
Sex and Brain: The Role of Sex Chromosomes and Hormones in Brain Development and Parkinson’s Disease

Abstract
:1. Sex Hormones and Sex Chromosomes as Players in the Sexual Differentiation of the Mammalian Brain
1.1. Steroid Hormones in the Central Nervous System

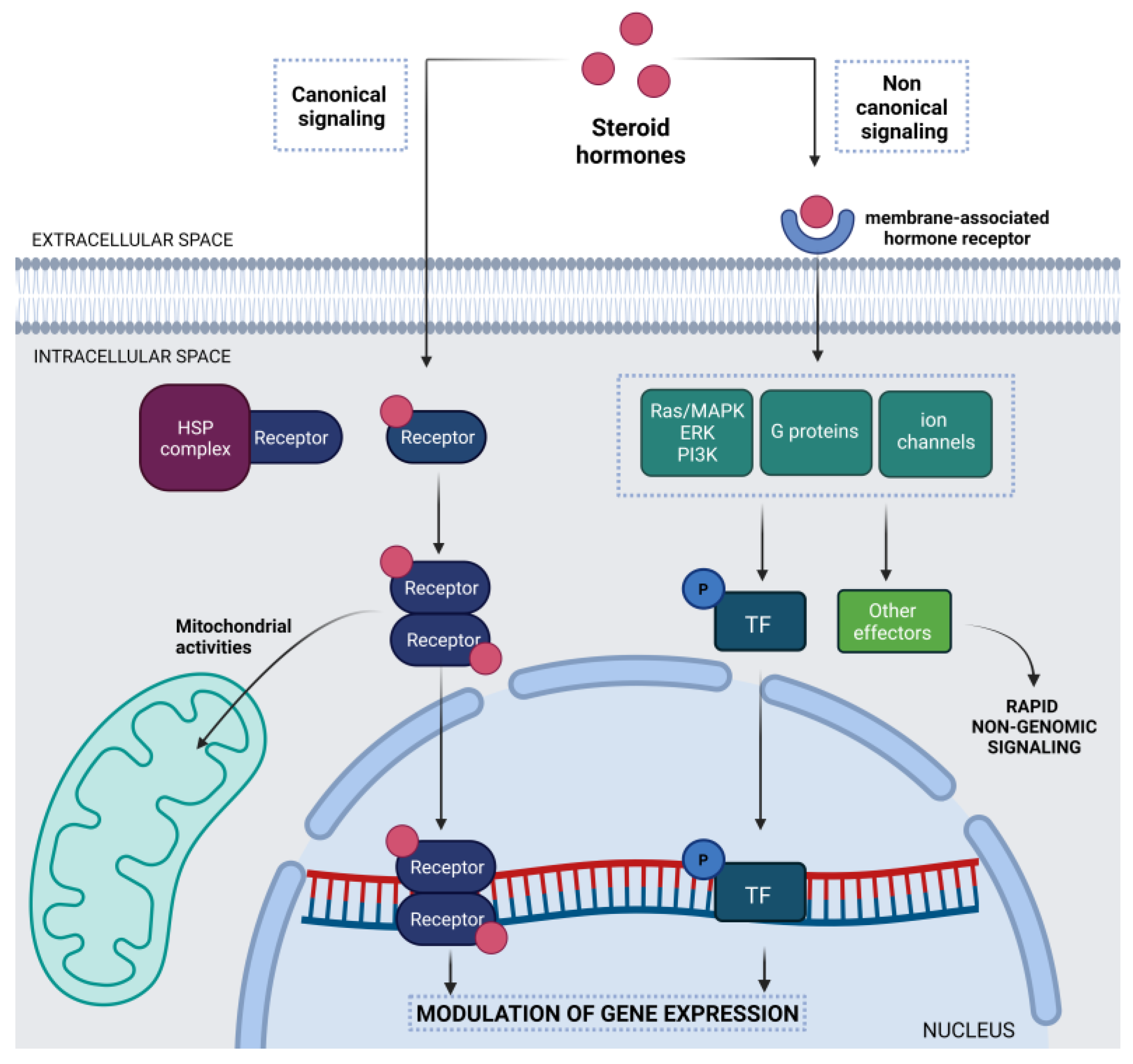{kind=link}
{kind=link}
| Steroid Hormone | Activated Receptor | Functions | Targets | References |
|---|---|---|---|---|
| Estrogen | ERα, ERβ | Neuroprotection | Antiapoptotic genes | [20,21,22,23] |
| Proliferative genes | ||||
| Progesterone | PR-A, PR-B | Neuroprotection | Promyelinization genes | [24,25] |
| Anti-inflammatory genes | ||||
| Testosterone | AR | Neuroprotection | Remyelination process by oligodendrocytes | [26,27,28] |
| Astrocyte recruitment | ||||
| Sex phenotype | Development and maintenance of male phenotype in CNS | [29,30,31] |
| Steroid Hormone | Activated Receptor | Functions | References |
|---|---|---|---|
| Estrogen | ERα, ERβ, mERs, GPER1 | Oligodendrocyte differentiation, survival, and function | [32] |
| Spine plasticity | [33,34,35,36] | ||
| Behavior | |||
| Progesterone | PR-A, PR-B, mPRs, PGRMC1 | Female reproductive behavior | [37,38,39,40] |
| Cerebellum cortical formation | |||
| Neuroprotection | |||
| Testosterone | AR, GPCRs | Sexual differentiation | [41,42,43,44,45,46] |
| Reproductive and aggressive behaviors | |||
| Neuroendocrine response |
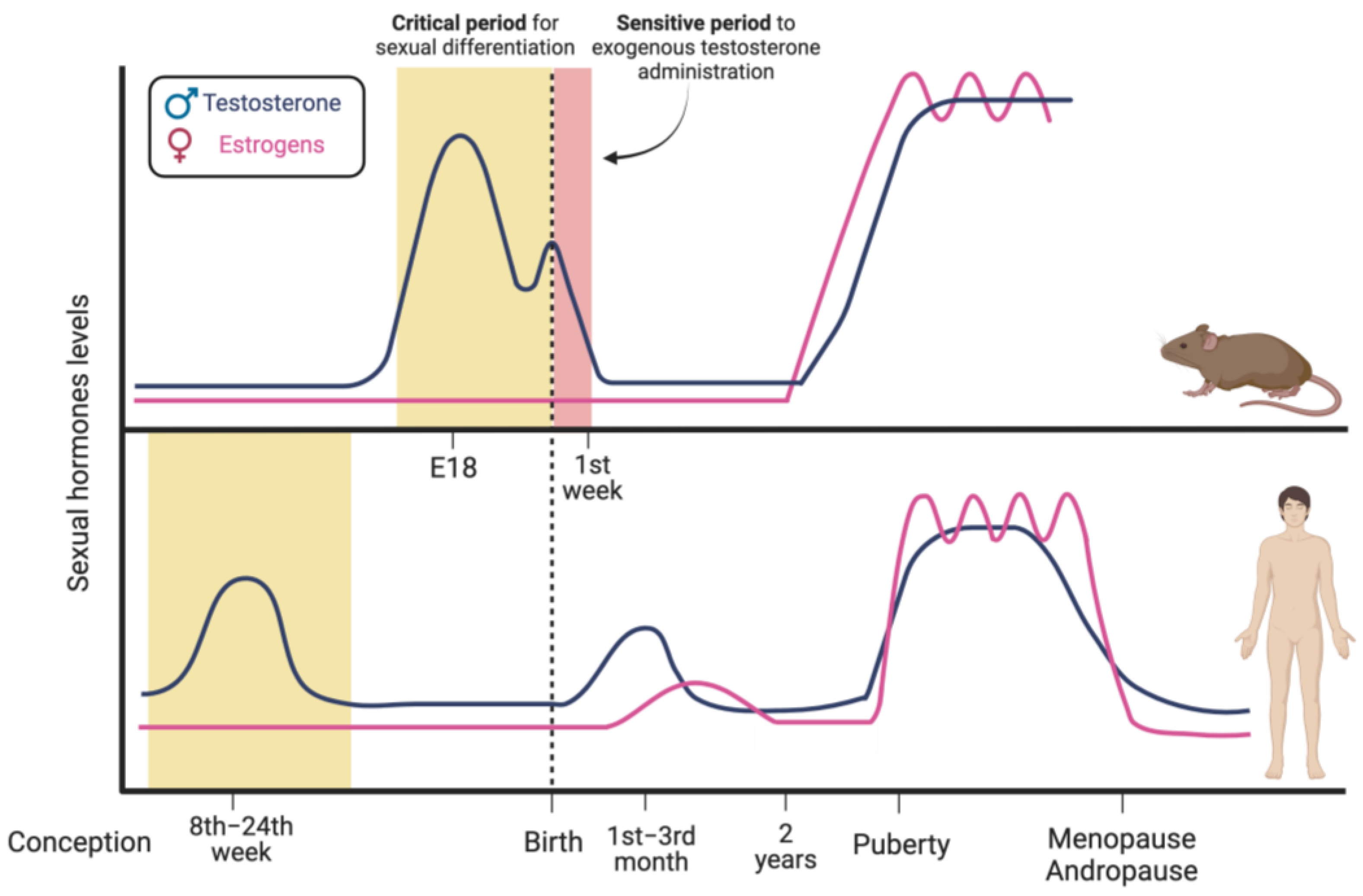
1.2. Sex Hormones’ Modulation of Brain Sexual Differentiation
1.3. Sex Hormone-Dependent Brain Sexual Dimorphism
1.4. Sex Chromosomes’ Impact on Brain Sexual Differentiation
1.5. Epigenetic Regulation of Brain Sexual Differentiation
2. Sex-Related Differences in Neurodegeneration
2.1. Focus on Parkinson’s Disease
2.2. Sex-Related Differences in Microglia and Their Implication on Neuroinflammation in PD
2.3. Sex Bias in Parkinson’s Disease and the Role of Sex Hormones
2.3.1. Estrogens
2.3.2. Progesterone
2.3.3. Androgens
2.4. Effects of Estrogen-Based Pharmacological Treatments on Parkinson’s Disease
2.5. Contribution of Genes Located on the Sex Chromosomes to Parkinson’s Disease Etiology
2.6. Sex Differences in Immortalized and Primary Cells Relevant to Parkinson’s Disease Research
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Naftolin, F.; Ryan, K.J.; Davies, I.J. The Formation of Estrogens by Central Neuroendocrine Tissues. Recent Prog. Horm. Res. 1975, 31, 295–319. [Google Scholar] [CrossRef]
- Arnold, A.P. Sex Chromosomes and Brain Gender. Nat. Rev. Neurosci. 2004, 5, 701–708. [Google Scholar] [CrossRef]
- Arnold, A.P.; Xu, J.; Grisham, W.; Chen, X.; Kim, Y.H.; Itoh, Y. Minireview: Sex Chromosomes and Brain Sexual Differentiation. Endocrinology 2004, 145, 1057–1062. [Google Scholar] [CrossRef]
- Menger, Y.; Bettscheider, M.; Murgatroyd, C.; Spengler, D. Sex Differences in Brain Epigenetics. Epigenomics 2010, 2, 807–821. [Google Scholar] [CrossRef]
- Mccarthy, M.M.; Nugent, B.M. Epigenetic Contributions to Hormonally-Mediated Sexual Differentiation of the Brain. J. Neuroendocrinol. 2013, 25, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, C.; Karali, K.; Fodelianaki, G.; Gravanis, A.; Chavakis, T.; Charalampopoulos, I.; Alexaki, V.I. Neurosteroids as Regulators of Neuroinflammation. Front. Neuroendocrinol. 2019, 55, 788. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Waller-Evans, H. Biosynthesis and Signalling Functions of Central and Peripheral Nervous System Neurosteroids in Health and Disease. Essays Biochem. 2020, 64, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Baulieu, E.E. Neurosteroids: A Novel Function of the Brain. Psychoneuroendocrinology 1998, 23, 963–987. [Google Scholar] [CrossRef]
- Do Rego, J.L.; Seong, J.Y.; Burel, D.; Leprince, J.; Luu-The, V.; Tsutsui, K.; Tonon, M.C.; Pelletier, G.; Vaudry, H. Neurosteroid Biosynthesis: Enzymatic Pathways and Neuroendocrine Regulation by Neurotransmitters and Neuropeptides. Front. Neuroendocrinol. 2009, 30, 259–301. [Google Scholar] [CrossRef]
- Diotel, N.; Charlier, T.D.; Lefebvre d’Hellencourt, C.; Couret, D.; Trudeau, V.L.; Nicolau, J.C.; Meilhac, O.; Kah, O.; Pellegrini, E. Steroid Transport, Local Synthesis, and Signaling within the Brain: Roles in Neurogenesis, Neuroprotection, and Sexual Behaviors. Front. Neurosci. 2018, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S. Neurosteroids. Endogenous Role in the Human Brain and Therapeutic Potentials. Prog. Brain Res. 2010, 186, 113–137. [Google Scholar] [CrossRef]
- Akk, G.; Covey, D.F.; Evers, A.S.; Steinbach, J.H.; Zorumski, C.F.; Mennerick, S. The Influence of the Membrane on Neurosteroid Actions at GABAA Receptors. Psychoneuroendocrinology 2009, 34, 20. [Google Scholar] [CrossRef]
- Kawata, M. Roles of Steroid Hormones and Their Receptors in Structural Organization in the Nervous System. Neurosci. Res. 1995, 24, 1–46. [Google Scholar] [CrossRef]
- Panzica, G.C.; Melcangi, R.C. The Endocrine Nervous System: Source and Target for Neuroactive Steroids. Brain Res. Rev. 2008, 57, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, D. Hormone-Driven Mechanisms in the Central Nervous System Facilitate the Analysis of Mammalian Behaviours. J. Endocrinol. 2005, 184, 447–453. [Google Scholar] [CrossRef]
- Tetel, M.J. Nuclear Receptor Coactivators: Essential Players for Steroid Hormone Action in the Brain and in Behaviour. J. Neuroendocrinol. 2009, 21, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Takahashi, H.; Matsuda, K.; Nishi, M.; Takanami, K.; Ogoshi, M.; Sakamoto, T.; Kawata, M. Rapid Signaling of Steroid Hormones in the Vertebrate Nervous System. Front. Biosci. 2012, 17, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Psarra, A.M.G.; Solakidi, S.; Sekeris, C.E. The Mitochondrion as a Primary Site of Action of Steroid and Thyroid Hormones: Presence and Action of Steroid and Thyroid Hormone Receptors in Mitochondria of Animal Cells. Mol. Cell. Endocrinol. 2006, 246, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Gaignard, P.; Liere, P.; Thérond, P.; Schumacher, M.; Slama, A.; Guennoun, R. Role of Sex Hormones on Brain Mitochondrial Function, with Special Reference to Aging and Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 406. [Google Scholar] [CrossRef]
- Vegeto, E.; Villa, A.; Della Torre, S.; Crippa, V.; Rusmini, P.; Cristofani, R.; Galbiati, M.; Maggi, A.; Poletti, A. The Role of Sex and Sex Hormones in Neurodegenerative Diseases. Endocr. Rev. 2020, 41, 273–319. [Google Scholar] [CrossRef]
- Meda, C.; Vegeto, E.; Pollio, G.; Ciana, P.; Patrone, C.; Pellicciari, C.; Maggi, A. Oestrogen Prevention of Neural Cell Death Correlates with Decreased Expression of MRNA for the Pro-Apoptotic Protein Nip-2. J. Neuroendocrinol. 2000, 12, 1051–1059. [Google Scholar] [CrossRef]
- Brusadelli, A.; Sialino, H.; Piepoli, T.; Pollio, G.; Maggi, A. Expression of the Estrogen-Regulated Gene Nip2 during Rat Brain Maturation. Int. J. Dev. Neurosci. 2000, 18, 317–320. [Google Scholar] [CrossRef]
- Maggi, A.; Ciana, P.; Belcredito, S.; Vegeto, E. Estrogens in the Nervous System: Mechanisms and Nonreproductive Functions. Annu. Rev. Physiol. 2004, 66, 291–313. [Google Scholar] [CrossRef]
- De Nicola, A.F.; Labombarda, F.; Deniselle, M.C.G.; Gonzalez, S.L.; Garay, L.; Meyer, M.; Gargiulo, G.; Guennoun, R.; Schumacher, M. Progesterone Neuroprotection in Traumatic CNS Injury and Motoneuron Degeneration. Front. Neuroendocrinol. 2009, 30, 173–187. [Google Scholar] [CrossRef] [PubMed]
