
Mitochondrial Unfolded Protein Response and Integrated Stress Response as Promising Therapeutic Targets for Mitochondrial Diseases
Abstract
:1. Introduction
2. UPRmt
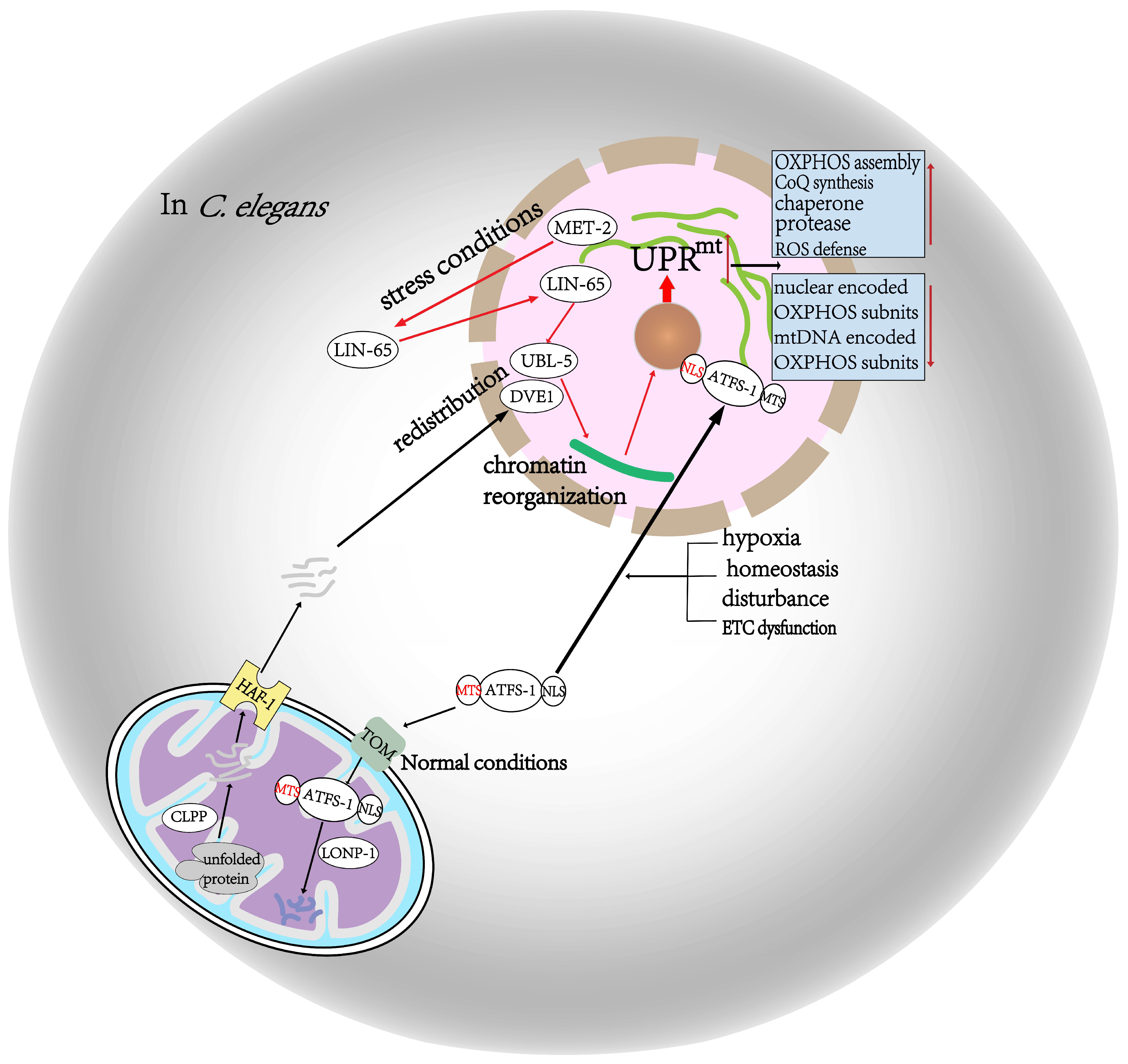
2.1. In C. Elegans
2.2. In Mammals
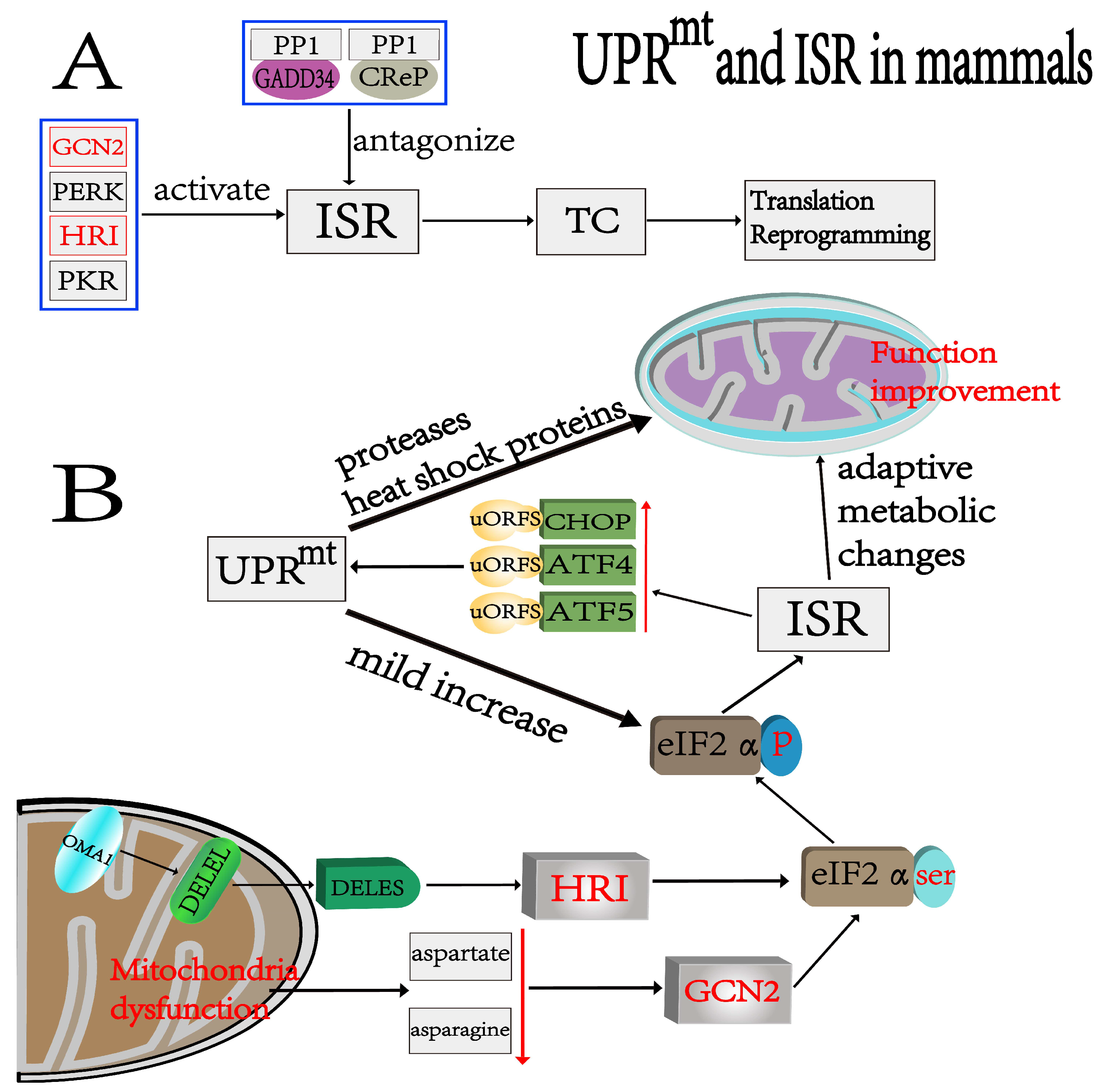
3. ISR
4. Relationship between UPRmt and ISR
5. Mitochondrial Diseases
5.1. MDs Caused by Mutations in mtDNA
5.2. MDs Caused by Mutations in nDNA
6. Roles of UPRmt and ISR in Mitochondrial Diseases
7. Treatment of Mitochondrial Diseases
7.1. Current Treatments for Mitochondrial Diseases
7.2. Potential Therapies for Mitochondrial Diseases Targeting UPRmt and ISR
7.3. Advantages of UPRmt and ISR in the Treatment of Mitochondrial Diseases
8. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nature 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Berendzen, K.M.; Durieux, J.; Shao, L.W.; Tian, Y.; Kim, H.E.; Wolff, S.; Liu, Y.; Dillin, A. Neuroendocrine Coordination of Mitochondrial Stress Signaling and Proteostasis. Cell 2016, 166, 1553–1563.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, V.S.; Morgan, M.M.; Scott, R.; Clements, L.S.; Butow, R.A. The mitochondrial genotype can influence nuclear gene expression in yeast. Science 1987, 235, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Hoj, P.B.; Hoogenraad, N.J. Selective induc-tion of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 1996, 240, 98–103. [Google Scholar] [CrossRef]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial Import Efficiency of ATFS-1 Regulates Mitochondrial UPR Activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [Green Version]
- DiMauro, S.; Schon, E.A. Mitochondrial Respiratory-Chain Diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef]
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V.; et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Janer, A.; Prudent, J.; Paupe, V.; Fahiminiya, S.; Majewski, J.; Sgarioto, N.; Rosiers, C.D.; Forest, A.; Lin, Z.Y.; Gingras, A.C.; et al. SLC25A46 is required for mitochondrial lipid homeostasis and cristae maintnance and is responsible for Leigh syndrome. EMBO Mol. Med. 2016, 8, 1019–1038. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, X.; Li, C.; Li, Q.; An, Y.A.; Luo, X.; Deng, Y.; Gillette, T.G.; Scherer, P.E.; Wang, Z.V. Integrated Stress Response Couples Mitochondrial Protein Translation with Oxidative Stress Control. Circulation 2021, 144, 1500–1515. [Google Scholar] [CrossRef]
- Anderson, N.S.; Haynes, C.M. Folding the Mitochondrial UPR into the Integrated Stress Response. Trends Cell Biol. 2020, 30, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Benedetti, C.; Urano, F.; Clark, S.G.; Harding, H.P.; Ron, D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 2004, 117, 4055–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaskaran, S.; Pharaoh, G.; Ranjit, R.; Murphy, A.; Matsuzaki, S.; Nair, B.C.; Forbes, B.; Gispert, S.; Auburger, G.; Humphries, K.M.; et al. Loss of mitochondrial protease ClpP protects mice from diet-induced obesity and insulin resistance. EMBO Rep. 2018, 19, e45009. [Google Scholar] [CrossRef]
- Haynes, C.M.; Yang, Y.; Blais, S.P.; Neubert, T.A.; Ron, D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 2010, 37, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPRmt. Cell 2016, 165, 1197–1208. [Google Scholar] [CrossRef] [Green Version]
