

Cellular Senescence in Hepatocellular Carcinoma: The Passenger or the Driver?

Abstract
:1. Introduction
2. Cellular Senescence in Hepatocarcinogenesis
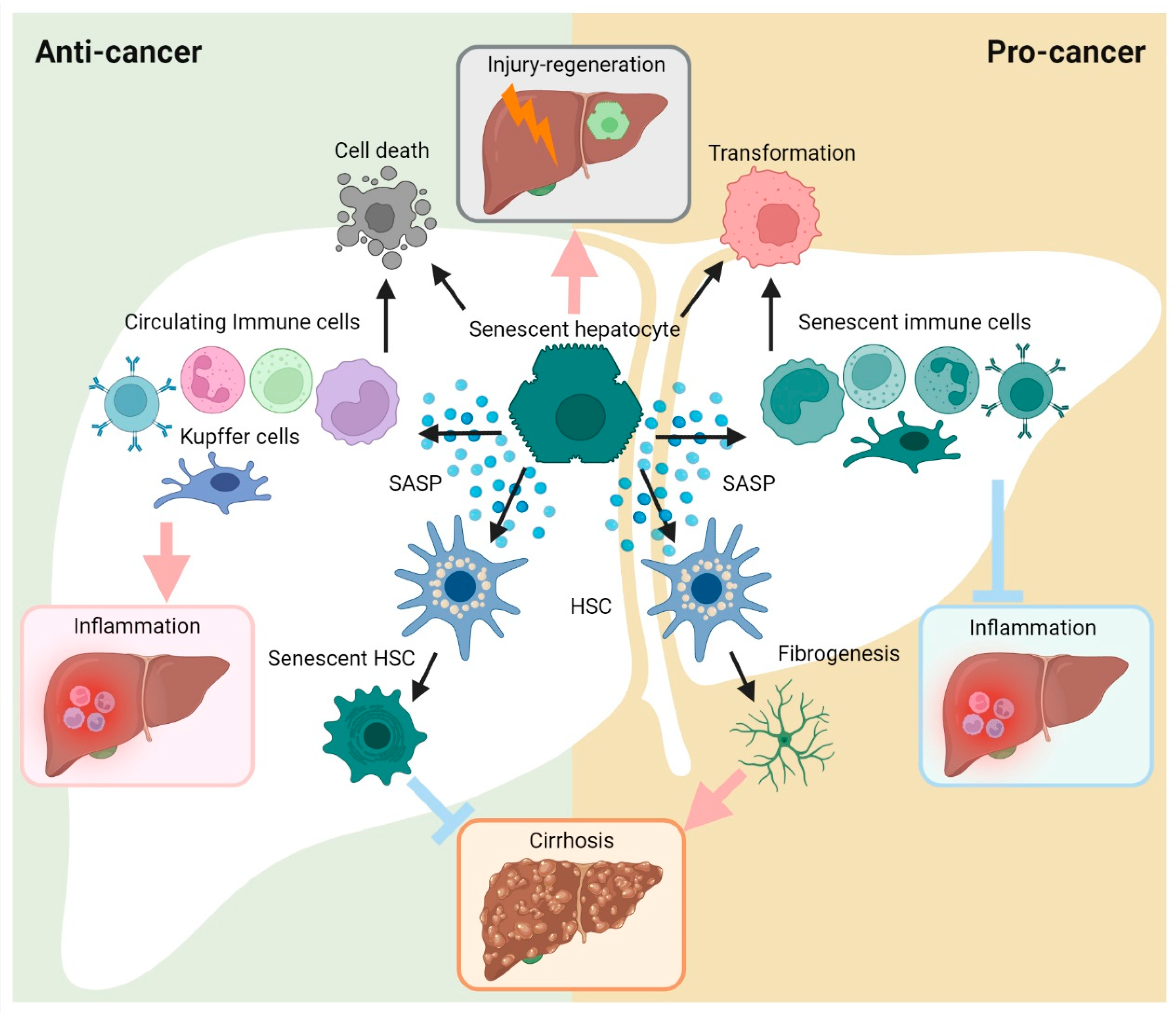
2.1. Cellular Senescence in Hepatocyte Injury
2.2. Cellular Senescence in Hepatic Fibrosis/Cirrhosis
2.3. Cellular Senescence in Hepatic Inflammation
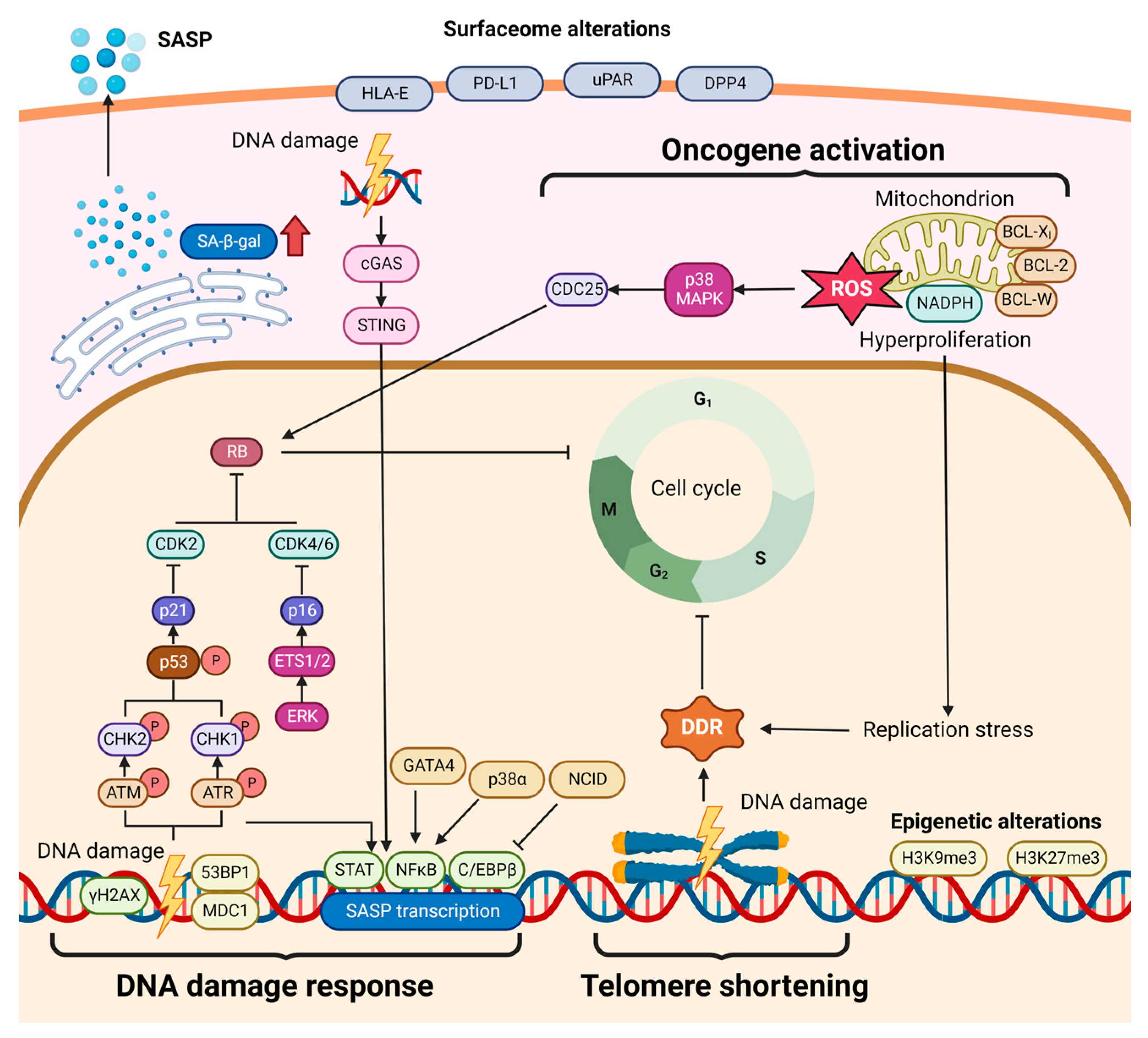
2.4. Cellular Senescence in HCC
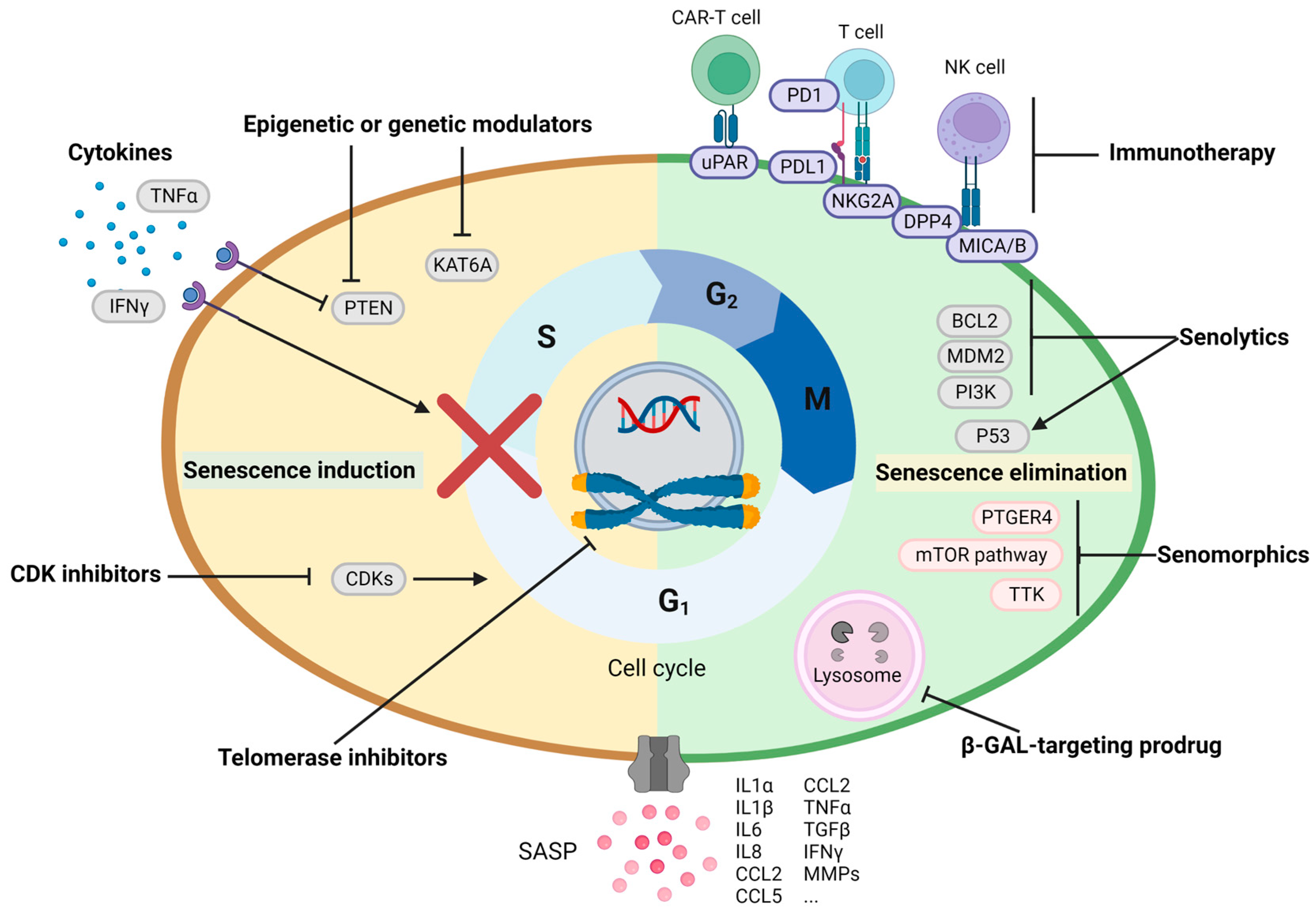
3. Cellular Senescence in HCC Therapies
3.1. Senescence Induction as an Anti-Cancer Therapy
3.1.1. Cell-Cycle Inhibitors
3.1.2. Telomerase Inhibitors
3.1.3. Cytokines
3.1.4. Epigenetic or Genetic Modulators
3.1.5. Other Senescence Inducers
3.2. Elimination of Detrimental Senescence as an Anti-Cancer Therapy
3.2.1. Senolytics
3.2.2. Senomorphics
3.2.3. Senescence-Targeted Immunotherapy
4. Discussion and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski, A. Hepatocellular carcinoma. Lancet 2022, 400, 1345–1362. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Garrido, A.; Djouder, N. Cirrhosis: A questioned risk factor for hepatocellular carcinoma. Trends Cancer 2021, 7, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fabrega, J.; Burrel, M.; Garcia-Criado, A.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. Bclc strategy for prognosis prediction and treatment recommendation: The 2022 update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef]
- Lee, T.K.; Guan, X.Y.; Ma, S. Cancer stem cells in hepatocellular carcinoma—From origin to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 26–44. [Google Scholar] [CrossRef]
- Huang, A.; Yang, X.R.; Chung, W.Y.; Dennison, A.R.; Zhou, J. Targeted therapy for hepatocellular carcinoma. Signal Transduct. Target. Ther. 2020, 5, 146. [Google Scholar] [CrossRef]
- Shay, J.W. Role of telomeres and telomerase in aging and cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16ink4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Perez-Mancera, P.A.; Young, A.R.; Narita, M. Inside and out: The activities of senescence in cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.W.; Barradas, M.; Stone, J.C.; van Aelst, L.; Serrano, M.; Lowe, S.W. Premature senescence involving p53 and p16 is activated in response to constitutive mek/mapk mitogenic signaling. Genes Dev. 1998, 12, 3008–3019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Rai, T.S.; Cole, J.J.; Nelson, D.M.; Dikovskaya, D.; Faller, W.J.; Vizioli, M.G.; Hewitt, R.N.; Anannya, O.; McBryan, T.; Manoharan, I.; et al. Hira orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014, 28, 2712–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, T.; Kirschner, K.; Thuret, J.Y.; Pope, B.D.; Ryba, T.; Newman, S.; Ahmed, K.; Samarajiwa, S.A.; Salama, R.; Carroll, T.; et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol. Cell 2012, 47, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Hoare, M.; Narita, M. Spatial and temporal control of senescence. Trends Cell Biol. 2017, 27, 820–832. [Google Scholar] [CrossRef] [Green Version]