- González, S.L.; Coronel, M.F.; Raggio, M.C.; Labombarda, F. Progesterone Receptor-Mediated Actions and the Treatment of Central Nervous System Disorders: An up-Date of the Known and the Challenge of the Unknown. Steroids 2020, 153, 108525. [Google Scholar] [CrossRef] [PubMed]
- Fanaei, H.; Karimian, S.M.; Sadeghipour, H.R.; Hassanzade, G.; Kasaeian, A.; Attari, F.; Khayat, S.; Ramezani, V.; Javadimehr, M. Testosterone Enhances Functional Recovery after Stroke through Promotion of Antioxidant Defenses, BDNF Levels and Neurogenesis in Male Rats. Brain Res. 2014, 1558, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Bielecki, B.; Mattern, C.; Ghoumari, A.M.; Javaid, S.; Smietanka, K.; Ghanem, C.A.; Mhaouty-Kodja, S.; Ghandour, M.S.; Baulieu, E.E.; Franklin, R.J.M.; et al. Unexpected Central Role of the Androgen Receptor in the Spontaneous Regeneration of Myelin. Proc. Natl. Acad. Sci. USA 2016, 113, 14829–14834. [Google Scholar] [CrossRef]
- Hussain, R.; Ghoumari, A.M.; Bielecki, B.; Steibel, J.; Boehm, N.; Liere, P.; MacKlin, W.B.; Kumar, N.; Habert, R.; Mhaouty-Kodja, S.; et al. The Neural Androgen Receptor: A Therapeutic Target for Myelin Repair in Chronic Demyelination. Brain 2013, 136, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Zorrilla Veloz, R.I.; McKenzie, T.; Palacios, B.E.; Hu, J. Nuclear Hormone Receptors in Demyelinating Diseases. J. Neuroendocrinol. 2022, 34, 1–15. [Google Scholar] [CrossRef]
- Sato, T.; Matsumoto, T.; Kawano, H.; Watanabe, T.; Uematsu, Y.; Sekine, K.; Fukuda, T.; Aihara, K.I.; Krust, A.; Yamada, T.; et al. Brain Masculinization Requires Androgen Receptor Function. Proc. Natl. Acad. Sci. USA 2004, 101, 1673–1678. [Google Scholar] [CrossRef]
- Perrin, J.S.; Hervé, P.Y.; Leonard, G.; Perron, M.; Pike, G.B.; Pitiot, A.; Richer, L.; Veillette, S.; Pausova, Z.; Paus, T. Growth of White Matter in the Adolescent Brain: Role of Testosterone and Androgen Receptor. J. Neurosci. 2008, 28, 9519–9524. [Google Scholar] [CrossRef] [PubMed]
- Hirahara, Y.; Matsuda, K.I.; Gao, W.; Arvanitis, D.N.; Kawata, M.; Boggs, J.M. The Localization and Non-Genomic Function of the Membrane-Associated Estrogen Receptor in Oligodendrocytes. Glia 2009, 57, 153–165. [Google Scholar] [CrossRef]
- Mermelstein, P.G.; Micevych, P.E. Nervous System Physiology Regulated by Membrane Estrogen Receptors. Rev. Neurosci. 2008, 19, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, N.; Pfaff, D.W. Non-Genomic Actions of Estrogens and Their Interaction with Genomic Actions in the Brain. Front. Neuroendocrinol. 2008, 29, 238–257. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.; Akama, K.; Alves, S.; Brake, W.G.; Bulloch, K.; Lee, S.; Li, C.; Yuen, G.; Milner, T.A. Tracking the Estrogen Receptor in Neurons: Implications for Estrogen-Induced Synapse Formation. Proc. Natl. Acad. Sci. USA 2001, 98, 7093–7100. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Rønnekleiv, O.K.; Kelly, M.J. Modulation of Hypothalamic Neuronal Activity through a Novel G-Protein-Coupled Estrogen Membrane Receptor. Steroids 2008, 73, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Su, C.; Ng, S. Non-Genomic Mechanisms of Progesterone Action in the Brain. Front. Neurosci. 2013, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Krebs, C.J.; Jarvis, E.D.; Chan, J.; Lydon, J.P.; Ogawa, S.; Pfaff, D.W. A Membrane-Associated Progesterone-Binding Protein, 25-Dx, Is Regulated by Progesterone in Brain Regions Involved in Female Reproductive Behaviors. Proc. Natl. Acad. Sci. USA 2000, 97, 12816–12821. [Google Scholar] [CrossRef]
- Sakamoto, H.; Ukena, K.; Takemori, H.; Okamoto, M.; Kawata, M.; Tsutsui, K. Expression and Localization of 25-Dx, a Membrane-Associated Putative Progesterone-Binding Protein, in the Developing Purkinje Cell. Neuroscience 2004, 126, 325–334. [Google Scholar] [CrossRef]
- Labombarda, F.; Meffre, D.; Delespierre, B.; Krivokapic-Blondiaux, S.; Chastre, A.; Thomas, P.; Pang, Y.; Lydon, J.P.; Gonzalez, S.L.; De Nicola, A.F.; et al. Membrane Progesterone Receptors Localization in the Mouse Spinal Cord. Neuroscience 2010, 166, 94–106. [Google Scholar] [CrossRef]
- Deng, Q.; Zhang, Z.; Wu, Y.; Yu, W.Y.; Zhang, J.; Jiang, Z.M.; Zhang, Y.; Liang, H.; Gui, Y.T. Non-Genomic Action of Androgens Is Mediated by Rapid Phosphorylation and Regulation of Androgen Receptor Trafficking. Cell. Physiol. Biochem. 2017, 43, 223–236. [Google Scholar] [CrossRef]
- Michels, G.; Hoppe, U.C. Rapid Actions of Androgens. Front. Neuroendocrinol. 2008, 29, 182–198. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.A.; Jordan, C.L.; Breedlove, S.M. Sexual Differentiation of the Vertebrate Nervous System. Nat. Neurosci. 2004, 7, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Sakamoto, H.; Kawata, M. Androgen Action in the Brain and Spinal Cord for the Regulation of Male Sexual Behaviors. Curr. Opin. Pharmacol. 2008, 8, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Breedlove, S.M.; Arnold, A.P. Hormonal Control of a Developing Neuromuscular System. I. Complete Demasculinization of the Male Rat Spinal Nucleus of the Bulbocavernosus Using the Anti-Androgen Flutamide. J. Neurosci. 1983, 3, 417–423. [Google Scholar] [CrossRef]
- Breedlove, S.M.; Arnold, A.P. Hormonal Control of a Developing Neuromuscular System. II. Sensitive Periods for the Androgen-Induced Masculinization of the Rat Spinal Nucleus of the Bulbocavernosus. J. Neurosci. 1983, 3, 424–432. [Google Scholar] [CrossRef]
- Tanaka, S.S.; Nishinakamura, R. Regulation of Male Sex Determination: Genital Ridge Formation and Sry Activation in Mice. Cell. Mol. Life Sci. 2014, 71, 4781–4802. [Google Scholar] [CrossRef]
- She, Z.Y.; Yang, W.X. Sry and SoxE Genes: How They Participate in Mammalian Sex Determination and Gonadal Development? Semin. Cell Dev. Biol. 2017, 63, 13–22. [Google Scholar] [CrossRef]
- Spiller, C.; Koopman, P.; Bowles, J. Sex Determination in the Mammalian Germline. Annu. Rev. Genet. 2017, 51, 265–287. [Google Scholar] [CrossRef]
- Stévant, I.; Nef, S. Genetic Control of Gonadal Sex Determination and Development. Trends Genet. 2019, 35, 346–358. [Google Scholar] [CrossRef]
- Lundgaard Riis, M.; Jørgensen, A. Deciphering Sex-Specific Differentiation of Human Fetal Gonads: Insight From Experimental Models. Front. Cell Dev. Biol. 2022, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Weisz, J.; Ward, I.L. Plasma Testosterone and Progesterone Titers of Pregnant Rats, Their Male and Female Fetuses, and Neonatal Offspring. Endocrinology 1980, 106, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Corbier, P.; Edwards, D.A.; Roffi, J. The Neonatal Testosterone Surge: A Comparative Study. Arch. Physiol. Biochem. 1992, 100, 127–131. [Google Scholar] [CrossRef]
- Arnold, A.P.; Chen, X. What Does the “Four Core Genotypes” Mouse Model Tell Us about Sex Differences in the Brain and Other Tissues? Front. Neuroendocrinol. 2009, 30, 1–9. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Lieberburg, I.; Chaptal, C.; Krey, L.C. Aromatization: Important for Sexual Differentiation of the Neonatal Rat Brain. Horm. Behav. 1977, 9, 249–263. [Google Scholar] [CrossRef]
- Lenz, K.M.; Nugent, B.M.; McCarthy, M.M. Sexual Differentiation of the Rodent Brain: Dogma and Beyond. Front. Neurosci. 2012, 6, 26. [Google Scholar] [CrossRef]
- Negri-Cesi, P.; Poletti, A.; Celotti, F. Metabolism of Steroids in the Brain: A New Insight into the Role of 5α-Reductase and Aromatase in Brain Differentiation and Functions. J. Steroid Biochem. Mol. Biol. 1996, 58, 455–466. [Google Scholar] [CrossRef]