- Gao, K.; Li, Y.; Hu, S.; Liu, Y. SUMO peptidase ULP-4 regulates mitochondrial UPR-mediated innate immunity and lifespan extension. Elife 2019, 8, e41792. [Google Scholar] [CrossRef]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Shpilka, T.; Du, Y.; Yang, Q.; Melber, A.; Uma, N.N.; Lavelle, J.; Kim, S.; Liu, P.; Weidberg, H.; Li, R.; et al. UPR(mt) scales mitochondrial network expansion with protein synthesis via mitochondrial import in Caenorhabditis elegans. Nat. Commun. 2021, 12, 479. [Google Scholar] [CrossRef]
- Nargund, A.M.; Fiorese, C.J.; Pellegrino, M.W.; Deng, P.; Haynes, C.M. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol. Cell 2015, 58, 123–133. [Google Scholar] [CrossRef]
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.-F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samluk, L.; Urbanska, M.; Kisielewska, K.; Mohanraj, K.; Kim, M.-J.; Machnicka, K.; Liszewska, E.; Jaworski, J.; Chacinska, A. Cytosolic translational responses differ under conditions of severe short-term and long-term mitochondrial stress. Mol. Biol. Cell 2019, 30, 1864–1877. [Google Scholar] [CrossRef] [PubMed]
- Forsstrom, S.; Jackson, C.B.; Carroll, C.J.; Kuronen, M.; Pirinen, E.; Pradhan, S.; Marmyleva, A.; Auranen, M.; Kleine, I.M.; Khan, N.A.; et al. Fibroblast Growth Factor 21 Drives Dynamics of Local and Systemic Stress Responses in Mitochondrial Myopathy with mtDNA Deletions. Cell Metab. 2019, 30, 1040–1054.e7. [Google Scholar] [CrossRef] [PubMed]
- Sayles, N.M.; Southwell, N.; McAvoy, K.; Kim, K.; Pesini, A.; Anderson, C.J.; Quinzii, C.; Cloonan, S.; Kawamata, H.; Manfredi, G. Mutant CHCHD10 causes an extensive metabolic rewiring that precedes OXPHOS dysfunction in a murine model of mitochondrial cardiomyopathy. Cell Rep. 2022, 38, 110475. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Nargund, A.M.; Sun, T.; Haynes, C.M. Protective coupling of mitochondrial function and protein syn-thesis via the eIF2alpha kinase GCN-2. PLoS Genet. 2012, 8, e1002760. [Google Scholar] [CrossRef] [Green Version]
- Weng, H.; Ma, Y.; Chen, L.; Cai, G.; Chen, Z.; Zhang, S.; Ye, Q. A New Vision of Mitochondrial Unfolded Protein Response to the Sirtuin Family. Curr. Neuropharmacol. 2020, 18, 613–623. [Google Scholar] [CrossRef]
- Khan, R.S.; Dine, K.; Das, S.J.; Shindler, K.S. SIRT1 activating compounds reduce oxidative stress mediated neuronal loss in viral induced CNS demyelinating disease. Acta Neuropathol. Commun. 2014, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-F.; Sam, J.; Evans, T. Sirt1 promotes tissue regeneration in zebrafish through regulating the mitochondrial unfolded protein response. iScience 2021, 24, 103118. [Google Scholar] [CrossRef]
- Gariani, K.; Ryu, D.; Menzies, K.J.; Yi, H.-S.; Stein, S.; Zhang, H.; Perino, A.; Lemos, V.; Katsyuba, E.; Jha, P.; et al. Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease. J. Hepatol. 2017, 66, 132–141. [Google Scholar] [CrossRef]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Hinnebusch, A.G.; Ivanov, I.P.; Sonenberg, N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 2016, 352, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.S.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [Green Version]
- Palam, L.R.; Baird, T.D.; Wek, R.C. Phosphorylation of eIF2 Facilitates Ribosomal Bypass of an Inhibitory Upstream ORF to Enhance CHOP Translation. J. Biol. Chem. 2011, 286, 10939–10949. [Google Scholar] [CrossRef] [Green Version]
