
RNF213 Loss-of-Function Promotes Angiogenesis of Cerebral Microvascular Endothelial Cells in a Cellular State Dependent Manner
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and CRISPR-Cas9-Mediated RNF213 Invalidation
2.2. Cell Proliferation Assays
2.3. In Vitro Scratch Assay
2.4. Tube Formation Assay
2.5. Microarray Analysis and Bioinformatics
2.6. Human Angiogenesis Proteome Profiler
2.7. ELISA
2.8. Immunoblotting
2.9. Flow Cytometry
2.10. Tri-Dimensional Capillary Network Formation
2.11. Immunofluorescence
2.12. Statistical Analysis
3. Results
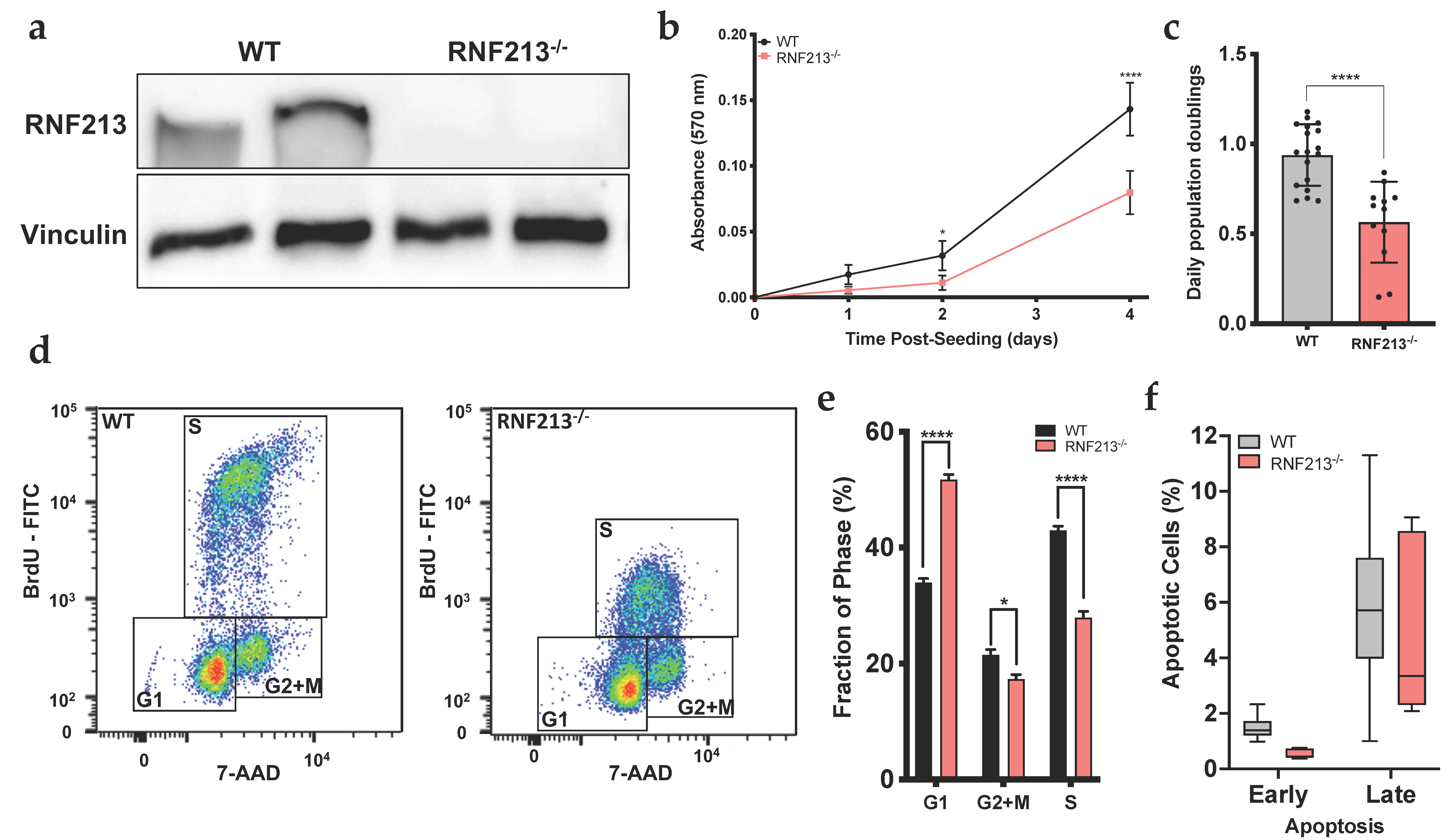
3.1. RNF213 Regulates Proliferation and Cell Cycle of Cerebral Endothelial Cells
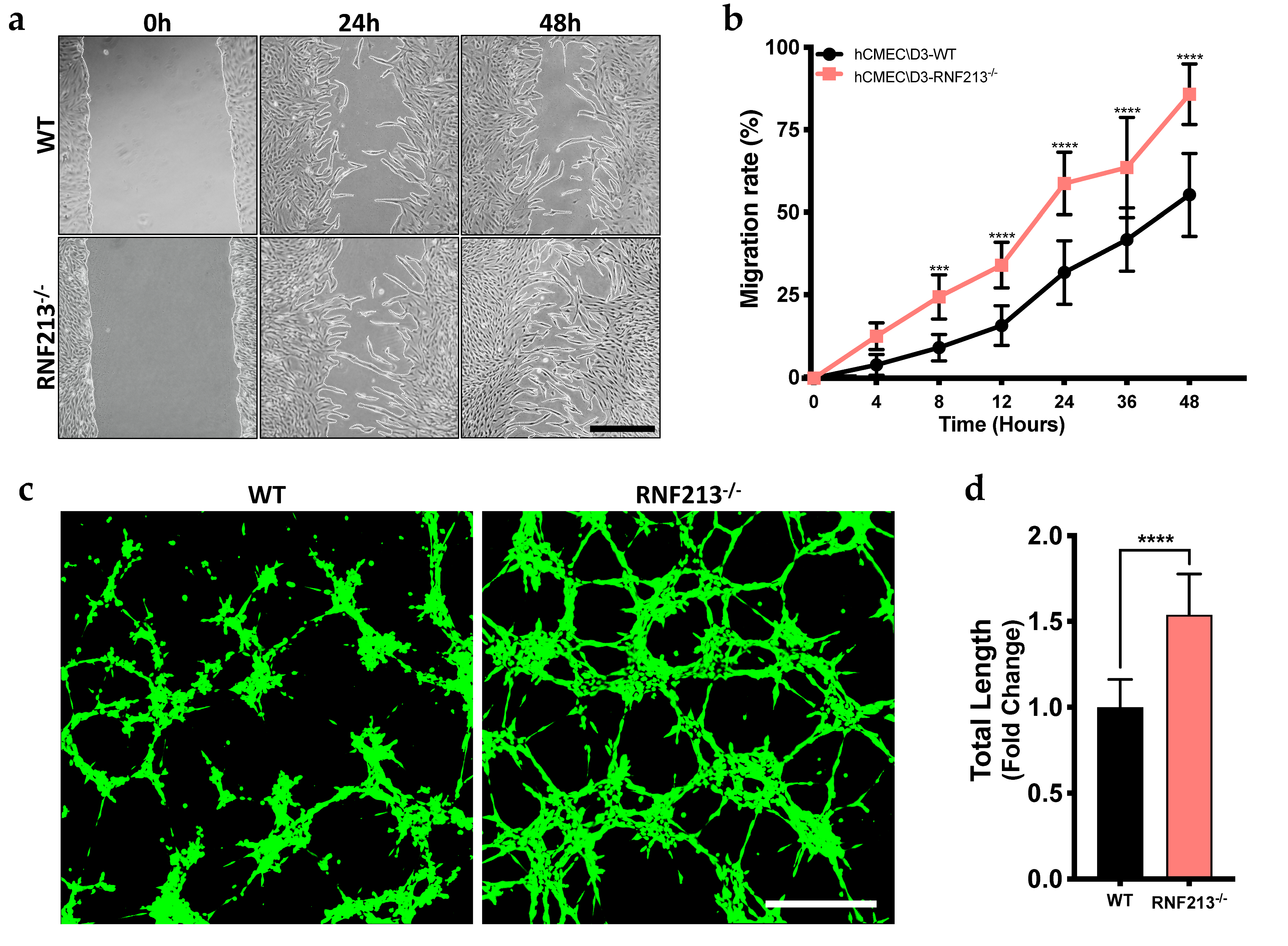
3.2. CRISPR/Cas9-Mediated RNF213 Knockout Drives Cellular Migration as Well as the Angiogenic Activity of Cerebral Endothelial Cells
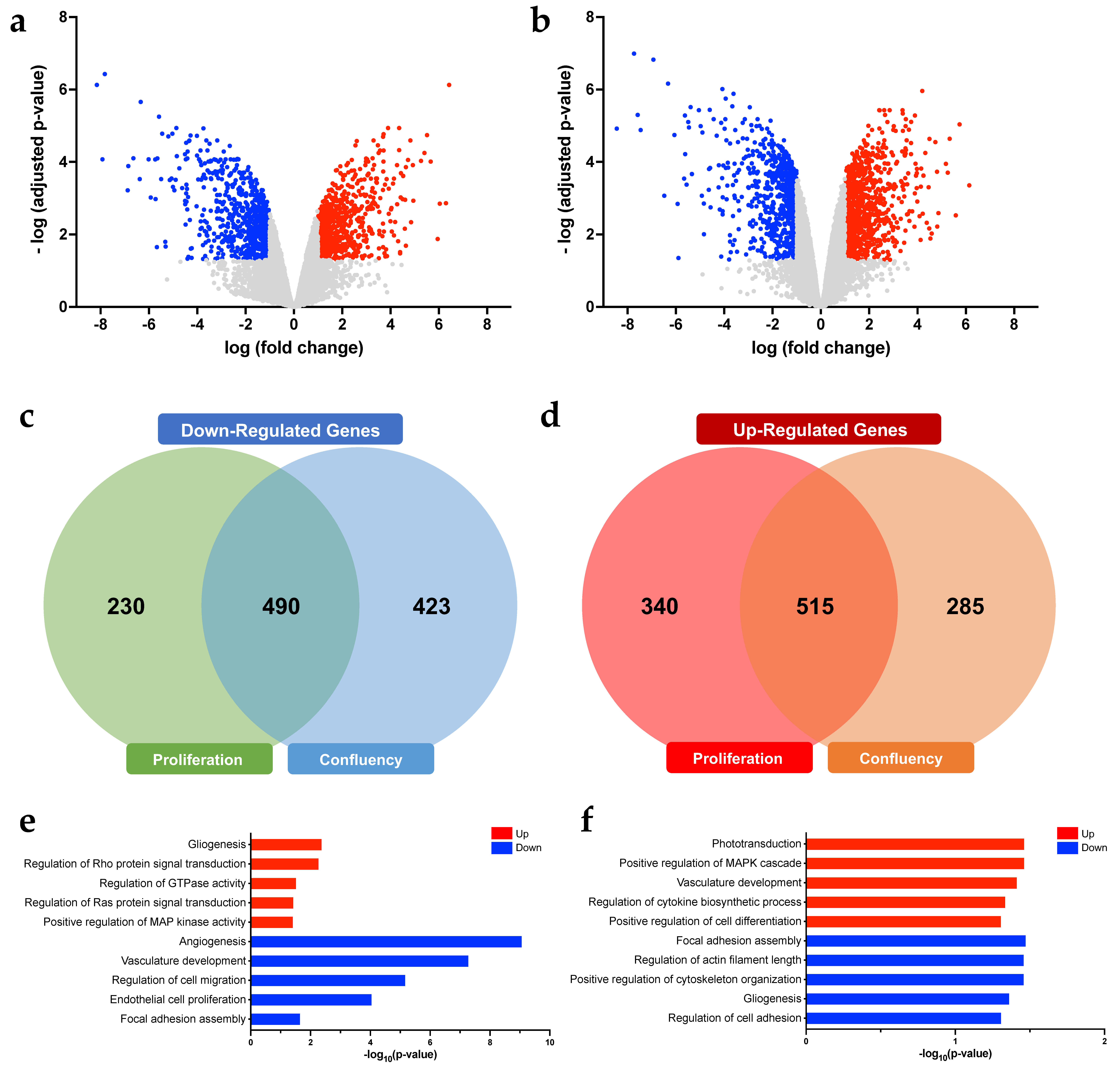
3.3. Gene Expression and Functionally Enriched Analyses in RNF213-Decficient hCMEC/D3 in A Proliferative or Confluent State
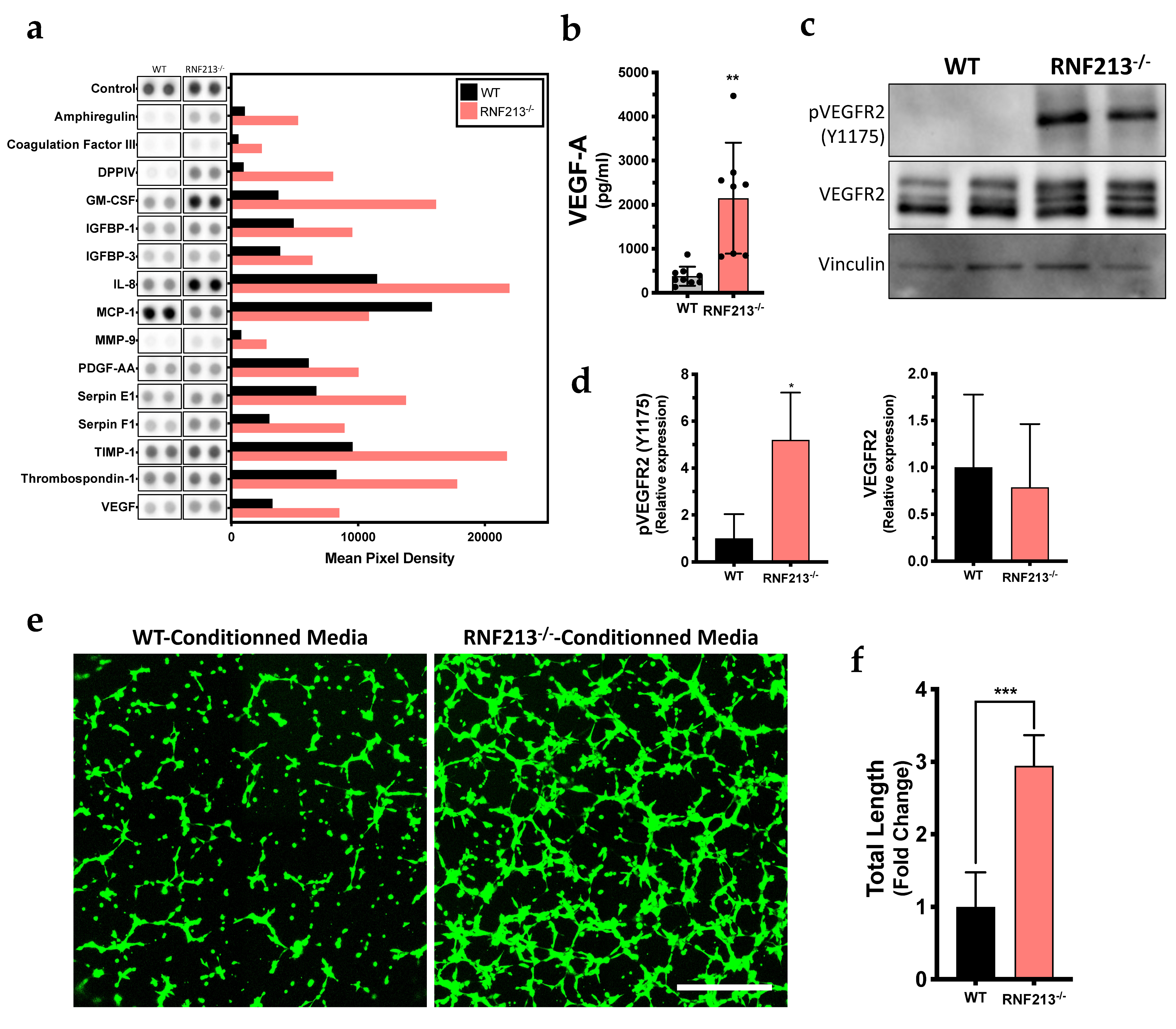
3.4. Inactivation of RNF213 Promotes Angiogenesis through Activation of VEGFR2
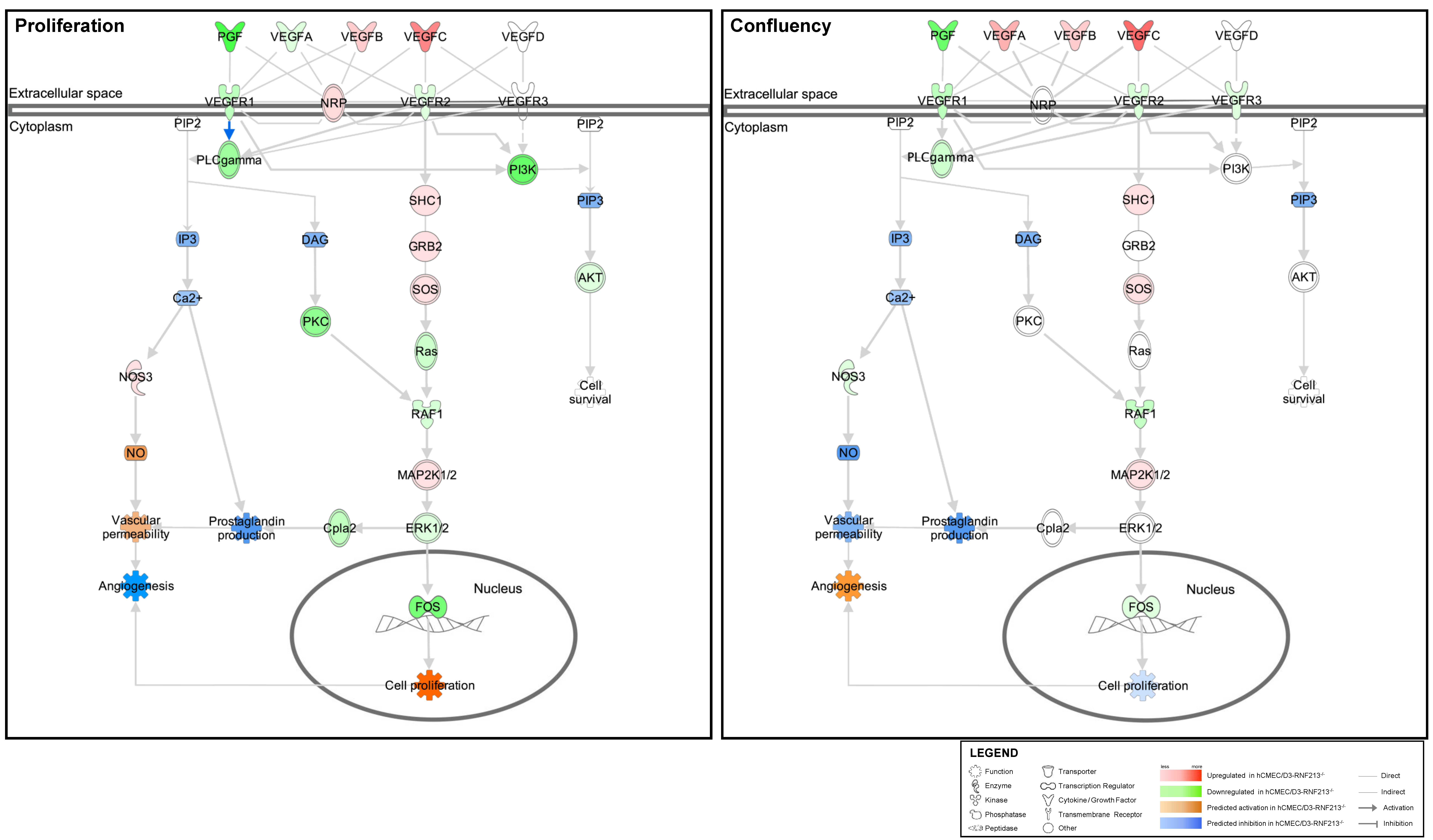
3.5. VEGF Signaling Pathway Prediction Analysis of Functions Associated with Angiogenesis
3.6. Inactivation of RNF213 Promotes the Formation of Abnormal Microvascular Brain Endothelial Cell Network in Tridimensional Tissue-Engineered Cell Culture Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weinberg, D.G.; Arnaout, O.M.; Rahme, R.J.; Aoun, S.G.; Batjer, H.H.; Bendok, B.R. Moyamoya disease: A review of histopathology, biochemistry, and genetics. Neurosurg. Focus 2011, 30, E20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, J.; Takaku, A. Cerebrovascular “moyamoya” disease. Disease showing abnormal net-like vessels in base of brain. Arch. Neurol. 