290–306. [Google Scholar] [CrossRef]
- Chandrasekher, G.; Ma, X.; Lallier, T.E.; Bazan, H.E.P. Delay of Corneal Epithelial Wound Healing and Induction of Keratocyte Apoptosis by Platelet-Activating Factor. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1422–1428. [Google Scholar]
- Bonanno, J.A. Molecular Mechanisms Underlying the Corneal Endothelial Pump. Exp. Eye Res. 2012, 95, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Jester, J.V.; Moller-Pedersen, T.; Huang, J.; Sax, C.M.; Kays, W.T.; Cavangh, H.D.; Petroll, W.M.; Piatigorsky, J. The Cellular Basis of Corneal Transparency: Evidence for “Corneal Crystallins”. J. Cell Sci. 1999, 112 Pt 5, 613–622. [Google Scholar] [CrossRef]
- Ljubimov, A.V.; Saghizadeh, M. Progress in Corneal Wound Healing HHS Public Access. Prog. Retin. Eye Res. 2015, 49, 17–45. [Google Scholar] [CrossRef] [Green Version]
- Bourne, W.M. Biology of the Corneal Endothelium in Health and Disease. Eye 2003, 17, 912–918. [Google Scholar] [CrossRef]
- Sinha, N.R.; Balne, P.K.; Bunyak, F.; Hofmann, A.C.; Lim, R.R.; Rajiv, R. Collagen Matrix Perturbations in Corneal Stroma of Ossabaw Mini Pigs with Type 2 Diabetes. Mol. Vis. 2021, 27, 666–678. [Google Scholar]
- Tandon, A.; Tovey, J.C.K.; Sharma, A.; Gupta, R.; Mohan, R.R. Role of Transforming Growth Factor Beta in Corneal Function, Biology and Pathology. Curr. Mol. Med. 2012, 10, 565–578. [Google Scholar] [CrossRef]
- Wilson, S.E.; Liu, J.J.; Mohan, R.R. Stromal-Epithelial Interactions in the Cornea. Prog. Retin. Eye Res. 1999, 18, 293–309. [Google Scholar] [CrossRef]
- Wilson, S.E.; Mohan, R.R.; Mohan, R.R.; Ambro, R.; Hong, J.; Lee, J. The corneal wound healing response: Cytokine-mediated interaction of the epithelium, stroma, and inflammatory cells. Prog. Retin. Eye Res. 2001, 20, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Zielins, E.R.; Atashroo, D.A.; Maan, Z.N.; Duscher, D.; Walmsley, G.G.; Hu, M.; Senarath-Yapa, K.; McArdle, A.; Tevlin, R.; Wearda, T.; et al. Wound Healing: An Update. Regen. Med. 2014, 9, 817–830. [Google Scholar] [CrossRef]
- Chen, L.; Mongan, M.; Meng, Q.; Wang, Q.; Kao, W.; Xia, Y. Corneal Wound Healing Requires IKB Kinase β Signaling in Keratocytes. PLoS ONE 2016, 11, e0151869. [Google Scholar] [CrossRef] [Green Version]
- Bazan, H.E.P. Cellular and Molecular Events in Corneal Wound Healing: Significance of Lipid Signalling. Exp. Eye Res. 2005, 80, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Reinach, P.S.; Pokorny, K.S. The Corneal Epithelium: Clinical Relevance of Cytokine-Mediated Responses to Maintenance of Corneal Health. Arq. Bras. Oftalmol. 2008, 71, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Andrades, M.; Alonso-Pastor, L.; Mauris, J.; Cruzat, A.; Dohlman, C.H.; Argüeso, P. Establishment of a Novel in Vitro Model of Stratified Epithelial Wound Healing with Barrier Function. Sci. Rep. 2016, 6, 19395. [Google Scholar] [CrossRef] [Green Version]
- Netto, M.V.; Mohan, R.R.; Sinha, S.; Sharma, A.; Gupta, P.C.; Wilson, S.E. Effect of Prophylactic and Therapeutic. J. Refract. Surg. 2006, 562–575. [Google Scholar] [CrossRef] [Green Version]