- Celotti, F.; Negri-Cesi, P.; Poletti, A. Steroid Metabolism in the Mammalian Brain: 5Alpha-Reduction and Aromatization. Brain Res. Bull. 1997, 44, 365–375. [Google Scholar] [CrossRef]
- Azcoitia, I.; Mendez, P.; Garcia-Segura, L.M. Aromatase in the Human Brain. Androgens 2021, 2, 189–202. [Google Scholar] [CrossRef]
- Tsutsui, K. Neurosteroid Biosynthesis and Action during Cerebellar Development. Cerebellum 2012, 11, 414–415. [Google Scholar] [CrossRef]
- Kudwa, A.E.; Bodo, C.; Gustafsson, J.Å.; Rissman, E.F. A Previously Uncharacterized Role for Estrogen Receptor β: Defeminization of Male Brain and Behavior. Proc. Natl. Acad. Sci. USA 2005, 102, 4608–4612. [Google Scholar] [CrossRef]
- Bakker, J.; De Mees, C.; Douhard, Q.; Balthazart, J.; Gabant, P.; Szpirer, J.; Szpirer, C. Alpha-Fetoprotein Protects the Developing Female Mouse Brain from Masculinization and Defeminization by Estrogens. Nat. Neurosci. 2006, 9, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; McCarthy, M.M. Steroid-Induced Sexual Differentiation of the Developing Brain: Multiple Pathways, One Goal. J. Neurochem. 2008, 105, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Bakker, J.; Honda, S.I.; Harada, N.; Balthazart, J. The Aromatase Knock-out Mouse Provides New Evidence That Estradiol Is Required during Development in the Female for the Expression of Sociosexual Behaviors in Adulthood. J. Neurosci. 2002, 22, 9104–9112. [Google Scholar] [CrossRef] [PubMed]
- Brock, O.; Baum, M.J.; Bakker, J. The Development of Female Sexual Behavior Requires Prepubertal Estradiol. J. Neurosci. 2011, 31, 5574–5578. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M.; Arnold, A.P. Reframing Sexual Differentiation of the Brain. Nat. Neurosci. 2011, 14, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Sisk, C.L.; Zehr, J.L. Pubertal Hormones Organize the Adolescent Brain and Behavior. Front. Neuroendocrinol. 2005, 26, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Reyes, F.I.; Boroditsky, R.S.; Winter, J.S.D.; Faiman, C. Studies on Human Sexual Development. II. Fetal and Maternal Serum Gonadotropin and Sex Steroid Concentrations. J. Clin. Endocrinol. Metab. 1974, 38, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Word, R.A.; George, F.W.; Wilson, J.D.; Carr, B.R. Testosterone Synthesis and Adenylate Cyclase Activity in the Early Human Fetal Testis Appear to Be Independent of Human Chorionic Gonadotropin Control. J. Clin. Endocrinol. Metab. 1989, 69, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Bao, A.M.; Swaab, D.F. Sexual Differentiation of the Human Brain: Relation to Gender Identity, Sexual Orientation and Neuropsychiatric Disorders. Front. Neuroendocrinol. 2011, 32, 214–226. [Google Scholar] [CrossRef]
- Kuiri-Hänninen, T.; Sankilampi, U.; Dunkel, L. Activation of the Hypothalamic-Pituitary-Gonadal Axis in Infancy: Minipuberty. Horm. Res. Paediatr. 2014, 82, 73–80. [Google Scholar] [CrossRef]
- Muller, M.; den Tonkelaar, I.; Thijssen, J.H.H.; Grobbee, D.E.; van der Schouw, Y.T. Endogenous Sex Hormones in Men Aged 40-80 Years. Eur. J. Endocrinol. 2003, 149, 583–589. [Google Scholar] [CrossRef]
- Morishima, A.; Grumbach, M.M.; Simpson, E.R.; Fisher, C.; Qin, K. Aromatase Deficiency in Male and Female Siblings Caused by a Novel Mutation and the Physiological Role of Estrogens. J. Clin. Endocrinol. Metab. 1995, 80, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Carani, C.; Rochira, V.; Faustini-Fustini, M.; Balestrieri, A.; Granata, A.R.M. Role of Oestrogen in Male Sexual Behaviour: Insights from the Natural Model of Aromatase Deficiency. Clin. Endocrinol. 1999, 51, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.E.; Boyd, J. Estrogen Resistance Caused by a Mutation in the Estrogen-Receptor Gene in a Man. N. Engl. J. Med. 1994, 331, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, A.B.; Migeon, C.J.; Meyer-Bahlburg, H.F.L.; Gearhart, J.P.; Berkovitz, G.D.; Brown, T.R.; Money, J. Complete Androgen Insensitivity Syndrome: Long-Term Medical, Surgical, and Psychosexual Outcome. J. Clin. Endocrinol. Metab. 2000, 85, 2664–2669. [Google Scholar] [CrossRef]
- Hines, M.; Ahmed, S.F.; Hughes, I.A. Psychological Outcomes and Gender-Related Development in Complete Androgen Insensitivity Syndrome. Arch. Sex. Behav. 2003, 32, 93–101. [Google Scholar] [CrossRef]
- VanRyzin, J.W.; Marquardt, A.E.; Pickett, L.A.; McCarthy, M.M. Microglia and Sexual Differentiation of the Developing Brain: A Focus on Extrinsic Factors. Glia 2019, 68, 1100–1113. [Google Scholar] [CrossRef]
- Davis, E.C.; Popper, P.; Gorski, R.A. The Role of Apoptosis in Sexual Differentiation of the Rat Sexually Dimorphic Nucleus of the Preoptic Area. Brain Res. 1996, 734, 10–18. [Google Scholar] [CrossRef]
- Gegenhuber, B.; Wu, M.V.; Bronstein, R.; Tollkuhn, J. Gene Regulation by Gonadal Hormone Receptors Underlies Brain Sex Differences. Nature 2022, 606, 153–159. [Google Scholar] [CrossRef]
- Simerly, R.B. Wired for Reproduction: Organization and Development of Sexually Dimorphic Circuits in the Mammalian Forebrain. Annu. Rev. Neurosci. 2002, 25, 507–536. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.C.; Shryne, J.E.; Gorski, R.A. Structural Sexual Dimorphisms in the Anteroventral Periventricular Nucleus of the Rat Hypothalamus Are Sensitive to Gonadal Steroids Perinatally, but Develop Peripubertally. Neuroendocrinology 1996, 63, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Tobet, S.; Knoll, J.G.; Hartshorn, C.; Aurand, E.; Stratton, M.; Kumar, P.; Searcy, B.; McClellan, K. Brain Sex Differences and Hormone Influences: A Moving Experience? J. Neuroendocrinol. 2009, 21, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Pérez, S.E.; Chen, E.Y.; Mufson, E.J. Distribution of Estrogen Receptor Alpha and Beta Immunoreactive Profiles in the Postnatal Rat Brain. Dev. Brain Res. 2003, 145, 117–139. [Google Scholar] [CrossRef]
- Wilson, M.E.; Rosewell, K.L.; Kashon, M.L.; Shughrue, P.J.; Merchenthaler, I.; Wise, P.M. Age Differentially Influences Estrogen Receptor-α (ERα) and Estrogen Receptor-β (ERβ) Gene Expression in Specific Regions of the Rat Brain. Mech. Ageing Dev. 2002, 123, 593–601. [Google Scholar] [CrossRef]
- Gillies, G.E.; McArthur, S. Estrogen Actions in the Brain and the Basis for Differential Action in Men and Women: A Case for Sex-Specific Medicines. Pharmacol. Rev. 2010, 62, 155–198. [Google Scholar] [CrossRef]
- Ogawa, S.; Eng, V.; Taylor, J.; Lubahn, D.B.; Korach, K.S.; Pfaff, D.W. Roles of Estrogen Receptor-α Gene Expression in Reproduction-Related Behaviors in Female Mice. Endocrinology 1998, 139, 5070–5081. [Google Scholar] [CrossRef]
- Ogawa, S.; Chester, A.E.; Hewitt, S.C.; Walker, V.R.; Gustafsson, J.Å.; Smithies, O.; Korach, K.S.; Pfaff, D.W. Abolition of Male Sexual Behaviors in Mice Lacking Estrogen Receptors α and β (AβERKO). Proc. Natl. Acad. Sci. USA 2000, 97, 14737–14741. [Google Scholar] [CrossRef]
- Hammes, S.R.; Levin, E.R. Extranuclear Steroid Receptors: Nature and Actions. Endocr. Rev. 2007, 28, 726–741. [Google Scholar] [CrossRef]
- Mong, J.A.; Nuñez, J.L.; McCarthy, M.M. GABA Mediates Steroid-Induced Astrocyte Differentiation in the Neonatal Rat Hypothalamus. J. Neuroendocrinol. 2002, 14, 45–55. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Liang, S.L.; Thompson, S.M.; McCarthy, M.M. Estradiol Induces Dendritic Spines by Enhancing Glutamate Release Independent of Transcription: A Mechanism for Organizational Sex Differences. Neuron 2008, 58, 584–598. [Google Scholar] [CrossRef]