1969, 20, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Czabanka, M.; Peña-Tapia, P.; Schubert, G.A.; Woitzik, J.; Vajkoczy, P.; Schmiedek, P. Characterization of cortical microvascularization in adult moyamoya disease. Stroke 2008, 39, 1703–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, O.Y.; Fujimura, M.; Kim, S.K. The Pathophysiology of Moyamoya Disease: An Update. J. Stroke 2016, 18, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Son, T.O.; Kim, K.H.; Jeon, P.; Hyun, S.H.; Lee, K.H.; Yeon, J.Y.; Kim, J.S.; Hong, S.C.; Shin, H.J.; et al. Neovascularization precedes occlusion in moyamoya disease: Angiographic findings in 172 pediatric patients. Eur. Neurol. 2014, 72, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Houkin, K.; Takahashi, A.; Abe, H. Angiogenic factors in moyamoya disease. Stroke 1996, 27, 2160–2165. [Google Scholar] [CrossRef] [PubMed]
- Bedini, G.; Blecharz, K.G.; Nava, S.; Vajkoczy, P.; Alessandri, G.; Ranieri, M.; Acerbi, F.; Ferroli, P.; Riva, D.; Esposito, S.; et al. Vasculogenic and Angiogenic Pathways in Moyamoya Disease. Curr. Med. Chem. 2016, 23, 315–345. [Google Scholar] [CrossRef]
- Baba, T.; Houkin, K.; Kuroda, S. Novel epidemiological features of moyamoya disease. J. Neurol. Neurosurg. Psychiatry 2008, 79, 900–904. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, L.; Feng, L. Epidemiology, diagnosis and treatment of moyamoya disease. Exp. Med. 2019, 17, 1977–1984. [Google Scholar] [CrossRef] [Green Version]
- Guey, S.; Tournier-Lasserve, E.; Hervé, D.; Kossorotoff, M. Moyamoya disease and syndromes: From genetics to clinical management. Appl. Clin. Genet. 2015, 8, 49–68. [Google Scholar] [CrossRef]
- Liu, W.; Morito, D.; Takashima, S.; Mineharu, Y.; Kobayashi, H.; Hitomi, T.; Hashikata, H.; Matsuura, N.; Yamazaki, S.; Toyoda, A.; et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS ONE 2011, 6, e22542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, F.; Aoki, Y.; Narisawa, A.; Abe, Y.; Komatsuzaki, S.; Kikuchi, A.; Kanno, J.; Niihori, T.; Ono, M.; Ishii, N.; et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J. Hum. Genet. 