- Chibaya, L.; Snyder, J.; Ruscetti, M. Senescence and the tumor-immune landscape: Implications for cancer immunotherapy. Semin. Cancer Biol. 2022, 86, 827–845. [Google Scholar] [CrossRef]
- Biran, A.; Perelmutter, M.; Gal, H.; Burton, D.G.; Ovadya, Y.; Vadai, E.; Geiger, T.; Krizhanovsky, V. Senescent cells communicate via intercellular protein transfer. Genes Dev. 2015, 29, 791–802. [Google Scholar] [CrossRef]
- Ferreira-Gonzalez, S.; Rodrigo-Torres, D.; Gadd, V.L.; Forbes, S.J. Cellular senescence in liver disease and regeneration. Semin. Liver Dis. 2021, 41, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Greten, T.F.; Eggert, T. Cellular senescence associated immune responses in liver cancer. Hepatic Oncol. 2017, 4, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Stanger, B.Z. Cellular homeostasis and repair in the mammalian liver. Annu. Rev. Physiol. 2015, 77, 179–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allaire, M.; Gilgenkrantz, H. The aged liver: Beyond cellular senescence. Clin. Res. Hepatol. Gastroenterol. 2020, 44, 6–11. [Google Scholar] [CrossRef]
- Gissen, P.; Arias, I.M. Structural and functional hepatocyte polarity and liver disease. J. Hepatol. 2015, 63, 1023–1037. [Google Scholar] [CrossRef] [Green Version]
- Bird, T.G.; Muller, M.; Boulter, L.; Vincent, D.F.; Ridgway, R.A.; Lopez-Guadamillas, E.; Lu, W.Y.; Jamieson, T.; Govaere, O.; Campbell, A.D.; et al. Tgfβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci. Transl. Med. 2018, 10, eaan1230. [Google Scholar] [CrossRef] [Green Version]
- Guo, M. Cellular senescence and liver disease: Mechanisms and therapeutic strategies. Biomed. Pharmacother. 2017, 96, 1527–1537. [Google Scholar] [CrossRef]
- Liu, P.; Tang, Q.; Chen, M.; Chen, W.; Lu, Y.; Liu, Z.; He, Z. Hepatocellular senescence: Immunosurveillance and future senescence-induced therapy in hepatocellular carcinoma. Front. Oncol. 2020, 10, 589908. [Google Scholar] [CrossRef]
- Karakousis, N.D.; Papatheodoridi, A.; Chatzigeorgiou, A.; Papatheodoridis, G. Cellular senescence and hepatitis b-related hepatocellular carcinoma: An intriguing link. Liver Int. 2020, 40, 2917–2927. [Google Scholar] [CrossRef]