- Schwarz, J.M.; McCarthy, M.M. The Role of Neonatal NMDA Receptor Activation in Defeminization and Masculinization of Sex Behavior in the Rat. Horm. Behav. 2008, 54, 662–668. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.; Kang, S.; Kim, S.H.; Kim, J.C.; Yang, M.; Moon, C. Brain-Derived Neurotropic Factor and GABAergic Transmission in Neurodegeneration and Neuroregeneration. Neural Regen. Res. 2017, 12, 1733–1741. [Google Scholar] [CrossRef]
- Melcangi, R.C.; Poletti, A.; Cavarretta, I.; Celotti, F.; Colciago, A.; Magnaghi, V.; Motta, M.; Negri-Cesi, P.; Martini, L. The 5α-Reductase in the Central Nervous System: Expression and Modes of Control. J. Steroid Biochem. Mol. Biol. 1998, 65, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.P.; Casti, A.; Casu, A.; Frau, R.; Bortolato, M.; Spiga, S.; Ennas, M.G. Regional Distribution of 5α-Reductase Type 2 in the Adult Rat Brain: An Immunohistochemical Analysis. Psychoneuroendocrinology 2013, 38, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Guennoun, R.; Robert, F.; Carelli, C.; Gago, N.; Ghoumari, A.; Gonzalez Deniselle, M.C.; Gonzalez, S.L.; Ibanez, C.; Labombarda, F.; et al. Local Synthesis and Dual Actions of Progesterone in the Nervous System: Neuroprotection and Myelination. Growth Horm. IGF Res. 2004, 14, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Guennoun, R. Progesterone in the Brain: Hormone, Neurosteroid and Neuroprotectant. Int. J. Mol. Sci. 2020, 21, 5271. [Google Scholar] [CrossRef]
- Gaignard, P.; Savouroux, S.; Liere, P.; Pianos, A.; Thérond, P.; Schumacher, M.; Slama, A.; Guennoun, R. Effect of Sex Differences on Brain Mitochondrial Function and Its Suppression by Ovariectomy and in Aged Mice. Endocrinology 2015, 156, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- González-Orozco, J.C.; Camacho-Arroyo, I. Progesterone Actions during Central Nervous System Development. Front. Neurosci. 2019, 13, 503. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.; Matthews, S.G. Glucocorticoids and Sex-Dependent Development of Brain Glucocorticoid and Mineralocorticoid Receptors. Endocrinology 2003, 144, 2775–2784. [Google Scholar] [CrossRef]
- Reinius, B.; Jazin, E. Prenatal Sex Differences in the Human Brain. Mol. Psychiatry 2009, 14, 988–991. [Google Scholar] [CrossRef] [PubMed]
- Lahr, G.; Maxson, S.C.; Mayer, A.; Just, W.; Pilgrim, C.; Reisert, I. Transcription of the Y Chromosomal Gene, Sry, in Adult Mouse Brain. Mol. Brain Res. 1995, 33, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.; Lahr, G.; Swaab, D.F.; Pilgrim, C.; Reisert, I. The Y-Chromosomal Genes SRY and ZFY Are Transcribed in Adult Human Brain. Neurogenetics 1998, 1, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Dewing, P.; Chiang, C.W.K.; Sinchak, K.; Sim, H.; Fernagut, P.O.; Kelly, S.; Chesselet, M.F.; Micevych, P.E.; Albrecht, K.H.; Harley, V.R.; et al. Direct Regulation of Adult Brain Function by the Male-Specific Factor SRY. Curr. Biol. 2006, 16, 415–420. [Google Scholar] [CrossRef]
- Wu, J.B.; Chen, K.; Li, Y.; Lau, Y.F.C.; Shih, J.C. Regulation of Monoamine Oxidase A by the SRY Gene on the Y Chromosome. FASEB J. 2009, 23, 4029–4038. [Google Scholar] [CrossRef]
- Czech, D.P.; Lee, J.; Sim, H.; Parish, C.L.; Vilain, E.; Harley, V.R. The Human Testis-Determining Factor SRY Localizes in Midbrain Dopamine Neurons and Regulates Multiple Components of Catecholamine Synthesis and Metabolism. J. Neurochem. 2012, 122, 260–271. [Google Scholar] [CrossRef]
- Voskuhl, R.; Itoh, Y. Sex Differences Series The X Factor in Neurodegeneration. J. Exp. Med. 2022, 219, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Migeon, B.R. X-Linked Diseases: Susceptible Females. Genet. Med. 2020, 22, 1156–1174. [Google Scholar] [CrossRef] [PubMed]
- Berletch, J.B.; Disteche, C.M. Genes the Escape X Inactivation. Hum. Genet. 2012, 130, 237–245. [Google Scholar] [CrossRef]
- De Vries, G.J.; Rissman, E.F.; Simerly, R.B.; Yang, L.Y.; Scordalakes, E.M.; Auger, C.J.; Swain, A.; Lovell-Badge, R.; Burgoyne, P.S.; Arnold, A.P. A Model System for Study of Sex Chromosome Effects on Sexually Dimorphic Neural and Behavioral Traits. J. Neurosci. 2002, 22, 9005–9014. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, P.S.; Arnold, A.P. A Primer on the Use of Mouse Models for Identifying Direct Sex Chromosome Effects That Cause Sex Differences in Non-Gonadal Tissues. Biol. Sex Differ. 2016, 7, 1–21. [Google Scholar] [CrossRef]
- Hong, D.S.; Hoeft, F.; Marzelli, M.J.; Lepage, J.F.; Roeltgen, D.; Ross, J.; Reiss, A.L. Influence of the X-Chromosome on Neuroanatomy: Evidence from Turner and Klinefelter Syndromes. J. Neurosci. 2014, 34, 3509–3516. [Google Scholar] [CrossRef]
- Zhang, X.; Hong, D.; Ma, S.; Ward, T.; Ho, M.; Pattni, R.; Duren, Z.; Stankov, A.; Shrestha, S.B.; Hallmayer, J.; et al. Integrated Functional Genomic Analyses of Klinefelter and Turner Syndromes Reveal Global Network Effects of Altered X Chromosome Dosage. Proc. Natl. Acad. Sci. USA 2020, 117, 4864–4873. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson ’ s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Cerri, S.; Mus, L.; Blandini, F. Parkinson’s Disease in Women and Men: What’s the Difference? J. Parkinsons. Dis. 2019, 9, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Shmidt, M.L.; Lee, M.-Y.V.; Trojanowski, J.; Jakes, R.; Goedert, M. A -Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Plotegher, N.; Greggio, E.; Bisaglia, M.; Bubacco, L. Biophysical Groundwork as a Hinge to Unravel the Biology of α-Synuclein Aggregation and Toxicity. Q. Rev. Biophys. 2014, 47, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Badanjak, K.; Fixemer, S.; Smajić, S.; Skupin, A.; Grünewald, A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4676. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef]
- Streubel-Gallasch, L.; Giusti, V.; Sandre, M.; Tessari, I.; Plotegher, N.; Giusto, E.; Masato, A.; Iovino, L.; Battisti, I.; Arrigoni, G.; et al. Parkinson’s Disease–Associated LRRK2 Interferes with Astrocyte-Mediated Alpha-Synuclein Clearance. Mol. Neurobiol. 2021, 58, 3119–3140. [Google Scholar] [CrossRef]
- Semchuk, K.M.; Love, E.J.; Lee, R.G. Parkinson’s Disease and Exposure to Agricultural Work and Pesticide Chemicals. Neurology 1992, 42, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Seidler, A.; Hellenbrand, W.; Robra, B.P.; Vieregge, P.; Nischan, P.; Joerg, J.; Oertel, W.H.; Ulm, G.; Schneider, E. Possible Environmental, Occupational, and Other Etiologic Factors for Parkinson’s Disease: A Case-Control Study in Germany. Neurology 1996, 46, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, A.; Theuns, J.; Van Broeckhoven, C. Progress in Unraveling the Genetic Etiology of Parkinson Disease in a Genomic Era. Trends Genet. 2015, 31, 140–149. [Google Scholar] [CrossRef]
- Jia, F.; Fellner, A.; Kumar, K.R. Monogenic Parkinson’s Disease: Genotype, Phenotype, Pathophysiology, and Genetic Testing. Genes 2022, 13, 471. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.; Anastacio, H.; Bardy, C. Genetic Predispositions of Parkinson’s Disease Revealed in Patient-Derived Brain Cells. NPJ Park. Dis. 2020, 6, 8. [Google Scholar] [CrossRef]
- Oliveira, L.M.A.; Gasser, T.; Edwards, R.; Zweckstetter, M.; Melki, R.; Stefanis, L.; Lashuel, H.A.; Sulzer, D.; Vekrellis, K.; Halliday, G.M.; et al. Alpha-Synuclein Research: Defining Strategic Moves in the Battle against Parkinson’s Disease. NPJ Park. Dis. 2021, 7, 39. [Google Scholar] [CrossRef]