- Watatani, Y.; Ichikawa, K.; Nakanishi, N.; Fujimoto, M.; Takeda, H.; Kimura, N.; Hirose, H.; Takahashi, S.; Takahashi, Y. Stress-induced Translation of ATF5 mRNA Is Regulated by the 5′-Untranslated Region. J. Biol. Chem. 2008, 283, 2543–2553. [Google Scholar] [CrossRef] [Green Version]
- Zyryanova, A.F.; Weis, F.; Faille, A.; Alard, A.A.; Crespillo-Casado, A.; Sekine, Y.; Harding, H.P.; Allen, F.; Parts, L.; Fromont, C.; et al. Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eIF2B. Science 2018, 359, 1533–1536. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 2020, 22, 54–70. [Google Scholar] [CrossRef]
- Mick, E.; Titov, D.V.; Skinner, O.S.; Sharma, R.; A Jourdain, A.; Mootha, V.K. Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. Elife 2020, 9, e49178. [Google Scholar] [CrossRef]
- Misra, J.; Holmes, M.J.; Mirek, E.T.; Langevin, M.; Kim, H.-G.; Carlson, K.R.; Watford, M.; Dong, X.C.; Anthony, T.G.; Wek, R.C. Discordant regulation of eIF2 kinase GCN2 and mTORC1 during nutrient stress. Nucleic Acids Res. 2021, 49, 5726–5742. [Google Scholar] [CrossRef] [PubMed]
- Young, S.K.; Wek, R.C. Upstream Open Reading Frames Differentially Regulate Gene-specific Translation in the Integrated Stress Response. J. Biol. Chem. 2016, 291, 16927–16935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaspar, S.; Oertlin, C.; Szczepanowska, K.; Kukat, A.; Senft, K.; Lucas, C.; Brodesser, S.; Hatzoglou, M.; Larsson, O.; Topisirovic, I.; et al. Adaptation to mitochondrial stress requires CHOP-directed tuning of ISR. Sci. Adv. 2021, 7, eabf0971. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.; Eckl, E.-M.; Schmitt, S.; Mancilla, I.A.; Meyer-Bender, M.F.; Hanf, M.; Philippou-Massier, J.; Krebs, S.; Zischka, H.; Jae, L.T. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 2020, 579, 433–437. [Google Scholar] [CrossRef]
- Hao, L.; Zhong, W.; Dong, H.; Guo, W.; Sun, X.; Zhang, W.; Yue, R.; Li, T.; Griffiths, A.; Ahmadi, A.R.; et al. ATF4 activation promotes hepatic mitochondrial dysfunction by repressing NRF1–TFAM signalling in alcoholic steatohepatitis. Gut 2020, 70, 1933–1945. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Jiang, Z.; Yan, X.; Liu, J.; Li, F.; Liu, P.; Li, J.; Wei, Y.; Sun, Y.E.; Zhang, Y.; et al. ATF5, a putative therapeutic target for the mitochondrial DNA 3243A > G mutation-related disease. Cell Death Dis. 2021, 12, 701. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.Y.W.; Wai, T.; Simonsen, A. Quality control of the mitochondrion. Dev. Cell 2021, 56, 881–905. [Google Scholar] [CrossRef] [PubMed]
- Eckl, E.-M.; Ziegemann, O.; Krumwiede, L.; Fessler, E.; Jae, L.T. Sensing, signaling and surviving mitochondrial stress. Cell Mol. Life Sci. 2021, 78, 5925–5951. [Google Scholar] [CrossRef] [PubMed]
- Urbina-Varela, R.; Castillo, N.; Videla, L.A.; Del Campo, A. Impact of Mitophagy and Mitochondrial Unfolded Protein Response as New Adaptive Mechanisms Underlying Old Pathologies: Sarcopenia and Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 7704. [Google Scholar] [CrossRef]
- Münch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef]