2011, 56, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morito, D.; Nishikawa, K.; Hoseki, J.; Kitamura, A.; Kotani, Y.; Kiso, K.; Kinjo, M.; Fujiyoshi, Y.; Nagata, K. Moyamoya disease-associated protein mysterin/RNF213 is a novel AAA+ ATPase, which dynamically changes its oligomeric state. Sci. Rep. 2014, 4, 4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollaci, G.; Gorla, G.; Potenza, A.; Carrozzini, T.; Canavero, I.; Bersano, A.; Gatti, L. Novel Multifaceted Roles for RNF213 Protein. Int. J. Mol. Sci. 2022, 23, 4492. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, T.; Habu, T.; Kobayashi, H.; Okuda, H.; Harada, K.H.; Osafune, K.; Taura, D.; Sone, M.; Asaka, I.; Ameku, T.; et al. Downregulation of Securin by the variant RNF213 R4810K (rs112735431, G>A) reduces angiogenic activity of induced pluripotent stem cell-derived vascular endothelial cells from moyamoya patients. Biochem. Biophys. Res. Commun. 2013, 438, 13–19. [Google Scholar] [CrossRef]
- Hitomi, T.; Habu, T.; Kobayashi, H.; Okuda, H.; Harada, K.H.; Osafune, K.; Taura, D.; Sone, M.; Asaka, I.; Ameku, T.; et al. The moyamoya disease susceptibility variant RNF213 R4810K (rs112735431) induces genomic instability by mitotic abnormality. Biochem. Biophys. Res. Commun. 2013, 439, 419–426. [Google Scholar] [CrossRef]
- Ohkubo, K.; Sakai, Y.; Inoue, H.; Akamine, S.; Ishizaki, Y.; Matsushita, Y.; Sanefuji, M.; Torisu, H.; Ihara, K.; Sardiello, M.; et al. Moyamoya disease susceptibility gene RNF213 links inflammatory and angiogenic signals in endothelial cells. Sci. Rep. 2015, 5, 13191. [Google Scholar] [CrossRef] [Green Version]
- Sonobe, S.; Fujimura, M.; Niizuma, K.; Nishijima, Y.; Ito, A.; Shimizu, H.; Kikuchi, A.; Arai-Ichinoi, N.; Kure, S.; Tominaga, T. Temporal profile of the vascular anatomy evaluated by 9.4-T magnetic resonance angiography and histopathological analysis in mice lacking RNF213: A susceptibility gene for moyamoya disease. Brain Res. 2014, 1552, 64–71. [Google Scholar] [CrossRef]