- Rietdijk, C.D.; Perez-Pardo, P.; Garssen, J.; van Wezel, R.J.A.; Kraneveld, A.D. Exploring Braak’s Hypothesis of Parkinson’s Disease. Front. Neurol. 2017, 8, 37. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Analysis of Genome-Wide Association Studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Konnova, E.A.; Swanberg, M. Animal Models of Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; ISBN 978-0-9944381-6-4. [Google Scholar]
- Isik, S.; Yeman Kiyak, B.; Akbayir, R.; Seyhali, R.; Arpaci, T. Microglia Mediated Neuroinflammation in Parkinson’s Disease. Cells 2023, 12, 1012. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s Disease. Park. Relat. Disord. 2012, 18, 210–212. [Google Scholar] [CrossRef]
- Castillo-Rangel, C.; Marin, G.; Hernández-Contreras, K.A.; Vichi-Ramírez, M.M.; Zarate-Calderon, C.; Torres-Pineda, O.; Diaz-Chiguer, D.L.; De la Mora González, D.; Gómez Apo, E.; Teco-Cortes, J.A.; et al. Neuroinflammation in Parkinson’s Disease: From Gene to Clinic: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 5792. [Google Scholar] [CrossRef]
- Mishra, A.; Bandopadhyay, R.; Singh, P.K.; Mishra, P.S.; Sharma, N.; Khurana, N. Neuroinflammation in Neurological Disorders: Pharmacotherapeutic Targets from Bench to Bedside; Springer: New York, NY, USA, 2021; Volume 36, ISBN 0123456789. [Google Scholar]
- Teleanu, D.M.; Niculescu, A.G.; Lungu, I.I.; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex Differences in Immune Responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Aparicio, I.; Paris, I.; Sierra-Torre, V.; Plaza-Zabala, A.; Rodríguez-Iglesias, N.; Márquez-Ropero, M.; Beccari, S.; Huguet, P.; Abiega, O.; Alberdi, E.; et al. Microglia Actively Remodel Adult Hippocampal Neurogenesis through the Phagocytosis Secretome. J. Neurosci. 2020, 40, 1453–1482. [Google Scholar] [CrossRef]
- Chowen, J.A.; Garcia-Segura, L.M. Role of Glial Cells in the Generation of Sex Differences in Neurodegenerative Diseases and Brain Aging. Mech. Ageing Dev. 2021, 196, 111473. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Gelosa, P.; Castiglioni, L.; Cimino, M.; Rizzi, N.; Pepe, G.; Lolli, F.; Marcello, E.; Sironi, L.; Vegeto, E.; et al. Sex-Specific Features of Microglia from Adult Mice. Cell Rep. 2018, 23, 3501–3511. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Ceasrine, A.M.; Bilbo, S.D. Microglia and Sexual Differentiation of the Developing Brain: A Focus on Ontogeny and Intrinsic Factors. Glia 2020, 68, 1085–1099. [Google Scholar] [CrossRef]
- Yanguas-Casás, N. Physiological Sex Differences in Microglia and Their Relevance in Neurological Disorders. Neuroimmunol. Neuroinflammation 2020, 7, 13–22. [Google Scholar] [CrossRef]
- Kodama, L.; Gan, L. Do Microglial Sex Differences Contribute to Sex Differences in Neurodegenerative Diseases? Trends Mol. Med. 2019, 25, 741–749. [Google Scholar] [CrossRef]
- Gatto, N.M.; Deapen, D.; Stoyanoff, S.; Pinder, R.; Narayan, S.; Bordelon, Y.; Ritz, B. Lifetime Exposure to Estrogens and Parkinson’s Disease in California Teachers. Park. Relat. Disord. 2014, 20, 1149–1156. [Google Scholar] [CrossRef]
- Kusters, C.D.J.; Paul, K.C.; Duarte Folle, A.; Keener, A.M.; Bronstein, J.M.; Bertram, L.; Hansen, J.; Horvath, S.; Sinsheimer, J.S.; Lill, C.M.; et al. Increased Menopausal Age Reduces the Risk of Parkinson’s Disease: A Mendelian Randomization Approach. Mov. Disord. 2021, 36, 2264–2272. [Google Scholar] [CrossRef]
- Ou, R.; Wei, Q.; Hou, Y.; Zhang, L.; Liu, K.; Lin, J.; Yang, T.; Yang, J.; Jiang, Z.; Song, W.; et al. Reproductive Lifespan and Motor Progression of Parkinson’s Disease. J. Clin. Med. 2022, 11, 6163. [Google Scholar] [CrossRef]
- Pesce, G.; Artaud, F.; Roze, E.; Degaey, I.; Portugal, B.; Nguyen, T.T.H.; Fournier, A.; Boutron-Ruault, M.-C.; Severi, G.; Elbaz, A.; et al. Reproductive Characteristics, Use of Exogenous Hormones and Parkinson Disease in Women from the E3N Study. Brain 2022, 2022, awac440. [Google Scholar] [CrossRef]
- Canonico, M.; Pesce, G.; Bonaventure, A.; Le Noan-Lainé, M.; Benatru, I.; Ranoux, D.; Moisan, F.; Elbaz, A. Increased Risk of Parkinson’s Disease in Women after Bilateral Oophorectomy. Mov. Disord. 2021, 36, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Simon, K.C.; Chen, H.; Gao, X.; Schwarszchild, M.A.; Ascherio, A. Reproductive Factors, Exogenous Estrogen Use, and Risk of Parkinson’s Disease. Mov. Disord. 2009, 24, 1359–1365. [Google Scholar] [CrossRef]
- Lv, M.; Zhang, Y.; Chen, G.C.; Li, G.; Rui, Y.; Qin, L.; Wan, Z. Reproductive Factors and Risk of Parkinson’s Disease in Women: A Meta-Analysis of Observational Studies. Behav. Brain Res. 2017, 335, 103–110. [Google Scholar] [CrossRef]
- Langston, W.J.; Ballard, P.; Tetrud, W.J.; Irwin, I. Chronic Parkinsonism in Humans Due to a Product of Meperidine-Analog Synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Joniec, I.; Ciesielska, A.; Kurkowska-Jastrzebska, I.; Przybylkowski, A.; Czlonkowska, A.; Czlonkowski, A. Age- and Sex-Differences in the Nitric Oxide Synthase Expression and Dopamine Concentration in the Murine Model of Parkinson’s Disease Induced by 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine. Brain Res. 2009, 1261, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Wang, V.; Huang, E.Y.K.; Chou, Y.C.; Kuo, T.T.; Olson, L.; Hoffer, B.J. Delayed Dopamine Dysfunction and Motor Deficits in Female Parkinson Model Mice. Int. J. Mol. Sci. 2019, 20, 6251. [Google Scholar] [CrossRef]
- Makav, M.; Eroğlu, H.A. Recuperative Effect of Estrogen on Rotenone-Induced Experimental Model of Parkinson’s Disease in Rats. Environ. Sci. Pollut. Res. 2021, 28, 21266–21275. [Google Scholar] [CrossRef]
- Morale, M.C.; Serra, P.A.; L’Episcopo, F.; Tirolo, C.; Caniglia, S.; Testa, N.; Gennuso, F.; Giaquinta, G.; Rocchitta, G.; Desole, M.S.; et al. Estrogen, Neuroinflammation and Neuroprotection in Parkinson’s Disease: Glia Dictates Resistance versus Vulnerability to Neurodegeneration. Neuroscience 2006, 138, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Tripanichkul, W.; Sripanichkulchai, K.; Finkelstein, D.I. Estrogen Down-Regulates Glial Activation in Male Mice Following 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Intoxication. Brain Res. 2006, 1084, 28–37. [Google Scholar] [CrossRef]
- Aguirre-Vidal, Y.; Morales-Montor, J.; Gómez de León, C.T.; Ostoa-Saloma, P.; Díaz-Zaragoza, M.; Montes, S.; Arteaga-Silva, M.; Monroy-Noyola, A. Protection Induced by Estradiol Benzoate in the MPP+ Rat Model of Parkinson’s Disease Is Associated with the Regulation of the Inflammatory Cytokine Profile in the Nigro Striatum. J. Neuroimmunol. 2020, 349, 577426. [Google Scholar] [CrossRef] [PubMed]
- Giordano, G.; Tait, L.; Furlong, C.E.; Cole, T.B.; Kavanagh, T.J.; Costa, L.G. Gender Differences in Brain Susceptibility to Oxidative Stress Are Mediated by Levels of Paraoxonase-2 Expression. Free Radic. Biol. Med. 2013, 58, 98–108. [Google Scholar] [CrossRef]
- Siani, F.; Greco, R.; Levandis, G.; Ghezzi, C.; Daviddi, F.; Demartini, C.; Vegeto, E.; Fuzzati-Armentero, M.T.; Blandini, F. Influence of Estrogen Modulation on Glia Activation in a Murine Model of Parkinson’s Disease. Front. Neurosci. 2017, 11, 306. [Google Scholar] [CrossRef] [PubMed]
- Varmazyar, R.; Noori-Zadeh, A.; Rajaei, F.; Darabi, S.; Bakhtiyari, S. 17 β-Estradiol Oxidative Stress Attenuation and Autophagy-Induced Dopaminergic Neuroprotection. Cell J. 2019, 21, 1–6. [Google Scholar] [CrossRef]
- Baker, A.E.; Brautigam, V.M.; Watters, J.J. Estrogen Modulates Microglial Inflammatory Mediator Production via Interactions with Estrogen Receptor β. Endocrinology 2004, 145, 5021–5032. [Google Scholar] [CrossRef] [PubMed]