- Deng, P.; Haynes, C.M. Mitochondrial dysfunction in cancer: Potential roles of ATF5 and the mitochondrial UPR. Semin. Cancer Biol. 2017, 47, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Durieux, J.; Wolff, S.; Dillin, A. The Cell-Non-Autonomous Nature of Electron Transport Chain-Mediated Longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.-B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR Inhibition Alleviates Mitochondrial Disease in a Mouse Model of Leigh Syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobava, R.; Mao, Y.; Guan, B.-J.; Hu, D.; Krokowski, D.; Chen, C.-W.; Shu, X.E.; Chukwurah, E.; Wu, J.; Gao, Z.; et al. Adaptive translational pausing is a hallmark of the cellular response to severe environmental stress. Mol. Cell 2021, 81, 4191–4208.e8. [Google Scholar] [CrossRef]
- Ng, Y.S.; Bindoff, L.A.; Gorman, G.S.; Klopstock, T.; Kornblum, C.; Mancuso, M.; McFarland, R.; Sue, C.M.; Suoma-lainen, A.; Taylor, R.W.; et al. Mitochondrial disease in adults: Recent advances and future promise. Lancet Neurol. 2021, 20, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Lightowlers, R.N.; Taylor, R.W.; Turnbull, D.M. Mutations causing mitochondrial disease: What is new and what challenges remain? Science 2015, 349, 1494–1499. [Google Scholar] [CrossRef] [PubMed]
- Molnar, M.J.; Kovacs, G.G. Mitochondrial diseases. Handb. Clin. Neurol. 2017, 145, 147–155. [Google Scholar]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Shammas, M.K.; Huang, X.; Wu, B.P.; Fessler, E.; Song, I.Y.; Randolph, N.P.; Li, Y.; Bleck, C.K.; Springer, D.A.; Fratter, C.; et al. OMA1 mediates local and global stress responses against protein misfolding in CHCHD10 mitochondrial myopathy. J. Clin. Investig. 2022, 132, e157504. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Renkema, G.H.; Bras, M.; van den Heuvel, L.P.; Hoischen, A.; Gilissen, C.; Nabuurs, S.B.; Huynen, M.A.; de Vries, M.C.; Smeitink, J.A.; et al. A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain 2013, 136, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Bonora, E.; Chakrabarty, S.; Kellaris, G.; Tsutsumi, M.; Bianco, F.; Bergamini, C.; Ullah, F.; Isidori, F.; Liparulo, I.; Diquigiovanni, C.; et al. Biallelic variants in LIG3 cause a novel mitochondrial neurogastrointestinal encephalomyopathy. Brain 2021, 144, 1451–1466. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.; Hirano, M.; DiMauro, S. Mitochondrial diseases. Neurol. Clin. 2002, 20, 809–839, vii–viii. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Copeland, W.C. POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol. 2018, 15, 40–52. [Google Scholar] [CrossRef]
- Yonova-Doing, E.; Calabrese, C.; Gomez-Duran, A.; Schon, K.; Wei, W.; Karthikeyan, S.; Chinnery, P.F.; Howson, J.M.M. An atlas of mitochondrial DNA genotype–phenotype associations in the UK Biobank. Nat. Genet. 2021, 53, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Duanmu, X.; Zeng, L.; Liu, B.; Song, Z. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells 2019, 8, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, G.A.; Gahlon, H.L. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020, 48, 11244–11258. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, R.; Nakahira, K.; Gu, Z. Mitochondrial DNA Mutation, Diseases, and Nutrient-Regulated Mitophagy. Annu. Rev. Nutr. 2019, 39, 201–226. [Google Scholar] [CrossRef]
- McCormick, E.M.; Lott, M.T.; Dulik, M.C.; Shen, L.; Attimonelli, M.; Vitale, O.; Karaa, A.; Bai, R.; Pineda-Alvarez, D.E.; Singh, L.N.; et al. Specifications of the ACMG/AMP standards and guidelines for mitochondrial DNA variant interpretation. Hum. Mutat. 2020, 41, 2028–2057. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef]