- Kobayashi, H.; Matsuda, Y.; Hitomi, T.; Okuda, H.; Shioi, H.; Matsuda, T.; Imai, H.; Sone, M.; Taura, D.; Harada, K.H.; et al. Biochemical and Functional Characterization of RNF213 (Mysterin) R4810K, a Susceptibility Mutation of Moyamoya Disease, in Angiogenesis In Vitro and In Vivo. J. Am. Heart Assoc. 2015, 4, e002146. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, T.; Enmi, J.I.; Hattori, Y.; Iguchi, S.; Saito, S.; Harada, K.H.; Okuda, H.; Mineharu, Y.; Takagi, Y.; Youssefian, S.; et al. Dysregulation of RNF213 promotes cerebral hypoperfusion. Sci. Rep. 2018, 8, 3607. [Google Scholar] [CrossRef]
- Ito, A.; Fujimura, M.; Niizuma, K.; Kanoke, A.; Sakata, H.; Morita-Fujimura, Y.; Kikuchi, A.; Kure, S.; Tominaga, T. Enhanced post-ischemic angiogenesis in mice lacking RNF213; a susceptibility gene for moyamoya disease. Brain Res. 2015, 1594, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Roy, V.; Ross, J.P.; Pépin, R.; Cortez Ghio, S.; Brodeur, A.; Touzel Deschênes, L.; Le-Bel, G.; Phillips, D.E.; Milot, G.; Dion, P.A.; et al. Moyamoya Disease Susceptibility Gene RNF213 Regulates Endothelial Barrier Function. Stroke 2022, 53, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, T.; Roy, V.; Bourget, J.-M.; Tsutsumi, T.; Picard-Deland, M.; Morin, J.-F.; Gauvin, R.; Ismail, A.A.; Auger, F.A.; Gros-Louis, F. Cell Seeding on UV-C-Treated 3D Polymeric Templates Allows for Cost-Effective Production of Small-Caliber Tissue-Engineered Blood Vessels. Biotechnol. J. 2019, 14, e1800306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komiyama, T.; Nakao, Y.; Toyama, Y.; Asou, H.; Vacanti, C.A.; Vacanti, M.P. A novel technique to isolate adult Schwann cells for an artificial nerve conduit. J. Neurosci. Methods 2003, 122, 195–200. [Google Scholar] [CrossRef]
- Cortez Ghio, S.; Cantin-Warren, L.; Guignard, R.; Larouche, D.; Germain, L. Are the Effects of the Cholera Toxin and Isoproterenol on Human Keratinocytes’ Proliferative Potential Dependent on Whether They Are Co-Cultured with Human or Murine Fibroblast Feeder Layers? Int. J. Mol. Sci. 2018, 19, 2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vang Mouritzen, M.; Jenssen, H. Optimized Scratch Assay for In Vitro Testing of Cell Migration with an Automated Optical Camera. J. Vis. Exp. 2018, 138, e57691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic. Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [Green Version]