- Benedusi, V.; Meda, C.; Della Torre, S.; Monteleone, G.; Vegeto, E.; Maggi, A. A Lack of Ovarian Function Increases Neuroinflammation in Aged Mice. Endocrinology 2012, 153, 2777–2788. [Google Scholar] [CrossRef]
- Morale, M.C.; L’Episcopo, F.; Tirolo, C.; Giaquinta, G.; Caniglia, S.; Testa, N.; Arcieri, P.; Serra, P.A.; Lupo, G.; Alberghina, M.; et al. Loss of Aromatase Cytochrome P450 Function as a Risk Factor for Parkinson’s Disease? Brain Res. Rev. 2008, 57, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Campos, F.L.; Cristovão, A.C.; Rocha, S.M.; Fonseca, C.P.; Baltazar, G. GDNF Contributes to Oestrogen-Mediated Protection of Midbrain Dopaminergic Neurones. J. Neuroendocrinol. 2012, 24, 1386–1397. [Google Scholar] [CrossRef]
- Küppers, E.; Ivanova, T.; Karolczak, M.; Beyer, C. Estrogen: A Multifunctional Messenger to Nigrostriatal Dopaminergic Neurons. J. Neurocytol. 2000, 29, 375–385. [Google Scholar] [CrossRef] [PubMed]
- D’Astous, M.; Morissette, M.; Di Paolo, T. Effect of Estrogen Receptor Agonists Treatment in MPTP Mice: Evidence of Neuroprotection by an ERα Agonist. Neuropharmacology 2004, 47, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Morissette, M.; Jourdain, S.; Al Sweidi, S.; Menniti, F.S.; Ramirez, A.D.; Di Paolo, T. Role of Estrogen Receptors in Neuroprotection by Estradiol against MPTP Toxicity. Neuropharmacology 2007, 52, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Al Sweidi, S.; Sánchez, M.G.; Bourque, M.; Morissette, M.; Dluzen, D.; di Paolo, T. Oestrogen Receptors and Signalling Pathways: Implications for Neuroprotective Effects of Sex Steroids in Parkinson’s Disease. J. Neuroendocrinol. 2012, 24, 48–61. [Google Scholar] [CrossRef]
- Bourque, M.; Morissette, M.; Di Paolo, T. Neuroprotection in Parkinsonian-Treated Mice via Estrogen Receptor α Activation Requires G Protein-Coupled Estrogen Receptor 1. Neuropharmacology 2015, 95, 343–352. [Google Scholar] [CrossRef]
- Barbati, A.; Corona, P.; Mattioli, W.; Quatrini, A. Biomassa Forestale per La Produzione Di Energia: Un Modello Di Analisi per l’Alta Valle Dell’Aniene. Italia For. Mont. 2012, 67, 329–336. [Google Scholar] [CrossRef]
- Marin, R.; Diaz, M. Estrogen Interactions with Lipid Rafts Related to Neuroprotection. Impact of Brain Ageing and Menopause. Front. Neurosci. 2018, 12, 128. [Google Scholar] [CrossRef]
- Litim, N.; Morissette, M.; Di Paolo, T. Effects of Progesterone Administered after MPTP on Dopaminergic Neurons of Male Mice. Neuropharmacology 2017, 117, 209–218. [Google Scholar] [CrossRef]
- Casas, S.; Giuliani, F.; Cremaschi, F.; Yunes, R.; Cabrera, R. Neuromodulatory Effect of Progesterone on the Dopaminergic, Glutamatergic, and GABAergic Activities in a Male Rat Model of Parkinson’s Disease. Neurol. Res. 2013, 35, 719–725. [Google Scholar] [CrossRef]
- Jarras, H.; Bourque, M.; Poirier, A.A.; Morissette, M.; Coulombe, K.; Di Paolo, T.; Soulet, D. Neuroprotection and Immunomodulation of Progesterone in the Gut of a Mouse Model of Parkinson’s Disease. J. Neuroendocrinol. 2019, 32, e12782. [Google Scholar] [CrossRef]
- Adeosun, S.O.; Hou, X.; Jiao, Y.; Zheng, B.; Henry, S.; Hill, R.; He, Z.; Pani, A.; Kyle, P.; Ou, X.; et al. Allopregnanolone Reinstates Tyrosine Hydroxylase Immunoreactive Neurons and Motor Performance in an MPTP-Lesioned Mouse Model of Parkinson’s Disease. PLoS ONE 2012, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Okun, M.S.; Crucian, G.P.; Fischer, L.; Walter, B.L.; Testa, C.M.; Vitek, J.L.; DeLong, M.R.; Hanfelt, J.; Huang, X. Testosterone Deficiency in a Parkinson’ s Disease Clinic: Results of a Survey LBP-1c / CP2 / LSF Gene Polymorphism and Risk of Sporadic Alzheimer’ s Disease. J. Neurol. Neurosurg. Psychiatry 2004, 288, 165–166. [Google Scholar]
- Mitchell, E.; Thomas, D.; Burnet, R. Testosterone Improves Motor Function in Parkinson’s Disease. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2006, 13, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.; Abbassi, B.; Su, C.; Singh, M.; Cunningham, R.L. Oxidative Stress Defines the Neuroprotective or Neurotoxic Properties of Androgens in Immortalized Female Rat Dopaminergic Neuronal Cells. Endocrinology 2013, 154, 4281–4292. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Day, A.E.; Ho, C.C.; Walker, Q.D.; Francis, R.; Kuhn, C.M. Androgen Decreases Dopamine Neurone Survival in Rat Midbrain. J. Neuroendocrinol. 2010, 22, 238–247. [Google Scholar] [CrossRef]
- Dluzen, D.E. Effects of Testosterone upon MPTP-Induced Neurotoxicity of the Nigrostriatal Dopaminergic System of C57/B1 Mice. Brain Res. 1996, 715, 113–118. [Google Scholar] [CrossRef]
- Ekue, A.; Boulanger, J.F.; Morissette, M.; Di Paolo, T. Lack of Effect of Testosterone and Dihydrotestosterone Compared to 17β-Oestradiol in 1-Methyl-4-Phenyl-1,2,3,6, Tetrahydropyridine-Mice. J. Neuroendocrinol. 2002, 14, 731–736. [Google Scholar] [CrossRef]
- Bispo, J.M.M.; Melo, J.E.C.; Gois, A.M.; Medeiros, K.A.A.L.; Silva, R.S.; Leal, P.C.; Franco, H.S.; Souza, M.F.; Lins, L.C.R.F.; Ribeiro, A.M.; et al. Testosterone Propionate Improves Motor Alterations and Dopaminergic Damage in the Reserpine-Induced Progressive Model of Parkinson’s Disease. Brain Res. Bull. 2022, 187, 162–168. [Google Scholar] [CrossRef]
- Khodamoradi, K.; Khosravizadeh, Z.; Parmar, M.; Kuchakulla, M.; Ramasamy, R.; Arora, H. Exogenous Testosterone Replacement Therapy versus Raising Endogenous Testosterone Levels: Current and Future Prospects. Physiol. Behav. 2021, 2, 32–42. [Google Scholar] [CrossRef]
- Lundberg, G.; Wu, P.; Wenger, N. Menopausal Hormone Therapy: A Comprehensive Review. Curr. Atheroscler. Rep. 2020, 22, 1–9. [Google Scholar] [CrossRef]
- Song, Y.; Li, S.; Li, X.; Chen, X.; Wei, Z.; Liu, Q.; Cheng, Y. The Effect of Estrogen Replacement Therapy on Alzheimer’s Disease and Parkinson’s Disease in Postmenopausal Women: A Meta-Analysis. Front. Neurosci. 2020, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Unda, S.R.; Marciano, S.; Milner, T.A.; Marongiu, R. State-of-the-Art Review of the Clinical Research on Menopause and Hormone Replacement Therapy Association with Parkinson’s Disease: What Meta-Analysis Studies Cannot Tell Us. Front. Aging Neurosci. 2022, 14. [Google Scholar] [CrossRef]
- Bourque, M.; Di Paolo, T. Neuroactive Steroids and Parkinson’s Disease. Curr. Opin. Endocr. Metab. Res. 2022, 22, 100312. [Google Scholar] [CrossRef]
- Bourque, M.; Morissette, M.; Di Paolo, T. Repurposing Sex Steroids and Related Drugs as Potential Treatment for Parkinson’s Disease. Neuropharmacology 2018, 147, 37–54. [Google Scholar] [CrossRef]
- Vaidya, B.; Dhamija, K.; Guru, P.; Sharma, S.S. Parkinson’s Disease in Women: Mechanisms Underlying Sex Differences. Eur. J. Pharmacol. 2021, 895, 173862. [Google Scholar] [CrossRef]
- Farkas, S.; Szabó, A.; Hegyi, A.E.; Török, B.; Fazekas, C.L.; Ernszt, D.; Kovács, T.; Zelena, D. Estradiol and Estrogen-like Alternative Therapies in Use: The Importance of the Selective and Non-Classical Actions. Biomedicines 2022, 10, 861. [Google Scholar] [CrossRef] [PubMed]
- Prokai-tatrai, K.; Prokai, L. A Novel Prodrug Approach for Central Nervous System-Selective Estrogen Therapy. Molecules 2019, 24, 4197. [Google Scholar] [CrossRef]
- Prokai, L.; Nguyen, V.; Szarka, S.; Garg, P.; Sabnis, G.; Bimonte-Nelson, H.A.; McLaughlin, K.J.; Talboom, J.S.; Conrad, C.D.; Shughrue, P.J.; et al. The Prodrug DHED Selectively Delivers 17β-Estradiol to the Brain for Treating Estrogen-Responsive Disorders. Sci. Transl. Med. 2015, 7, 297ra113. [Google Scholar] [CrossRef]