- Caporali, L.; Bello, L.; Tagliavini, F.; La Morgia, C.; Maresca, A.; Di Vito, L.; Liguori, R.; Valentino, M.L.; Cecchin, D.; Pegoraro, E.; et al. DGUOK recessive mutations in patients with CPEO, mitochondrial myopathy, parkinsonism and mtDNA deletions. Brain 2017, 141, e3. [Google Scholar] [CrossRef]
- Pickett, S.J.; Grady, J.P.; Ng, Y.S.; Gorman, G.S.; Schaefer, A.M.; Wilson, I.J.; Cordell, H.J.; Turnbull, D.M.; Taylor, R.W.; McFarland, R. Phenotypic heterogeneity in m.3243A>G mitochondrial disease: The role of nuclear factors. Ann. Clin. Transl. Neurol. 2018, 5, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Bourgeron, T.; Rustin, P.; Chretien, D.; Birch-Machin, M.; Bourgeois, M.; Viegas-Péquignot, E.; Munnich, A.; Rotig, A. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat. Genet. 1995, 11, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Berger, I.; Hershkovitz, E.; Shaag, A.; Edvardson, S.; Saada, A.; Elpeleg, O. Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann. Neurol. 2008, 63, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Caballero, L.; Ruzzenente, B.; Bianchi, L.; Assouline, Z.; Barcia, G.; Metodiev, M.D.; Rio, M.; Funalot, B.; van den Brand, M.A.; Guerrero-Castillo, S.; et al. Mutations in Complex I Assembly Factor TMEM126B Result in Muscle Weakness and Isolated Complex I Deficiency. Am. J. Hum. Genet. 2016, 99, 208–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, M.; Silvestri, G.; Blake, D.M.; Lombes, A.; Minetti, C.; Bonilla, E.; Hays, A.P.; Lovelace, R.E.; Butler, I.; Berto-rini, T.E.; et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): Clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology 1994, 44, 721–727. [Google Scholar] [CrossRef]
- Nishino, I.; Spinazzola, A.; Hirano, M. Thymidine Phosphorylase Gene Mutations in MNGIE, a Human Mitochondrial Disorder. Science 1999, 283, 689–692. [Google Scholar] [CrossRef]
- Güner, F.; Pozner, T.; Krach, F.; Prots, I.; Loskarn, S.; Schlötzer-Schrehardt, U.; Winkler, J.; Winner, B.; Regensburger, M. Axon-Specific Mitochondrial Pathology in SPG11 Alpha Motor Neurons. Front. Neurosci. 2021, 15, 680572. [Google Scholar] [CrossRef]
- Frazier, A.E.; Thorburn, D.R.; Compton, A.G. Mitochondrial energy generation disorders: Genes, mechanisms, and clues to pathology. J. Biol. Chem. 2019, 294, 5386–5395. [Google Scholar] [CrossRef] [Green Version]
- Thompson, K.; Collier, J.J.; Glasgow, R.I.C.; Robertson, F.M.; Pyle, A.; Blakely, E.L.; Alston, C.L.; Oláhová, M.; McFarland, R.; Taylor, R.W. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J. Inherit. Metab. Dis. 2019, 43, 36–50. [Google Scholar] [CrossRef] [Green Version]
- Repp, B.M.; Mastantuono, E.; Alston, C.L.; Schiff, M.; Haack, T.B.; Rotig, A.; Ardissone, A.; Lombes, A.; Catarino, C.B.; Diodato, D.; et al. Clinical, bio-chemical and genetic spectrum of 70 patients with ACAD9 deficiency: Is riboflavin supplementation effective? Orphanet. J. Rare Dis. 2018, 13, 120. [Google Scholar] [CrossRef]
- Hirano, M.; Emmanuele, V.; Quinzii, C.M. Emerging therapies for mitochondrial diseases. Essays Biochem. 2018, 62, 467–481. [Google Scholar] [CrossRef]