- Birch, J.; Gil, J. Senescence and the sasp: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, N.; Lowe, S.W. Senescent cells spread the word: Non-cell autonomous propagation of cellular senescence. EMBO J. 2013, 32, 1975–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, K.L.; Chang, S.; Millard, M.; Schreiber-Agus, N.; DePinho, R.A. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science 2000, 287, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, S.U.; Satyanarayana, A.; Tsahuridu, M.; Tillmann, H.L.; Zender, L.; Klempnauer, J.; Flemming, P.; Franco, S.; Blasco, M.A.; Manns, M.P.; et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J. 2002, 16, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Asp. Med. 2019, 65, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Gutierrez-Reyes, G.; del Carmen Garcia de Leon, M.; Varela-Fascinetto, G.; Valencia, P.; Perez Tamayo, R.; Rosado, C.G.; Labonne, B.F.; Rochilin, N.M.; Garcia, R.M.; Valadez, J.A.; et al. Cellular senescence in livers from children with end stage liver disease. PLoS ONE 2010, 5, e10231. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. Tgf-β in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef]
- Fabregat, I.; Caballero-Diaz, D. Transforming growth factor-β-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. Interleukin-17 signaling in inflammatory, kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012, 143, 765–776.e763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabre, T.; Molina, M.F.; Soucy, G.; Goulet, J.P.; Willems, B.; Villeneuve, J.P.; Bilodeau, M.; Shoukry, N.H. Type 3 cytokines il-17a and il-22 drive tgf-β-dependent liver fibrosis. Sci. Immunol. 2018, 3, eaar7754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Feng, D.; Wang, H.; Hong, F.; Bertola, A.; Wang, F.S.; Gao, B. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 2012, 56, 1150–1159. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Chen, M.H.; Guo, Q.L.; Chen, Z.X.; Chen, Q.D.; Wang, X.Z. Interleukin-10 induces senescence of activated hepatic stellate cells via stat3-p53 pathway to attenuate liver fibrosis. Cell Signal 2020, 66, 109445. [Google Scholar] [CrossRef]
- Guo, Q.; Chen, M.; Chen, Q.; Xiao, G.; Chen, Z.; Wang, X.; Huang, Y. Silencing p53 inhibits interleukin 10-induced activated hepatic stellate cell senescence and fibrotic degradation in vivo. Exp. Biol. Med. 2021, 246, 447–458. [Google Scholar] [CrossRef]
- Wang, F.; Li, Z.; Chen, L.; Yang, T.; Liang, B.; Zhang, Z.; Shao, J.; Xu, X.; Yin, G.; Wang, S.; et al. Inhibition of asct2 induces hepatic stellate cell senescence with modified proinflammatory secretome through an il-1alpha/nf-kappab feedback pathway to inhibit liver fibrosis. Acta Pharm. Sin. B 2022, 12, 3618–3638. [Google Scholar] [CrossRef]
- Luo, J.; Li, L.; Chang, B.; Zhu, Z.; Deng, F.; Hu, M.; Yu, Y.; Lu, X.; Chen, Z.; Zuo, D.; et al. Mannan-binding lectin via interaction with cell surface calreticulin promotes senescence of activated hepatic stellate cells to limit liver fibrosis progression. Cell Mol. Gastroenterol. Hepatol. 2022, 14, 75–99. [Google Scholar] [CrossRef]
