
High-Phosphate-Stimulated Macrophage-Derived Exosomes Promote Vascular Calcification via let-7b-5p/TGFBR1 Axis in Chronic Kidney Disease
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Patient Artery Samples
2.2. Cell Culture and Treatments
2.3. Animal Models
2.4. Statistical Analysis
3. Results
3.1. Increased Macrophage Infiltration in Media of CKD Patients with Aggravated Calcification and Increased Blood Phosphate
3.2. High-Phosphate-Stimulated Macrophages Releasing Exosomes Promotes Vascular Calcification
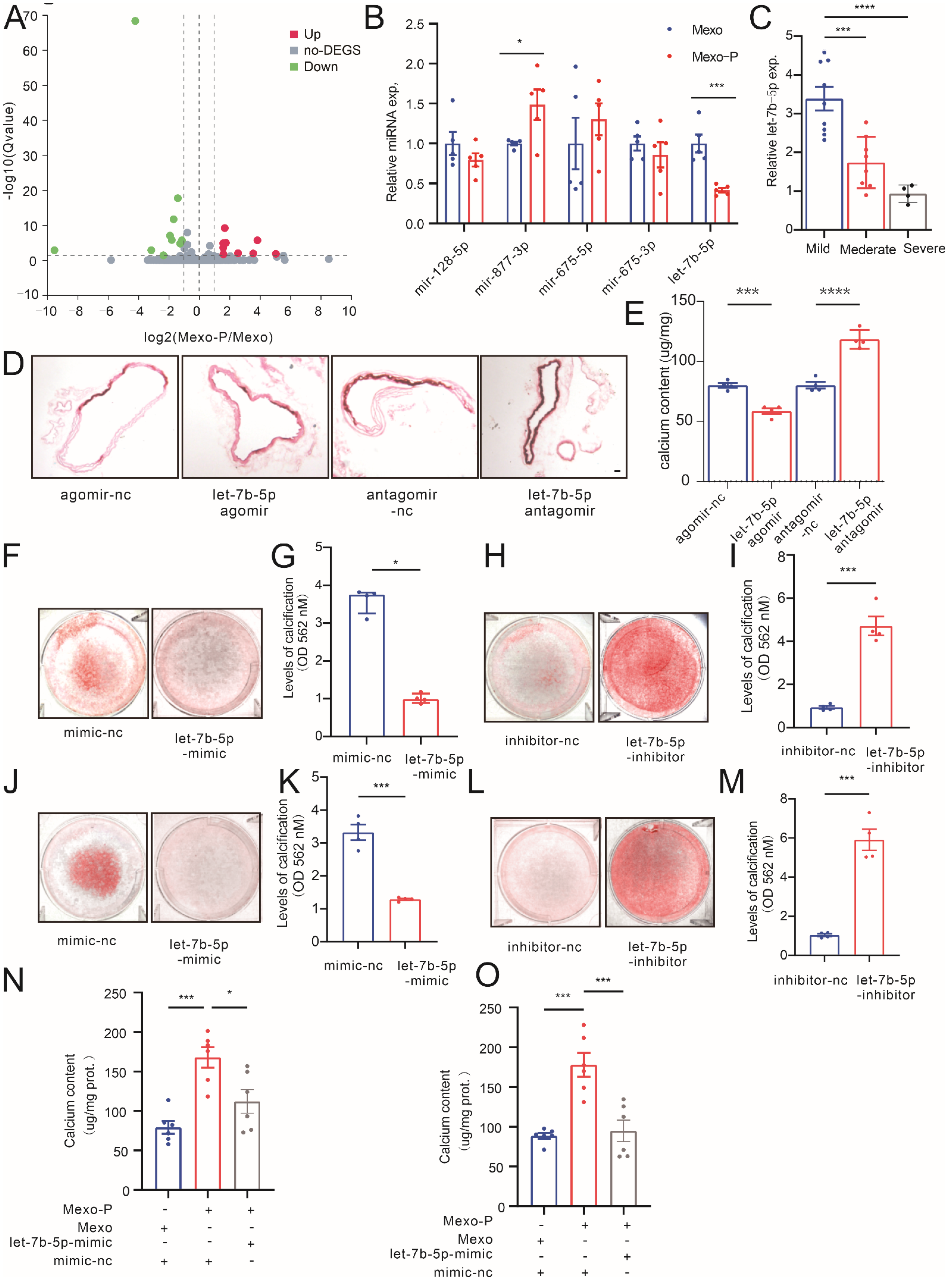
3.3. Mexo-P Regulates Vascular Calcification in a let-7b-5p-Dependent Manner
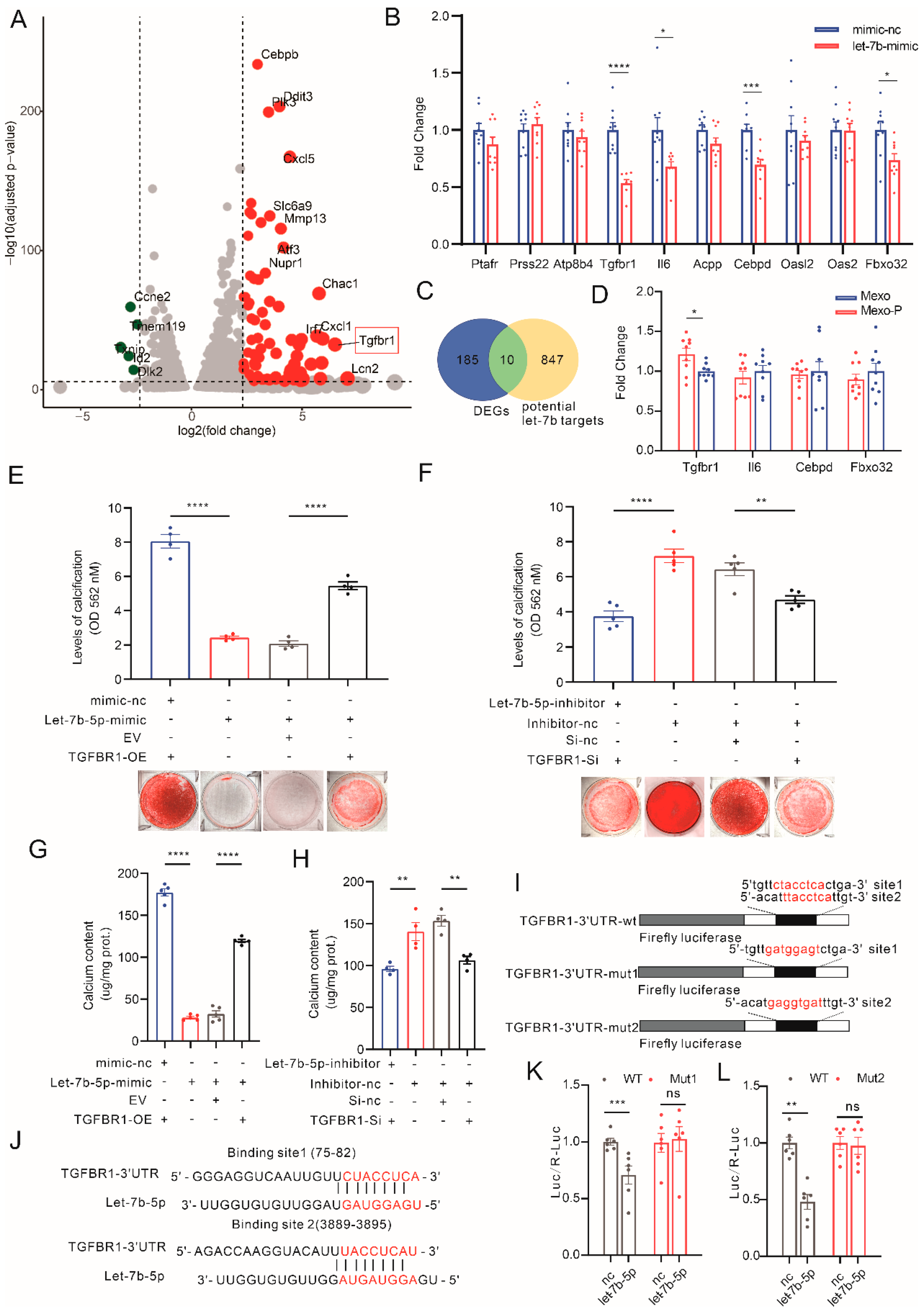
3.4. Mexo-P/let-7b-5p Regulate VC through a TGFBR1-Dependent Pathway
3.5. TGFBR1 Expression Upregulated in Calcified Media in CKD
3.6. Manipulating TGFBR1 Level Regulates Vascular Calcification in CKD
3.7. TGFBR1 Promotes Vascular Calcification through SMAD3/RUNX2 Pathway
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blacher, J.; Guerin, A.P.; Pannier, B.; Marchais, S.J.; London, G.M. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension 2001, 38, 938–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- London, G.M.; Guérin, A.P.; Marchais, S.J.; Métivier, F.; Pannier, B.; Adda, H. Arterial media calcification in end-stage renal disease: Impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transplant. 2003, 18, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.J.; Raggi, P.; Wolf, M.; Gold, A.M.; Chertow, G.M.; Roe, M.T. Targeting Vascular Calcification in Chronic Kidney Disease. JACC Basic Transl. Sci. 2020, 5, 398–412. [Google Scholar] [CrossRef] [PubMed]
- Lehto, S.; Niskanen, L.; Suhonen, M.; Rönnemaa, T.; Laakso, M. Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 978–983. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Lanzer, P.; Hannan, F.M.; Lanzer, J.D.; Janzen, J.; Raggi, P.; Furniss, D.; Schuchardt, M.; Thakker, R.; Fok, P.W.; Saez-Rodriguez, J.; et al. Medial Arterial Calcification: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 1145–1165. [Google Scholar] [CrossRef] [PubMed]