- Netto, M.V.; Mohan, R.R.; Sinha, S.; Sharma, A.; Dupps, W.; Wilson, S.E. Stromal Haze, Myofibroblasts, and Surface Irregularity after PRK. Exp. Eye Res. 2006, 82, 788–797. [Google Scholar] [CrossRef] [Green Version]
- Mohan, R.R.; Tovey, J.C.K.; Sharma, A.; Tandon, A. Gene Therapy in the Cornea: 2005-Present. Prog. Retin. Eye Res. 2012, 31, 43–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, S.E. Corneal Wound Healing. Exp. Eye Res. 2020, 197, 108089. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Balne, P.K.; Sinha, N.R.; Martin, L.M.; Kamil, S.; Landreneau, J.R.; Gupta, S.; Rodier, J.T.; Sinha, P.R.; Hesemann, N.P.; et al. A Novel Topical Ophthalmic Formulation to Mitigate Acute Mustard Gas Keratopathy in Vivo: A Pilot Study. Transl. Vis. Sci. Technol. 2020, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Giuliano, E.A.; Sinha, N.R.; Mohan, R.R. Ocular Toxicity of Mustard Gas: A Concise Review. Toxicol. Lett. 2021, 343, 21–27. [Google Scholar] [CrossRef]
- Mohan, R.R.; Martin, L.M.; Sinha, N.R. Novel Insights into Gene Therapy in the Cornea. Exp. Eye Res. 2020, 202, 108361. [Google Scholar] [CrossRef]
- Mohan, R.R.; Sharma, A.; Cebulko, T.C.; Tandon, A. Vector Delivery Technique Affects Gene Transfer in the Cornea in Vivo. Mol. Vis. 2010, 16, 2494–2501. [Google Scholar]
- Wang, T.; Zhou, X.; Yu, Y.; Zhu, J.; Dai, J.; Qu, X.; Le, Q.; Chu, R. Inhibition of Corneal Fibrosis by Smad7 in Rats after Photorefractive Keratectomy. Chin. Med. J. 2013, 126, 1445–1450. [Google Scholar]
- Wang, T.; Zhou, X.; Yu, Y.; Dai, J.; Qu, X.; LE, Q.; Chu, R. Expression of Smad7 Inhibits Fibrogenic Responses of Keratocytes to Transforming Growth Factor Β2. Chin. Med. J. 2011, 124, 1988–1993. [Google Scholar]
- Saika, S.; Ikeda, K.; Yamanaka, O.; Miyamoto, T.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Nakajima, Y.; Kao, W.W.-Y.; et al. Expression of Smad7 in Mouse Eyes Accelerates Healing of Corneal Tissue after Exposure to Alkali. Am. J. Pathol. 2005, 166, 1405–1418. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Rodier, J.T.; Sharma, A.; Giuliano, E.A.; Sinha, P.R.; Hesemann, N.P.; Ghosh, A.; Mohan, R.R. Targeted AAV5-Smad7 Gene Therapy Inhibits Corneal Scarring in Vivo. PLoS ONE 2017, 12, e0172928. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Sinha, N.R.; Siddiqui, S.; Mohan, R.R. Role of 5′TG3′-Interacting Factors (TGIFs) in Vorinostat (HDAC Inhibitor)-Mediated Corneal Fibrosis Inhibition. Mol. Vis. 2015, 21, 974–984. [Google Scholar] [PubMed]
- Marlo, T.L.; Giuliano, E.A.; Tripathi, R.; Sharma, A.; Mohan, R.R. Altering Equine Corneal Fibroblast Differentiation through Smad Gene Transfer. Vet. Ophthalmol. 2018, 21, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Poojya Anantharam, P.D. Sulfur Mustard Mitigation by Turbo Eye Drops (Chronic Efficacy and Safety Study). 2021. Available online: https://www.mriglobal.org/fda-approved-mustard-gas-burn-product/ (accessed on 21 December 2020).