- Jin, H.; Lian, N.; Zhang, F.; Chen, L.; Chen, Q.; Lu, C.; Bian, M.; Shao, J.; Wu, L.; Zheng, S. Activation of ppargamma/p53 signaling is required for curcumin to induce hepatic stellate cell senescence. Cell Death Dis. 2016, 7, e2189. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhou, Z.; Jin, Y.; Lu, J.; Feng, D.; Peng, R.; Sun, H.; Mu, X.; Li, C.; Chen, Y. Hepatic stellate cell activation and senescence induced by intrahepatic microbiota disturbances drive progression of liver cirrhosis toward hepatocellular carcinoma. J. Immunother. Cancer 2022, 10, e003069. [Google Scholar] [CrossRef]
- Bernard, M.; Yang, B.; Migneault, F.; Turgeon, J.; Dieude, M.; Olivier, M.A.; Cardin, G.B.; El-Diwany, M.; Underwood, K.; Rodier, F.; et al. Autophagy drives fibroblast senescence through mtorc2 regulation. Autophagy 2020, 16, 2004–2016. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, R.; Kamachi, F.; Nakamura, M.; Yamazaki, S.; Kamiya, T.; Takasugi, M.; Cheng, Y.; Nonaka, Y.; Yukawa-Muto, Y.; Thuy, L.T.T.; et al. Gasdermin d-mediated release of il-33 from senescent hepatic stellate cells promotes obesity-associated hepatocellular carcinoma. Sci. Immunol. 2022, 7, eabl7209. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cai, X.; Liu, Y.; Shen, Y.; Guillot, A.; Tacke, F.; Tang, L.; Liu, H. The macrophage-associated microrna-4715-3p/gasdermin d axis potentially indicates fibrosis progression in nonalcoholic fatty liver disease: Evidence from transcriptome and biological data. Bioengineered 2022, 13, 11740–11751. [Google Scholar] [CrossRef] [PubMed]
- Hoenicke, L.; Zender, L. Immune surveillance of senescent cells--biological significance in cancer- and non-cancer pathologies. Carcinogenesis 2012, 33, 1123–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [Green Version]
- Lian, J.; Yue, Y.; Yu, W.; Zhang, Y. Immunosenescence: A key player in cancer development. J. Hematol. Oncol. 2020, 13, 151. [Google Scholar] [CrossRef]
- Effros, R.B. Replicative senescence in the immune system: Impact of the hayflick limit on t-cell function in the elderly. Am. J. Hum. Genet. 1998, 62, 1003–1007. [Google Scholar] [CrossRef] [Green Version]
- Adibzadeh, M.; Pohla, H.; Rehbein, A.; Pawelec, G. Long-term culture of monoclonal human t lymphocytes: Models for immunosenescence? Mech. Ageing Dev. 1995, 83, 171–183. [Google Scholar] [CrossRef]
- Akbar, A.N.; Henson, S.M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 2011, 11, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Nixon, D.F.; Donahoe, S.M.; Gillespie, G.M.; Dong, T.; King, A.; Ogg, G.S.; Spiegel, H.M.; Conlon, C.; Spina, C.A.; et al. Hiv-specific cd8(+) t cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 2000, 192, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawelec, G.; Solana, R. Immunoageing—The cause or effect of morbidity. Trends Immunol. 2001, 22, 348–349. [Google Scholar] [CrossRef] [PubMed]