- Tschiffely, A.E.; Schuh, R.A.; Prokai-Tatrai, K.; Prokai, L.; Ottinger, M.A. A Comparative Evaluation of Treatments with 17β-Estradiol and Its Brain-Selective Prodrug in a Double-Transgenic Mouse Model of Alzheimer’s Disease. Horm. Behav. 2016, 83, 39–44. [Google Scholar] [CrossRef]
- Tschiffely, A.E.; Schuh, R.A.; Prokai-Tatrai, K.; Ottinger, M.A.; Prokai, L. An Exploratory Investigation of Brain-Selective Estrogen Treatment in Males Using a Mouse Model of Alzheimer’s Disease. Horm. Behav. 2018, 98, 16–21. [Google Scholar] [CrossRef]
- Rajsombath, M.M.; Nam, A.Y.; Ericsson, M.; Nuber, S. Female Sex and Brain-Selective Estrogen Benefit α-Synuclein Tetramerization and the PD-like Motor Syndrome in 3k Transgenic Mice. J. Neurosci. 2019, 39, 7628–7640. [Google Scholar] [CrossRef] [PubMed]
- Thadathil, N.; Xiao, J.; Hori, R.; Alway, S.E.; Khan, M.M. Brain Selective Estrogen Treatment Protects Dopaminergic Neurons and Preserves Behavioral Function in MPTP-Induced Mouse Model of Parkinson’s Disease. J. Neuroimmune Pharmacol. 2021, 16, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Cabrera Zapata, L.E.; Garcia-Segura, L.M.; Cambiasso, M.J.; Arevalo, M.A. Genetics and Epigenetics of the X and Y Chromosomes in the Sexual Differentiation of the Brain. Int. J. Mol. Sci. 2022, 23, 2288. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.H.; Zhang, S.Y.; Yang, Z.H.; Yang, J.; Shang, D.D.; Mao, C.Y.; Liu, H.; Hou, H.M.; Shi, M.M.; Wu, J.; et al. A Novel RAB39B Gene Mutation in X-Linked Juvenile Parkinsonism with Basal Ganglia Calcification. Mov. Disord. 2016, 31, 1905–1909. [Google Scholar] [CrossRef]
- Mata, I.F.; Jang, Y.; Kim, C.H.; Hanna, D.S.; Dorschner, M.O.; Samii, A.; Agarwal, P.; Roberts, J.W.; Klepitskaya, O.; Shprecher, D.R.; et al. The RAB39B p.G192R Mutation Causes X-Linked Dominant Parkinson’s Disease. Mol. Neurodegener. 2015, 10, 4–11. [Google Scholar] [CrossRef]
- Niu, M.; Zheng, N.; Wang, Z.; Gao, Y.; Luo, X.; Chen, Z.; Fu, X.; Wang, Y.; Wang, T.; Liu, M.; et al. RAB39B Deficiency Impairs Learning and Memory Partially Through Compromising Autophagy. Front. Cell Dev. Biol. 2020, 8, 598622. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered Synaptic Plasticity in a Mouse Model of Fragile X Mental Retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef]
- Kurz, M.W.; Melissa Schlitter, A.; Klenk, Y.; Mueller, T.; Larsen, J.P.; Aarsland, D.; Dekomien, G. FMR1 Alleles in Parkinson’s Disease: Relation to Cognitive Decline and Hallucinations, a Longitudinal Study. J. Geriatr. Psychiatry Neurol. 2007, 20, 89–92. [Google Scholar] [CrossRef]
- Salcedo-Arellano, M.J.; Wolf-Ochoa, M.W.; Hong, T.; Amina, S.; Tassone, F.; Lechpammer, M.; Hagerman, R.; Martínez-Cerdeño, V. Parkinsonism Versus Concomitant Parkinson’s Disease in Fragile X–Associated Tremor/Ataxia Syndrome. Mov. Disord. Clin. Pract. 2020, 7, 413–418. [Google Scholar] [CrossRef]
- Zeng, Q.; Pan, H.; Zhao, Y.; Wang, Y.; Xu, Q.; Tan, J.; Yan, X.; Li, J.; Tang, B.; Guo, J. Association Study of TAF1 Variants in Parkinson’s Disease. Front. Neurosci. 2022, 16, 1–6. [Google Scholar] [CrossRef]
- O’Rawe, J.A.; Wu, Y.; Dörfel, M.J.; Rope, A.F.; Au, P.Y.B.; Parboosingh, J.S.; Moon, S.; Kousi, M.; Kosma, K.; Smith, C.S.; et al. TAF1 Variants Are Associated with Dysmorphic Features, Intellectual Disability, and Neurological Manifestations. Am. J. Hum. Genet. 2015, 97, 922–932. [Google Scholar] [CrossRef]
- Plaitakis, A.; Latsoudis, H.; Kanavouras, K.; Ritz, B.; Bronstein, J.M.; Skoula, I.; Mastorodemos, V.; Papapetropoulos, S.; Borompokas, N.; Zaganas, I.; et al. Gain-of-Function Variant in GLUD2 Glutamate Dehydrogenase Modifies Parkinson’s Disease Onset. Eur. J. Hum. Genet. 2010, 18, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Le Guen, Y.; Napolioni, V.; Belloy, M.E.; Yu, E.; Krohn, L.; Ruskey, J.A.; Gan-Or, Z.; Kennedy, G.; Eger, S.J.; Greicius, M.D. Common X-Chromosome Variants Are Associated with Parkinson Disease Risk. Ann. Neurol. 2021, 90, 22–34. [Google Scholar] [CrossRef]
- Bourque, D.K.; Hartley, T.; Nikkel, S.M.; Pohl, D.; Tétreault, M.; Kernohan, K.D.; Dyment, D.A. A de Novo Mutation in RPL10 Causes a Rare X-Linked Ribosomopathy Characterized by Syndromic Intellectual Disability and Epilepsy: A New Case and Review of the Literature. Eur. J. Med. Genet. 2018, 61, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Gantz, S.C.; Ford, C.P.; Neve, K.A.; Williams, J.T. Loss of Mecp2 in Substantia Nigra Dopamine Neurons Compromises the Nigrostriatal Pathway. J. Neurosci. 2011, 31, 12629–12637. [Google Scholar] [CrossRef] [PubMed]
- Bach, S.; Ryan, N.M.; Guasoni, P.; Corvin, A.P.; El-Nemr, R.A.; Khan, D.; Sanfeliu, A.; Tropea, D. Methyl-CpG-Binding Protein 2 Mediates Overlapping Mechanisms across Brain Disorders. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Meira, J.G.C.; Magalhães, B.S.; Ferreira, I.B.B.; Tavares, D.F.; Kobayashi, G.S.; Leão, E.K.E.A. Novel USP9X Variant Associated with Syndromic Intellectual Disability in a Female: A Case Study and Review. Am. J. Med. Genet. Part A 2021, 185, 1569–1574. [Google Scholar] [CrossRef]
- Pravata, V.M.; Gundogdu, M.; Bartual, S.G.; Ferenbach, A.T.; Stavridis, M.; Õunap, K.; Pajusalu, S.; Žordania, R.; Wojcik, M.H.; van Aalten, D.M.F. A Missense Mutation in the Catalytic Domain of O-GlcNAc Transferase Links Perturbations in Protein O-GlcNAcylation to X-Linked Intellectual Disability. FEBS Lett. 2020, 594, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L.; Emmanouilidou, E.; Pantazopoulou, M.; Kirik, D.; Vekrellis, K.; Tofaris, G.K. How Is Alpha-Synuclein Cleared from the Cell? J. Neurochem. 2019, 150, 577–590. [Google Scholar] [CrossRef]
- Rott, R.; Szargel, R.; Haskin, J.; Bandopadhyay, R.; Lees, A.J.; Shani, V.; Engelender, S. α-Synuclein Fate Is Determined by USP9X-Regulated Monoubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 18666–18671. [Google Scholar] [CrossRef]
- Deng, X.; Berletch, J.B.; Nguyen, D.K.; Disteche, C.M. X Chromosome Regulation: Diverse Patterns in Development, Tissues and Disease. Nat. Rev. Genet. 2014, 15, 367–378. [Google Scholar] [CrossRef]
- Levine, P.M.; Galesic, A.; Balana, A.T.; Mahul-Mellier, A.L.; Navarro, M.X.; De Leon, C.A.; Lashuel, H.A.; Pratt, M.R. α-Synuclein O-GlcNAcylation Alters Aggregation and Toxicity, Revealing Certain Residues as Potential Inhibitors of Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2019, 116, 1511–1519. [Google Scholar] [CrossRef]
- Lee, B.E.; Kim, H.Y.; Kim, H.J.; Jeong, H.; Kim, B.G.; Lee, H.E.; Lee, J.; Kim, H.B.; Lee, S.E.; Yang, Y.R.; et al. O-GlcNAcylation Regulates Dopamine Neuron Function, Survival and Degeneration in Parkinson Disease. Brain 2020, 143, 3699–3716. [Google Scholar] [CrossRef] [PubMed]
- Milsted, A.; Serova, L.; Sabban, E.L.; Dunphy, G.; Turner, M.E.; Ely, D.L. Regulation of Tyrosine Hydroxylase Gene Transcription by Sry. Neurosci. Lett. 2004, 369, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Czech, D.P.; Lee, J.; Correia, J.; Loke, H.; Möller, E.K.; Harley, V.R. Transient Neuroprotection by SRY Upregulation in Dopamine Cells Following Injury in Males. Endocrinology 2014, 155, 2602–2612. [Google Scholar] [CrossRef]
- Lee, J.; Pinares-Garcia, P.; Loke, H.; Ham, S.; Vilain, E.; Harley, V.R. Sex-Specific Neuroprotection by Inhibition of the Y-Chromosome Gene, SRY, in Experimental Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2019, 116, 16577–16582. [Google Scholar] [CrossRef]