- Fullerton, M.; McFarland, R.; Taylor, R.W.; Alston, C.L. The genetic basis of isolated mitochondrial complex II deficiency. Mol. Genet. Metab. 2020, 131, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Gusic, M.; Schottmann, G.; Feichtinger, R.G.; Du, C.; Scholz, C.; Wagner, M.; Mayr, J.A.; Lee, C.Y.; Yépez, V.A.; Lorenz, N.; et al. Bi-Allelic UQCRFS1 Variants Are Associated with Mitochondrial Complex III Deficiency, Cardiomyopathy, and Alopecia Totalis. Am. J. Hum. Genet. 2020, 106, 102–111. [Google Scholar] [CrossRef]
- Wei, X.; Du, M.; Li, D.; Wen, S.; Xie, J.; Li, Y.; Chen, A.; Zhang, K.; Xu, P.; Jia, M.; et al. Mutations in FASTKD2 are associated with mitochondrial disease with multi-OXPHOS deficiency. Hum. Mutat. 2020, 41, 961–972. [Google Scholar] [CrossRef]
- Edvardson, S.; Porcelli, V.; Jalas, C.; Soiferman, D.; Kellner, Y.; Shaag, A.; Korman, S.H.; Pierri, C.L.; Scarcia, P.; Fraenkel, N.D.; et al. Agenesis of corpus callosum and optic nerve hypoplasia due to mutations in SLC25A1 encoding the mitochondrial citrate transporter. J. Med. Genet. 2013, 50, 240–245. [Google Scholar] [CrossRef]
- Rahman, J.; Rahman, S. Mitochondrial medicine in the omics era. Lancet 2018, 391, 2560–2574. [Google Scholar] [CrossRef] [Green Version]
- Mito, T.; Vincent, A.E.; Faitg, J.; Taylor, R.W.; Khan, N.A.; McWilliams, T.G.; Suomalainen, A. Mosaic dysfunction of mitophagy in mitochondrial muscle disease. Cell Metab. 2022, 34, 197–208.e5. [Google Scholar] [CrossRef]
- Ahola, S.; Mejías, P.R.; Hermans, S.; Chandragiri, S.; Giavalisco, P.; Nolte, H.; Langer, T. OMA1-mediated inte-grated stress response protects against ferroptosis in mitochondrial cardiomyopathy. Cell Metab. 2022, 34, 1875–1891. [Google Scholar] [CrossRef]
- Lin, Y.F.; Schulz, A.M.; Pellegrino, M.W.; Lu, Y.; Shaham, S.; Haynes, C.M. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 2016, 533, 416–419. [Google Scholar] [CrossRef] [Green Version]
- Gitschlag, B.L.; Kirby, C.S.; Samuels, D.C.; Gangula, R.D.; Mallal, S.A.; Patel, M.R. Homeostatic Responses Regulate Selfish Mitochondrial Genome Dynamics in C. elegans. Cell Metab. 2016, 24, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Weidling, I.; Swerdlow, R.H. Mitochondrial Dysfunction and Stress Responses in Alzheimer’s Disease. Biology 2019, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Onat, U.I.; Yildirim, A.D.; Tufanli, O.; Çimen, I.; Kocatürk, B.; Veli, Z.; Hamid, S.M.; Shimada, K.; Chen, S.; Sin, J.; et al. Intercepting the Lipid-Induced Integrated Stress Response Reduces Atherosclerosis. J. Am. Coll. Cardiol. 2019, 73, 1149–1169. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, R.P.; Chandel, N.S. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem. Cell 2021, 28, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Avula, S.; Parikh, S.; Demarest, S.; Kurz, J.; Gropman, A. Treatment of mitochondrial disorders. Curr. Treat Options Neurol. 2014, 16, 292. [Google Scholar] [CrossRef] [PubMed]
- Klopstock, T.; Metz, G.; Yu-Wai-Man, P.; Büchner, B.; Gallenmüller, C.; Bailie, M.; Nwali, N.; Griffiths, P.G.; Von Livonius, B.; Reznicek, L.; et al. Persistence of the treatment effect of idebenone in Leber’s hereditary optic neuropathy. Brain 2013, 136, e230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmalak, M.; Lew, A.; Ramezani, R.; Shroads, A.L.; Coats, B.S.; Langaee, T.; Shankar, M.N.; Neiberger, R.E.; Subramony, S.; Stacpoole, P.W. Long-term safety of dichloroacetate in congenital lactic acidosis. Mol. Genet. Metab. 2013, 109, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Douiev, L.; Sheffer, R.; Horvath, G.; Saada, A. Bezafibrate Improves Mitochondrial Fission and Function in DNM1L-Deficient Patient Cells. Cells 2020, 9, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Jang, I.A.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Resveratrol, an Nrf2 activator, ameliorates aging-related progressive renal injury. Aging 2018, 10, 83–99. [Google Scholar] [CrossRef] [Green Version]