- Waring, O.J.; Skenteris, N.T.; Biessen, E.A.L.; Donners, M. Two-faced Janus: The dual role of macrophages in atherosclerotic calcification. Cardiovasc. Res. 2021, 118, 2768–2777. [Google Scholar] [CrossRef]
- Li, C.; Qu, L.; Matz, A.J.; Murphy, P.A.; Liu, Y.; Manichaikul, A.W.; Aguiar, D.; Rich, S.S.; Herrington, D.M.; Vu, D.; et al. AtheroSpectrum Reveals Novel Macrophage Foam Cell Gene Signatures Associated With Atherosclerotic Cardiovascular Disease Risk. Circulation 2022, 145, 206–218. [Google Scholar] [CrossRef]
- Pluchino, S.; Smith, J.A. Explicating Exosomes: Reclassifying the Rising Stars of Intercellular Communication. Cell 2019, 177, 225–227. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Das, S.; Halushka, M.K. Extracellular vesicle microRNA transfer in cardiovascular disease. Cardiovasc. Pathol. 2015, 24, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Nazarenko, I.; Rupp, A.K.; Altevogt, P. Exosomes as a potential tool for a specific delivery of functional molecules. Methods Mol. Biol. 2013, 1049, 495–511. [Google Scholar] [PubMed]
- Ismail, N.; Wang, Y.; Dakhlallah, D.; Moldovan, L.; Agarwal, K.; Batte, K.; Shah, P.; Wisler, J.; Eubank, T.D.; Tridandapani, S.; et al. Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood 2013, 121, 984–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaker, L.; Tebani, A.; Lesueur, C.; Dias, C.; Jung, V.; Bekri, S.; Guerrera, I.C.; Kamel, S.; Ausseil, J.; Boullier, A. Extracellular Vesicles From LPS-Treated Macrophages Aggravate Smooth Muscle Cell Calcification by Propagating Inflammation and Oxidative Stress. Front. Cell Dev. Biol. 2022, 10, 823450. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wan, Z.; Yang, L.; Song, S.; Fu, Z.; Tang, K.; Chen, L.; Song, Y. Exosomes derived from reparative M2-like macrophages prevent bone loss in murine periodontitis models via IL-10 mRNA. J. Nanobiotechnol. 2022, 20, 110. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, B.; Wang, Z.; Wang, D.; Ni, H.; Zhang, L.; Wang, Y. Exosomes from nicotine-stimulated macrophages accelerate atherosclerosis through miR-21-3p/PTEN-mediated VSMC migration and proliferation. Theranostics 2019, 9, 6901–6919. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, H.; Shi, H.; Zhao, H.; Gao, R.; Weng, X.; Liu, R.; Li, X.; Zou, Y.; Hu, K.; et al. Exosomes derived from M1 macrophages aggravate neointimal hyperplasia following carotid artery injuries in mice through miR-222/CDKN1B/CDKN1C pathway. Cell Death Dis. 2019, 10, 422. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Li, T.; Yin, T.; Hou, Z.; Qu, K.; Wang, N.; Durkan, C.; Dong, L.; Qiu, J.; Gregersen, H.; et al. M2 macrophage-derived exosomes promote the c-KIT phenotype of vascular smooth muscle cells during vascular tissue repair after intravascular stent implantation. Theranostics 2020, 10, 10712–10728. [Google Scholar] [CrossRef]
- Li, R.; Li, D.; Wang, H.; Chen, K.; Wang, S.; Xu, J.; Ji, P. Exosomes from adipose-derived stem cells regulate M1/M2 macrophage phenotypic polarization to promote bone healing via miR-451a/MIF. Stem Cell Res. Ther. 2022, 13, 149. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, Y.; Ma, L.; Chen, Y.; Liu, J.; Guo, Y.; Yu, T.; Zhang, L.; Zhu, L.; Shu, Y. Role of exosomal non-coding RNAs from tumor cells and tumor-associated macrophages in the tumor microenvironment. Mol. Ther. J. Am. Soc. Gene Ther. 2022, 30, 3133–3154. [Google Scholar] [CrossRef]
- Bao, M.H.; Feng, X.; Zhang, Y.W.; Lou, X.Y.; Cheng, Y.; Zhou, H.H. Let-7 in cardiovascular diseases, heart development and cardiovascular differentiation from stem cells. Int. J. Mol. Sci. 2013, 14, 23086–23102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, K.; Chen, X. Inflammation-regulatory microRNAs: Valuable targets for intracranial atherosclerosis. J. Neurosci. Res. 2019, 97, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zhang, H.; Xiao, J.; Wang, Z. Regulation of human cardiac ion channel genes by microRNAs: Theoretical perspective and pathophysiological implications. Cell. Physiol. Biochem. 