- Pohl, T.T.; Hörnberg, H. Neuroligins in Neurodevelopmental Conditions: How Mouse Models of de Novo Mutations Can Help Us Link Synaptic Function to Social Behavior. Neuronal Signal. 2022, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Burré, J. α-Synuclein in Synaptic Function and Dysfunction. Trends Neurosci. 2022, 46, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Wu, K.; Pandey, S.; Lehr, A.W.; Li, Y.; Bemben, M.A.; Badger, J.D.; Lauzon, J.L.; Wang, T.; Zaghloul, K.A.; et al. A Cluster of Autism-Associated Variants on X-Linked NLGN4X Functionally Resemble NLGN4Y. Neuron 2020, 106, 759–768.e7. [Google Scholar] [CrossRef]
- Kim, J.Y.; Min, K.; Paik, H.Y.; Lee, S.K. Sex Omission and Male Bias Are Still Widespread in Cell Experiments. Am. J. Physiol. Cell Physiol. 2021, 320, C742–C749. [Google Scholar] [CrossRef]
- Shah, K.; McCormack, C.E.; Bradbury, N.A. Do You Know the Sex of Your Cells? Am. J. Physiol. Cell Physiol. 2014, 306, 2013. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Bradbury, N.A. Did You Forget Your Cell Sex? An Update on the Inclusion of Sex as a Variable in AJP-Cell Physiology. Am. J. Physiol. Cell Physiol. 2023, 324, C910–C926. [Google Scholar] [CrossRef]
- Mamlouk, G.; Dorris, D.; Barrett, L.; Meitzen, J. Sex Bias and Omission in Neuroscience Research Is Influenced by Research Model and Journal, but Not Reported NIH Funding. Physiol. Behav. 2020, 57, 100835. [Google Scholar] [CrossRef] [PubMed]
- James, B.D.; Guerin, P.; Allen, J.B. Let’s Talk About Sex—Biological Sex Is Underreported in Biomaterial Studies. Adv. Healthc. Mater. 2021, 10, 2001034. [Google Scholar] [CrossRef]
- Miguel-Aliaga, I. Let’s Talk about (Biological) Sex. Nat. Rev. Mol. Cell Biol. 2022, 23, 227–228. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sifuentes, Y.; Maney, D.L. Reporting and Misreporting of Sex Differences in the Biological Sciences. Elife 2021, 10, e70817. [Google Scholar] [CrossRef] [PubMed]
- Reisert, I.; Engele, J.; Pilgrim, C. Early Sexual Differentiation of Diencephalic Dopaminergic Neurons of the Rat in Vitro. Cell Tissue Res. 1989, 255, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Hickey, R.W.; Bayir, H.; Watkins, S.C.; Tyurin, V.A.; Guo, F.; Kochanek, P.M.; Jenkins, L.W.; Ren, J.; Gibson, G.; et al. Starving Neurons Show Sex Difference in Autophagy. J. Biol. Chem. 2009, 284, 2383–2396. [Google Scholar] [CrossRef]
- Gutleb, H.N.R.; Gutleb, A.C. A Short History of the Consideration of Sex Differences in Biomedical Research—Lessons for the In Vitro Community from Animal Models and Human Clinical Trials. Altern. Lab. Anim. 2023, 2023, 6720. [Google Scholar] [CrossRef]
- De Souza Santos, R.; Frank, A.P.; Palmer, B.F.; Clegg, D.J. Sex and Media: Considerations for Cell Culture Studies. ALTEX 2018, 35, 435–440. [Google Scholar] [CrossRef]
- Carvajal-Oliveros, A.; Uriostegui-Arcos, M.; Zurita, M.; Melchy-Perez, E.I.; Narváez-Padilla, V.; Reynaud, E. The BE (2)-M17 Cell Line Has a Better Dopaminergic Phenotype than the Traditionally Used for Parkinson’s Research SH-SY5Y, Which Is Mostly Serotonergic. IBRO Neurosci. Rep. 2022, 13, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Biedler, J.L.; Roffler-Tarlov, S.; Schachner, M.; Freedman, L.S. Multiple Neurotransmitter Synthesis by Human Neuroblastoma Cell Lines and Clones. Cancer Res. 1978, 38, 3751–3757. [Google Scholar] [PubMed]
- Filograna, R.; Civiero, L.; Ferrari, V.; Codolo, G.; Greggio, E.; Bubacco, L.; Beltramini, M.; Bisaglia, M. Analysis of the Catecholaminergic Phenotype in Human SH-SY5Y and BE(2)-M17 Neuroblastoma Cell Lines upon Differentiation. PLoS ONE 2015, 10, e0136769. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y Cell Line in Parkinson’s Disease Research: A Systematic Review. Mol. Neurodegener. 2017, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, E.; Cardinale, A.; Picconi, B.; Gardoni, F. From Cell Lines to Pluripotent Stem Cells for Modelling Parkinson’s Disease. J. Neurosci. Methods 2020, 340, 108741. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Pilgrim, C.; Reisert, I. Dopamine Content and Metabolism in Mesencephalic and Diencephalic Cell Cultures: Sex Differences and Effects of Sex Steroids. J. Neurosci. 1991, 11, 1325–1333. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine Neurons Derived from Human ES Cells Efficiently Engraft in Animal Models of Parkinson’s Disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef]
- Stephens, M.C.; Brandt, V.; Botas, J. The Developmental Roots of Neurodegeneration. Neuron 2022, 110, 1–3. [Google Scholar] [CrossRef]


| Gene | Gene Product | Function | PD Inheritance |
|---|---|---|---|
| Well established | |||
| SNCA | Alpha-synuclein | Synaptic vesicle exocytosis; DA neurotransmission; chaperone activity | Dominant |
| LRRK2 | Leucine-rich repeat kinase 2 | Neuronal plasticity; autophagy; vesicle trafficking; neuroinflammation | Dominant |
| VPS35 | Vacuolar protein sorting 35 | Membrane protein recycling | Dominant |
| PRKN | Parkin | Mitochondrial quality control | Recessive |
| PINK1 | PTEN-induced kinase 1 | Mitochondrial quality control | Recessive |
| DJ-1 | DJ-1 | Protection against oxidative stress; mitochondrial function | Recessive |
| Atypical complex PD | |||
| ATP13A2 | Cation-transporting ATPase 13A2 | Lysosomal cation and polyamine transporter | Recessive |
| DNAJC6 | DNAJ subfamily C member 6 | Endocytosis of clathrin-coated vesicles | Recessive |
| FBXO7 | F-box protein 7 | Ubiquitination; proteasome degradation | Recessive |
| SYNJ1 | Synaptojanin-1 | Synaptic vesicle endocytosis; actin filament rearrangements | Recessive |
| PLA2G6 | Phospholipase A2, group 6 | Membrane homeostasis; mitochondrial integrity; signal transduction | Recessive |
| Risk factors | |||
| GBA1 | Glucosylceramidase beta | Lysosomal function; lipid metabolism | Risk factor |
| MAPT | Microtubule-associated protein Tau | Axonal stability; axonal transport | Risk factor |
| X-linked genes associated with parkinsonism | |||
| RAB39B | Ras-related protein Rab-39B | Vesicular trafficking | X-linked parkinsonism |
| FMR1 | Fragile X messenger ribonucleoprotein 1 | Synaptic plasticity; negative role in translation | FXTAS with parkinsonism |
| TAF1 | TATA-box-binding protein associated factor 1 | Initiation of transcription | X-linked dystonia-parkinsonism |
| GLUD2 | Glutamate dehydrogenase 2 | Glutamate metabolism; neurotransmission | Polymorphism |
| PD Model | Effect | Phenotype |
|---|---|---|
| Acute MPTP model | Inhibition of complex I | Motor deficit; DA neuron death; No α-syn aggregates. |
| Subacute/chronic MPTP model | Inhibition of complex I | Progressive model; No motor deficit; No DA neuron death; α-syn aggregates. |
| 6-OHDA | Inhibition of complex I and oxidative stress | Asymmetric motor deficit; DA neuron death; No α-syn aggregates. |
| Rotenone | Inhibition of complex I | Limited motor deficit; Moderate DA neuron death; α-syn aggregates. |
| Paraquat | Oxidative stress | Limited motor deficit; Limited DA neuron death; α-syn aggregates. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terrin, F.; Tesoriere, A.; Plotegher, N.; Dalla Valle, L. Sex and Brain: The Role of Sex Chromosomes and Hormones in Brain Development and Parkinson’s Disease. Cells 2023, 12, 1486. https://doi.org/10.3390/cells12111486
Terrin F, Tesoriere A, Plotegher N, Dalla Valle L. Sex and Brain: The Role of Sex Chromosomes and Hormones in Brain Development and Parkinson’s Disease. Cells. 2023; 12(11):1486. https://doi.org/10.3390/cells12111486
Chicago/Turabian StyleTerrin, Francesca, Annachiara Tesoriere, Nicoletta Plotegher, and Luisa Dalla Valle. 2023. "Sex and Brain: The Role of Sex Chromosomes and Hormones in Brain Development and Parkinson’s Disease" Cells 12, no. 11: 1486. https://doi.org/10.3390/cells12111486