- Feuer, W.J.; Schiffman, J.C.; Davis, J.L.; Porciatti, V.; Gonzalez, P.; Koilkonda, R.D.; Yuan, H.; Lalwani, A.; Lam, B.L.; Guy, J. Gene Therapy for Leber Hereditary Optic Neuropathy: Initial Results. Ophthalmology 2016, 123, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Biousse, V.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Barboni, P.; et al. Efficacy and Safety of Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy Treated within 6 Months of Disease Onset. Ophthalmology 2021, 128, 649–660. [Google Scholar] [CrossRef]
- Wan, X.; Pei, H.; Zhao, M.J.; Yang, S.; Hu, W.K.; He, H.; Ma, S.Q.; Zhang, G.; Dong, X.Y.; Chen, C.; et al. Efficacy and Safety of rAAV2-ND4 Treatment for Leber’s Hereditary Optic Neuropathy. Sci. Rep. 2016, 6, 21587. [Google Scholar] [CrossRef] [Green Version]
- Enns, G.M. Treatment of mitochondrial disorders: Antioxidants and beyond. J. Child Neurol. 2014, 29, 1235–1240. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [Green Version]
- El-Hattab, A.W.; Emrick, L.T.; Hsu, J.W.; Chanprasert, S.; Almannai, M.; Craigen, W.J.; Jahoor, F.; Scaglia, F. Impaired nitric oxide production in children with MELAS syndrome and the effect of arginine and citrulline supplementation. Mol. Genet. Metab. 2016, 117, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Gomez, C.; Levy, R.J.; Sanchez-Quintero, M.J.; Juanola-Falgarona, M.; Barca, E.; Garcia-Diaz, B.; Tadesse, S.; Garone, C.; Hirano, M. Deoxycytidine and Deoxythymidine Treatment for Thymidine Kinase 2 Deficiency. Ann. Neurol. 2017, 81, 641–652. [Google Scholar] [CrossRef] [Green Version]
- Suomalainen, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2018, 19, 77–92. [Google Scholar] [CrossRef]
- Palmeira, C.; Teodoro, J.; Amorim, J.A.; Steegborn, C.; Sinclair, D.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free. Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef]
- Lanzillotta, C.; Di Domenico, F. Stress Responses in Down Syndrome Neurodegeneration: State of the Art and Therapeutic Molecules. Biomolecules 2021, 11, 266. [Google Scholar] [CrossRef]
- Zhang, B.; Tan, Y.; Zhang, Z.; Feng, P.; Ding, W.; Wang, Q.; Liang, H.; Duan, W.; Wang, X.; Yu, S.; et al. Novel PGC-1alpha/ATF5 Axis Partly Activates UPR(mt) and Mediates Cardioprotective Role of Tetrahydrocurcumin in Pathological Cardiac Hypertrophy. Oxid Med. Cell Longev. 2020, 2020, 9187065. [Google Scholar] [CrossRef]
- Xu, M.; Xue, R.-Q.; Lu, Y.; Yong, S.-Y.; Wu, Q.; Cui, Y.-L.; Zuo, X.-T.; Yu, X.-J.; Zhao, M.; Zang, W.-J. Choline ameliorates cardiac hypertrophy by regulating metabolic remodelling and UPRmt through SIRT3-AMPK pathway. Cardiovasc. Res. 2018, 115, 530–545. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Romero-Gonzalez, A.; Gomez-Fernandez, D.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; et al. Pterostilbene in Combination with Mitochondrial Cofactors Improve Mitochondrial Function in Cellular Models of Mitochondrial Diseases. Front. Pharmacol. 2022, 13, 862085. [Google Scholar] [CrossRef] [PubMed]