2010, 25, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Galuppo, P.; Wolf, C.; Fiedler, J.; Kneitz, S.; van Laake, L.W.; Doevendans, P.A.; Mummery, C.L.; Borlak, J.; Haverich, A.; et al. MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation 2007, 116, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Topkara, V.K.; Mann, D.L. Role of microRNAs in cardiac remodeling and heart failure. Cardiovasc. Drugs Ther. 2011, 25, 171–182. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, X.; Meng, T. Overexpression of let-7b exerts beneficial effects on the functions of human placental trophoblasts by activating the ERK1/2 signaling pathway. Mol. Reprod. Dev. 2022, 89, 39–53. [Google Scholar] [CrossRef]
- Liu, X.; Li, S.; Yang, Y.; Sun, Y.; Yang, Q.; Gu, N.; Li, J.; Huang, T.; Liu, Y.; Dong, H.; et al. The lncRNA ANRIL regulates endothelial dysfunction by targeting the let-7b/TGF-βR1 signalling pathway. J. Cell. Physiol. 2021, 236, 2058–2069. [Google Scholar] [CrossRef]
- Vander Ark, A.; Cao, J.; Li, X. TGF-β receptors: In and beyond TGF-β signaling. Cell. Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef]
- Li, C.; Zhang, S.; Chen, X.; Ji, J.; Yang, W.; Gui, T.; Gai, Z.; Li, Y. Farnesoid X receptor activation inhibits TGFBR1/TAK1-mediated vascular inflammation and calcification via miR-135a-5p. Commun. Biol. 2020, 3, 327. [Google Scholar] [CrossRef]
- Schminke, B.; Kauffmann, P.; Schubert, A.; Altherr, M.; Gelis, T.; Miosge, N. SMURF1 and SMURF2 in Progenitor Cells from Articular Cartilage and Meniscus during Late-Stage Osteoarthritis. Cartilage 2021, 13 (Suppl. 2), 117s–128s. [Google Scholar] [CrossRef]