- Clément, V.; Roy, V.; Paré, B.; Goulet, C.R.; Deschênes, L.T.; Berthod, F.; Bolduc, S.; Gros-Louis, F. Tridimensional cell culture of dermal fibroblasts promotes exosome-mediated secretion of extracellular matrix proteins. Sci. Rep. 2022, 12, 19786. [Google Scholar] [CrossRef]
- Roy, V.; Lamontagne, R.; Talagas, M.; Touzel-Deschênes, L.; Khuong, H.T.; Saikali, S.; Dupré, N.; Gros-Louis, F. Biofabrication of a three dimensional human-based personalized neurofibroma model. Biotechnol. J. 2021, 16, e2000250. [Google Scholar] [CrossRef]
- Cukierman, E.; Pankov, R.; Stevens, D.R.; Yamada, K.M. Taking cell-matrix adhesions to the third dimension. Science 2001, 294, 1708–1712. [Google Scholar] [CrossRef]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension: How 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthod, F.; Symes, J.; Tremblay, N.; Medin, J.A.; Auger, F.A. Spontaneous fibroblast-derived pericyte recruitment in a human tissue-engineered angiogenesis model in vitro. J. Cell. Physiol. 2012, 227, 2130–2137. [Google Scholar] [CrossRef] [PubMed]
- Sonobe, S.; Fujimura, M.; Niizuma, K.; Fujimura, T.; Furudate, S.; Nishijima, Y.; Kure, S.; Tominaga, T. Increased vascular MMP-9 in mice lacking RNF213: Moyamoya disease susceptibility gene. Neuroreport 2014, 25, 1442–1446. [Google Scholar] [CrossRef] [PubMed]
- Kanoke, A.; Fujimura, M.; Niizuma, K.; Ito, A.; Sakata, H.; Sato-Maeda, M.; Morita-Fujimura, Y.; Kure, S.; Tominaga, T. Temporal profile of the vascular anatomy evaluated by 9.4-tesla magnetic resonance angiography and histological analysis in mice with the R4859K mutation of RNF213, the susceptibility gene for moyamoya disease. Brain Res. 2015, 1624, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.T.; Chang, H.Y.; Haraldsen, G.; Jahnsen, F.L.; Troyanskaya, O.G.; Chang, D.S.; Wang, Z.; Rockson, S.G.; van de Rijn, M.; Botstein, D.; et al. Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. USA 2003, 100, 10623–10628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urich, E.; Lazic, S.E.; Molnos, J.; Wells, I.; Freskgård, P.O. Transcriptional profiling of human brain endothelial cells reveals key properties crucial for predictive in vitro blood–brain barrier models. PLoS ONE 2012, 7, e38149. [Google Scholar] [CrossRef] [Green Version]
- Wolburg, H.; Lippoldt, A. Tight junctions of the blood–brain barrier: Development, composition and regulation. Vasc. Pharm. 2002, 38, 323–337. [Google Scholar] [CrossRef]
- Zhang, L.; Rashad, S.; Zhou, Y.; Niizuma, K.; Tominaga, T. RNF213 loss of function reshapes vascular transcriptome and spliceosome leading to disrupted angiogenesis and aggravated vascular inflammatory responses. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2022, 11, 271678x221110679. [Google Scholar] [CrossRef]
- Kim, D.S.; Lee, M.W.; Lee, T.H.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Cell culture density affects the stemness gene expression of adipose tissue-derived mesenchymal stem cells. Biomed Rep. 2017, 6, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, M. Vascular endothelial growth factor (VEGF)-Receptor2: Its biological functions, major signaling pathway, and specific ligand VEGF-E. Endothelium 2006, 13, 63–69. [Google Scholar] [CrossRef]
- Wang, X.; Bove, A.M.; Simone, G.; Ma, B. Molecular Bases of VEGFR-2-Mediated Physiological Function and Pathological Role. Front. Cell Dev. Biol. 2020, 8, 599281. [Google Scholar] [CrossRef] [PubMed]
- Clegg, L.W.; Mac Gabhann, F. Site-Specific Phosphorylation of VEGFR2 Is Mediated by Receptor Trafficking: Insights from a Computational Model. PLoS Comput. Biol. 2015, 11, e1004158. [Google Scholar] [CrossRef] [PubMed]