- Mossanen, J.C.; Kohlhepp, M.; Wehr, A.; Krenkel, O.; Liepelt, A.; Roeth, A.A.; Mockel, D.; Heymann, F.; Lammers, T.; Gassler, N.; et al. Cxcr6 inhibits hepatocarcinogenesis by promoting natural killer t- and cd4(+) t-cell-dependent control of senescence. Gastroenterology 2019, 156, 1877–1889.e1874. [Google Scholar] [CrossRef]
- Schirdewahn, T.; Grabowski, J.; Sekyere, S.O.; Bremer, B.; Wranke, A.; Lunemann, S.; Schlaphoff, V.; Kirschner, J.; Hardtke, S.; Manns, M.P.; et al. The third signal cytokine interleukin 12 rather than immune checkpoint inhibitors contributes to the functional restoration of hepatitis d virus-specific t cells. J. Infect. Dis. 2017, 215, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Li, G.Y.; Ren, J.P.; Wang, L.; Zhao, J.; Ning, S.B.; Zhang, Y.; Lian, J.Q.; Huang, C.X.; Jia, Z.S.; et al. Protection of cd4+ t cells from hepatitis c virus infection-associated senescence via deltanp63-mir-181a-sirt1 pathway. J. Leukoc Biol. 2016, 100, 1201–1211. [Google Scholar] [CrossRef]
- Barathan, M.; Mohamed, R.; Saeidi, A.; Vadivelu, J.; Chang, L.Y.; Gopal, K.; Ram, M.R.; Ansari, A.W.; Kamarulzaman, A.; Velu, V.; et al. Increased frequency of late-senescent t cells lacking cd127 in chronic hepatitis c disease. Eur. J. Clin. Investig. 2015, 45, 466–474. [Google Scholar] [CrossRef]
- Hoare, M.; Gelson, W.T.; Das, A.; Fletcher, J.M.; Davies, S.E.; Curran, M.D.; Vowler, S.L.; Maini, M.K.; Akbar, A.N.; Alexander, G.J. Cd4+ t-lymphocyte telomere length is related to fibrosis stage, clinical outcome and treatment response in chronic hepatitis c virus infection. J. Hepatol. 2010, 53, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Liu, J.; Shi, L.; Liu, C.; Wang, G.; et al. Tim-3/galectin-9 signaling pathway mediates t-cell dysfunction and predicts poor prognosis in patients with hepatitis b virus-associated hepatocellular carcinoma. Hepatology 2012, 56, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of tim-3 and pd-1 expression is associated with tumor antigen-specific cd8+ t cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Bai, X.; Cao, Y.; Wu, J.; Huang, M.; Tang, D.; Tao, S.; Zhu, T.; Liu, Y.; Yang, Y.; et al. Lymphoma endothelium preferentially expresses tim-3 and facilitates the progression of lymphoma by mediating immune evasion. J. Exp. Med. 2010, 207, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Heffner, M.; Fearon, D.T. Loss of t cell receptor-induced bmi-1 in the klrg1(+) senescent cd8(+) t lymphocyte. Proc. Natl. Acad. Sci. USA 2007, 104, 13414–13419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenchley, J.M.; Karandikar, N.J.