- Hao, J.; Tang, J.; Zhang, L.; Li, X.; Hao, L. The Crosstalk between Calcium Ions and Aldosterone Contributes to Inflammation, Apoptosis, and Calcification of VSMC via the AIF-1/NF-κB Pathway in Uremia. Oxidative Med. Cell. Longev. 2020, 2020, 3431597. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem. Cell Biol. 2018, 149, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, C.; Li, L.; Liang, X.; Cheng, P.; Li, Q.; Chang, X.; Wang, K.; Huang, S.; Li, Y.; et al. Lymphangiogenesis in renal fibrosis arises from macrophages via VEGF-C/VEGFR3-dependent autophagy and polarization. Cell Death Dis. 2021, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Yang, Y.; Wang, Y.N.; Li, Q.; Xing, X.; Cheng, A.Y.; Zhan, X.N.; Li, J.; Xu, G.; He, F. Oxidative phosphorylation promotes vascular calcification in chronic kidney disease. Cell Death Dis. 2022, 13, 229. [Google Scholar] [CrossRef]
- Long, X.; Yao, X.; Jiang, Q.; Yang, Y.; He, X.; Tian, W.; Zhao, K.; Zhang, H. Astrocyte-derived exosomes enriched with miR-873a-5p inhibit neuroinflammation via microglia phenotype modulation after traumatic brain injury. J. Neuroinflamm. 2020, 17, 89. [Google Scholar] [CrossRef]
- Jiao, Y.; Li, W.; Wang, W.; Tong, X.; Xia, R.; Fan, J.; Du, J.; Zhang, C.; Shi, X. Platelet-derived exosomes promote neutrophil extracellular trap formation during septic shock. Crit. Care 2020, 24, 380. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Zhang, C.; Shi, J.; Yang, Y.; Xing, X.; Wang, Y.; Zhan, X.; Wang, L.; Xu, G.; He, F. High-Phosphate-Stimulated Macrophage-Derived Exosomes Promote Vascular Calcification via let-7b-5p/TGFBR1 Axis in Chronic Kidney Disease. Cells 2023, 12, 161. https://doi.org/10.3390/cells12010161
Li Q, Zhang C, Shi J, Yang Y, Xing X, Wang Y, Zhan X, Wang L, Xu G, He F. High-Phosphate-Stimulated Macrophage-Derived Exosomes Promote Vascular Calcification via let-7b-5p/TGFBR1 Axis in Chronic Kidney Disease. Cells. 2023; 12(1):161. https://doi.org/10.3390/cells12010161
Chicago/Turabian StyleLi, Qing, Cailin Zhang, Jia Shi, Yi Yang, Xue Xing, Yanan Wang, Xiaona Zhan, Le Wang, Gang Xu, and Fan He. 2023. "High-Phosphate-Stimulated Macrophage-Derived Exosomes Promote Vascular Calcification via let-7b-5p/TGFBR1 Axis in Chronic Kidney Disease" Cells 12, no. 1: 161. https://doi.org/10.3390/cells12010161