- Sase, H.; Watabe, T.; Kawasaki, K.; Miyazono, K.; Miyazawa, K. VEGFR2-PLCgamma1 axis is essential for endothelial specification of VEGFR2+ vascular progenitor cells. J. Cell Sci. 2009, 122, 3303–3311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Yamaguchi, S.; Chida, K.; Shibuya, M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. Embo J. 2001, 20, 2768–2778. [Google Scholar] [CrossRef] [Green Version]
- Woodfin, A.; Voisin, M.B.; Nourshargh, S. PECAM-1: A multi-functional molecule in inflammation and vascular biology. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2514–2523. [Google Scholar] [CrossRef] [Green Version]
- Cao, G.; O’Brien, C.D.; Zhou, Z.; Sanders, S.M.; Greenbaum, J.N.; Makrigiannakis, A.; DeLisser, H.M. Involvement of human PECAM-1 in angiogenesis and in vitro endothelial cell migration. Am. J. Physiology. Cell Physiol. 2002, 282, C1181–C1190. [Google Scholar] [CrossRef]
- Stevens, H.Y.; Melchior, B.; Bell, K.S.; Yun, S.; Yeh, J.C.; Frangos, J.A. PECAM-1 is a critical mediator of atherosclerosis. Dis. Model Mech. 2008, 1, 175–181; discussion 179. [Google Scholar] [CrossRef] [Green Version]
- Lertkiatmongkol, P.; Paddock, C.; Newman, D.K.; Zhu, J.; Thomas, M.J.; Newman, P.J. The Role of Sialylated Glycans in Human Platelet Endothelial Cell Adhesion Molecule 1 (PECAM-1)-mediated Trans Homophilic Interactions and Endothelial Cell Barrier Function. J. Biol. Chem. 2016, 291, 26216–26225. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Liu, A.; Miranda-Ribera, A.; Hyun, S.W.; Lillehoj, E.P.; Cross, A.S.; Passaniti, A.; Grimm, P.R.; Kim, B.Y.; Welling, P.A.; et al. NEU1 sialidase regulates the sialylation state of CD31 and disrupts CD31-driven capillary-like tube formation in human lung microvascular endothelia. J. Biol. Chem. 2014, 289, 9121–9135. [Google Scholar] [CrossRef] [Green Version]
- Imamaki, R.; Ogawa, K.; Kizuka, Y.; Komi, Y.; Kojima, S.; Kotani, N.; Honke, K.; Honda, T.; Taniguchi, N.; Kitazume, S. Glycosylation controls cooperative PECAM-VEGFR2-β3 integrin functions at the endothelial surface for tumor angiogenesis. Oncogene 2018, 37, 4287–4299. [Google Scholar] [CrossRef]