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Crotty, L.E.; Casazza, J.P.; Kuruppu, J.; Migueles, S.A.; Connors, M.; et al. Expression of cd57 defines replicative senescence and antigen-induced apoptotic death of cd8+ t cells. Blood 2003, 101, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.I.; et al. P16(ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 2017, 9, 1867–1884. [Google Scholar] [CrossRef] [Green Version]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.; et al. Aging of mice is associated with p16(ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging 2016, 8, 1294–1315. [Google Scholar] [CrossRef] [Green Version]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.A.; McGowan, S.J.; et al. An aged immune system drives senescence and ageing of solid organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 2021, 18, 45–56. [Google Scholar] [CrossRef]
- Guillot, A.; Tacke, F. Liver macrophages: Old dogmas and new insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef] [Green Version]
- Ovadya, Y.; Krizhanovsky, V. Senescent cells: Saspected drivers of age-related pathologies. Biogerontology 2014, 15, 627–642. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Adams, P.D. Healing and hurting: Molecular mechanisms, functions, and pathologies of cellular senescence. Mol. Cell 2009, 36, 2–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnabl, B.; Purbeck, C.A.; Choi, Y.H.; Hagedorn, C.H.; Brenner, D. Replicative senescence of activated human hepatic stellate cells is accompanied by a pronounced inflammatory but less fibrogenic phenotype. Hepatology 2003, 37, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Feng, D.; Mathews, S.; Gao, B. Hepatoprotective and anti-fibrotic functions of interleukin-22: Therapeutic potential for the treatment of alcoholic liver disease. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. 1), 56–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serna-Salas, S.A.; Arroyave-Ospina, J.C.; Zhang, M.; Damba, T.; Buist-Homan, M.; Munoz-Ortega, M.H.; Ventura-Juarez, J.; Moshage, H. α-1 adrenergic receptor antagonist doxazosin reverses hepatic stellate cells activation via induction of senescence. Mech. Ageing Dev. 2022, 201, 111617. [Google Scholar] [CrossRef]
- Sturmlechner, I.; Zhang, C.; Sine, C.C.; van Deursen, E.J.; Jeganathan, K.B.; Hamada, N.; Grasic, J.; Friedman, D.; Stutchman, J.T.; Can, I.; et al. P21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science 2021, 374, eabb3420. [Google Scholar] [CrossRef]