- Balne, P.K.; Sinha, N.R.; Hofmann, A.C.; Martin, L.M.; Mohan, R.R. Characterization of Hydrogen Sulfide Toxicity to Human Corneal Stromal Fibroblasts. Ann. N. Y. Acad. Sci. 2020, 1480, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.R.; Morgan, B.R.; Anumanthan, G.; Sharma, A.; Chaurasia, S.S.; Rieger, F.G. Characterization of Inhibitor of Differentiation (Id) Proteins in Human Cornea. Exp. Eye Res. 2016, 146, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Lim, R.R.; Tan, A.; Liu, Y.-C.C.; Barathi, V.A.; Mohan, R.R.; Mehta, J.S.; Chaurasia, S.S. ITF2357 Transactivates Id3 and Regulate TGFβ/BMP7 Signaling Pathways to Attenuate Corneal Fibrosis. Sci. Rep. 2016, 6, 20841. [Google Scholar] [CrossRef] [Green Version]
- Sinha, N.R.; Tripathi, R.; Balne, P.K.; Green, S.L.; Sinha, P.R.; Bunyak, F.; Giuliano, E.A.; Chaurasia, S.S.; Mohan, R.R. Time-Dependent in Situ Structural and Cellular Aberrations in Rabbit Cornea in Vivo after Mustard Gas Exposure. Exp. Eye Res. 2022, 224, 109247. [Google Scholar] [CrossRef]
- Gupta, S.; Fink, M.K.; Martin, L.M.; Sinha, P.R.; Rodier, J.T.; Sinha, N.R.; Hesemann, N.P.; Chaurasia, S.S.; Mohan, R.R. A Rabbit Model for Evaluating Ocular Damage from Acrolein Toxicity in Vivo. Ann. N. Y. Acad. Sci. 2020, 1480, 233–245. [Google Scholar] [CrossRef]
- Jester, J.V.; Petroll, W.M.; Barry, P.A.; Cavanagh, H.D. Expression of Alpha-Smooth Muscle (Alpha-SM) Actin during Corneal Stromal Wound Healing. Investig. Ophthalmol. Vis. Sci. 1995, 36, 809–819. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Species | Accession # | Forward Sequence | Reverse Sequence |
|---|---|---|---|---|
| GAPDH | rabbit, human | NM_002046.3 | GCCTCAAGATCATCAGCAATGCCT | TGTGGTCATGAGTCCTTCCACGAT |
| αSMA | rabbit, human | NM_001613 | AAGATCCTGACTGAGCGT | CAAAGTCCAGAGCGACATAG |
| Smad 2 | human | XM_002713521.3 | CGAAACGCCACAGTAGAA | GCACTATCACTTAGGCACTC |
| Smad 3 | human | NM_001145102.2 | CCCAGAGCAATATTCCAGAG | GTCCATGCTGTGGTTCAT |
| Smad 4 | human | XM_002713541.3 | TGGATGTTCAGGTAGGAGAG | CTGTGGACATTGGAGAGTTG |
| Smad 7 | human | XM_017344169.1 | ATCACCTTAGCCGACTCT | GCACAGCATCTGGACAAT |
| TGFβ1 | rabbit, human | NM_003242 | CGACTACTACGCCAAGGA | GAGAGCAACACGGGTTCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, N.R.; Tripathi, R.; Balne, P.K.; Suleiman, L.; Simkins, K.; Chaurasia, S.S.; Mohan, R.R. Mustard Gas Exposure Actuates SMAD2/3 Signaling to Promote Myofibroblast Generation in the Cornea. Cells 2023, 12, 1533. https://doi.org/10.3390/cells12111533
Sinha NR, Tripathi R, Balne PK, Suleiman L, Simkins K, Chaurasia SS, Mohan RR. Mustard Gas Exposure Actuates SMAD2/3 Signaling to Promote Myofibroblast Generation in the Cornea. Cells. 2023; 12(11):1533. https://doi.org/10.3390/cells12111533
Chicago/Turabian StyleSinha, Nishant R., Ratnakar Tripathi, Praveen K. Balne, Laila Suleiman, Katherine Simkins, Shyam S. Chaurasia, and Rajiv R. Mohan. 2023. "Mustard Gas Exposure Actuates SMAD2/3 Signaling to Promote Myofibroblast Generation in the Cornea" Cells 12, no. 11: 1533. https://doi.org/10.3390/cells12111533