- Abuoliat, Z.A.; AlFarhan, B.A.; Alshahrani, A.A.; AlFarhan, A.A.; Almuntashri, M.A.; Alotaibi, N. Atypical Location of Intracerebral Hemorrhage in Moyamoya Disease. Cureus 2017, 9, e1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yang, Y.; Li, X.; Zhou, F.; Wu, Z.; Liang, Q.; Liu, Y.; Wang, Y.; Na, S.; Chen, X.; et al. Lateral Posterior Choroidal Collateral Anastomosis Predicts Recurrent Ipsilateral Hemorrhage in Adult Patients with Moyamoya Disease. AJNR. Am. J. Neuroradiol. 2019, 40, 1665–1671. [Google Scholar] [CrossRef]
- Lv, X. Hemorrhagic Factors of Moyamoya Disease. AJNR. Am. J. Neuroradiol. 2019, 40, 1672–1673. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Shen, M.; Tong, F.; Cong, M.; Zhang, S.; Gong, Y.; Ding, F. Differential Gene Expression in Primary Cultured Sensory and Motor Nerve Fibroblasts. Front. Neurosci. 2018, 12, 1016. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro. Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ye, M.; Wu, M.; Fang, H.; Xiao, B.; Xie, L.; Zhu, X. RNF213 suppresses carcinogenesis in glioblastoma by affecting MAPK/JNK signaling pathway. Clin. Transl. Oncol. 2020, 22, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial influence on the blood brain barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, V.; Brodeur, A.; Touzel Deschênes, L.; Dupré, N.; Gros-Louis, F. RNF213 Loss-of-Function Promotes Angiogenesis of Cerebral Microvascular Endothelial Cells in a Cellular State Dependent Manner. Cells 2023, 12, 78. https://doi.org/10.3390/cells12010078
Roy V, Brodeur A, Touzel Deschênes L, Dupré N, Gros-Louis F. RNF213 Loss-of-Function Promotes Angiogenesis of Cerebral Microvascular Endothelial Cells in a Cellular State Dependent Manner. Cells. 2023; 12(1):78. https://doi.org/10.3390/cells12010078
Chicago/Turabian StyleRoy, Vincent, Alyssa Brodeur, Lydia Touzel Deschênes, Nicolas Dupré, and François Gros-Louis. 2023. "RNF213 Loss-of-Function Promotes Angiogenesis of Cerebral Microvascular Endothelial Cells in a Cellular State Dependent Manner" Cells 12, no. 1: 78. https://doi.org/10.3390/cells12010078