- Wang, C.; Chen, W.J.; Wu, Y.F.; You, P.; Zheng, S.Y.; Liu, C.C.; Xiang, D.; Wang, M.J.; Cai, Y.C.; Zhao, Q.H.; et al. The extent of liver injury determines hepatocyte fate toward senescence or cancer. Cell Death Dis. 2018, 9, 575. [Google Scholar] [CrossRef] [Green Version]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular senescence: Aging, cancer, and injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Tordella, L.; Khan, S.; Hohmeyer, A.; Banito, A.; Klotz, S.; Raguz, S.; Martin, N.; Dhamarlingam, G.; Carroll, T.; Meljem, J.M.G.; et al. Swi/snf regulates a transcriptional program that induces senescence to prevent liver cancer. Genes Dev. 2016, 30, 2187–2198. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.; Mok, M.T.; Kang, W.; Lau, S.S.; Yip, W.K.; Chen, Y.; Lai, P.B.; Wong, V.W.; To, K.F.; Sung, J.J.; et al. Truncated hbx-dependent silencing of gas2 promotes hepatocarcinogenesis through deregulation of cell cycle, senescence and p53-mediated apoptosis. J. Pathol. 2015, 237, 38–49. [Google Scholar] [CrossRef]
- Sasaki, M.; Nakanuma, Y. New concept: Cellular senescence in pathophysiology of cholangiocarcinoma. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Rivera, R.; Fattel-Fazenda, S.; Arellanes-Robledo, J.; Silva-Olivares, A.; Aleman-Lazarini, L.; Rodriguez-Segura, M.; Perez-Carreon, J.; Villa-Trevino, S.; Shibayama, M.; Serrano-Luna, J. Double staining of β-galactosidase with fibrosis and cancer markers reveals the chronological appearance of senescence in liver carcinogenesis induced by diethylnitrosamine. Toxicol. Lett. 2016, 241, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Mudbhary, R.; Hoshida, Y.; Chernyavskaya, Y.; Jacob, V.; Villanueva, A.; Fiel, M.I.; Chen, X.; Kojima, K.; Thung, S.; Bronson, R.T.; et al. Uhrf1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell 2014, 25, 196–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuestefeld, A.; Iakovleva, V.; Yap, S.X.L.; Ong, A.B.L.; Huang, D.Q.; Shuen, T.W.H.; Toh, H.C.; Dan, Y.Y.; Zender, L.; Wuestefeld, T. A pro-regenerative environment triggers premalignant to malignant transformation of senescent hepatocytes. Cancer Res. 2022. ahead of print. [Google Scholar] [CrossRef]
- Karabicici, M.; Alptekin, S.; Firtina Karagonlar, Z.; Erdal, E. Doxorubicin-induced senescence promotes stemness and tumorigenicity in epcam-/cd133- nonstem cell population in hepatocellular carcinoma cell line, huh-7. Mol. Oncol. 2021, 15, 2185–2202. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, X.; Meng, Y.; Shao, C.; Liao, J.; Li, F.; Li, R.; Jing, Y.; Huang, A. The hepatic senescence-associated secretory phenotype promotes hepatocarcinogenesis through bcl3-dependent activation of macrophages. Cell Biosci. 2021, 11, 173. [Google Scholar] [CrossRef]