- Germain, D. Sirtuins and the Estrogen Receptor as Regulators of the Mammalian Mitochondrial UPR in Cancer and Aging. Adv. Cancer Res. 2016, 130, 211–256. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhu, L.; Qiu, W.; Liu, Y.; Yang, F.; Chen, W.; Xu, R. Nicotinamide Riboside Enhances Mitochondrial Proteostasis and Adult Neurogenesis through Activation of Mitochondrial Unfolded Protein Response Signaling in the Brain of ALS SOD1(G93A) Mice. Int. J. Biol. Sci. 2020, 16, 284–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Ostrovsky, J.; Kwon, Y.J.; Polyak, E.; Licata, J.; Tsukikawa, M.; Marty, E.; Thomas, J.; Felix, C.A.; Xiao, R.; et al. Inhibiting cytosolic translation and autophagy improves health in mitochondrial disease. Hum. Mol. Genet. 2015, 24, 4829–4847. [Google Scholar] [CrossRef] [Green Version]
- Spaulding, E.L.; Hines, T.J.; Bais, P.; Tadenev, A.; Schneider, R.; Jewett, D.; Pattavina, B.; Pratt, S.L.; Morelli, K.H.; Stum, M.G.; et al. The integrated stress response contributes to tRNA synthetase-associated peripheral neuropathy. Science 2021, 373, 1156–1161. [Google Scholar] [CrossRef]
- Rodríguez-Nuevo, A.; Torres-Sanchez, A.; Duran, J.M.; De Guirior, C.; Martínez-Zamora, M.A.; Böke, E. Oocytes maintain ROS-free mitochondrial metabolism by suppressing complex I. Nature 2022, 607, 756–761. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Mutation Type | Related Diseases | Mutation Site or Gene |
|---|---|---|
| mtDNA mutation | ||
| Point mutation | MELAS syndrome, MERRF LHON Leigh’s syndrome | m.3243A>G m.8344A>G MT-TI, MT-TL1, MTTK, MT-TS1, and MT-TS2 m.11778G>A, m.3460G>A, and m.14484T>C MTFMT, MTTW, MTTV MTTL1, and MTTK |
| Large deletion | KSS, Pearson, CPEO | mtDNA deletion mtDNA deletion POLG mutation and mtDNA deletion |
| nDNA mutation | ||
| Respiratory chain subunits | Complex I deficiency Complex II deficiency Complex III deficiency Complex IV deficiency Complex V deficiency | TMEM126B mutation NDUF1V, NDUFV2, NDUFS1, NDUFS2, NDUFA12, NDUFAF2, NDUFAF5, NDUFAF6, FOXRED1, and SDHA SDHAF mutation [82] TTC19 and BCS1L UQCRFS1 mutation [83] SURF1, SCO1, and SCO2 FASTKD2 mutation [84] ATPAF2, TMEM70, and ATP5E |
| mtDNA replication or transcription | MNGIE MEMSA | TP gene mutation POLG mutation |
| Protein function | HSP Nerve system | SPG11 mutation SLC25A1 mutation [85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, H.; Wang, X.; Li, M.; Ji, D.; Liang, D.; Liang, C.; Liu, Y.; Zhang, Z.; Cao, Y.; Zou, W. Mitochondrial Unfolded Protein Response and Integrated Stress Response as Promising Therapeutic Targets for Mitochondrial Diseases. Cells 2023, 12, 20. https://doi.org/10.3390/cells12010020
Lu H, Wang X, Li M, Ji D, Liang D, Liang C, Liu Y, Zhang Z, Cao Y, Zou W. Mitochondrial Unfolded Protein Response and Integrated Stress Response as Promising Therapeutic Targets for Mitochondrial Diseases. Cells. 2023; 12(1):20. https://doi.org/10.3390/cells12010020
Chicago/Turabian StyleLu, Hedong, Xiaolei Wang, Min Li, Dongmei Ji, Dan Liang, Chunmei Liang, Yajing Liu, Zhiguo Zhang, Yunxia Cao, and Weiwei Zou. 2023. "Mitochondrial Unfolded Protein Response and Integrated Stress Response as Promising Therapeutic Targets for Mitochondrial Diseases" Cells 12, no. 1: 20. https://doi.org/10.3390/cells12010020