- Xiang, X.; Fu, Y.; Zhao, K.; Miao, R.; Zhang, X.; Ma, X.; Liu, C.; Zhang, N.; Qu, K. Cellular senescence in hepatocellular carcinoma induced by a long non-coding rna-encoded peptide pint87aa by blocking foxm1-mediated phb2. Theranostics 2021, 11, 4929–4944. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Hu, K.; Cao, J.; Wang, P.; Li, J.; Zeng, K.; He, X.; Tu, P.F.; Tong, T.; Han, L. Lncrna miat functions as a cerna to upregulate sirt1 by sponging mir-22-3p in hcc cellular senescence. Aging 2019, 11, 7098–7122. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. P53-dependent chemokine production by senescent tumor cells supports nkg2d-dependent tumor elimination by natural killer cells. J. Exp. Med. 2013, 210, 2057–2069. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Kohli, J.; Demaria, M. Senescent cells in cancer therapy: Friends or foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Bollard, J.; Miguela, V.; de Galarreta, M.R.; Venkatesh, A.; Bian, C.B.; Roberto, M.P.; Tovar, V.; Sia, D.; Molina-Sanchez, P.; Nguyen, C.B.; et al. Palbociclib (pd-0332991), a selective cdk4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2017, 66, 1286–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, F.P.; Denk, G.; Ziesch, A.; Ofner, A.; Wimmer, R.; Hohenester, S.; Schiergens, T.S.; Spampatti, M.; Ye, L.; Itzel, T.; et al. Predictors of ribociclib-mediated antitumour effects in native and sorafenib-resistant human hepatocellular carcinoma cells. Cell. Oncol. 2019, 42, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; He, Y.; Fu, H.; Li, Y.; Bao, X.; Wang, Q.; Wang, Y.; Xie, C.; Lou, L. Preclinical characterization of shr6390, a novel cdk 4/6 inhibitor, in vitro and in human tumor xenograft models. Cancer Sci. 2019, 110, 1420–1430. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, H.; Lieftink, C.; du Chatinier, A.; Gao, D.; Jin, G.; Jin, H.; Beijersbergen, R.L.; Qin, W.; Bernards, R. Cdk12 inhibition mediates DNA damage and is synergistic with sorafenib treatment in hepatocellular carcinoma. Gut 2020, 69, 727–736. [Google Scholar] [CrossRef]
- Cho, S.J.; Lee, S.S.; Kim, Y.J.; Park, B.D.; Choi, J.S.; Liu, L.; Ham, Y.M.; Kim, B.M.; Lee, S.K. Xylocydine, a novel cdk inhibitor, is an effective inducer of apoptosis in hepatocellular carcinoma cells in vitro and in vivo. Cancer Lett. 2010, 287, 196–206. [Google Scholar] [CrossRef]
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; de Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W.; et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 2019, 574, 268–272. [Google Scholar] [CrossRef]
- Jones, K.A.; Gilder, A.S.; Lam, M.S.; Du, N.; Banki, M.A.; Merati, A.; Pizzo, D.P.; VandenBerg, S.R.; Gonias, S.L. Selective coexpression of vegf receptor 2 in egfrviii-positive glioblastoma cells prevents cellular senescence and contributes to their aggressive nature. Neuro. Oncol. 2016, 18, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.X.; Ancukiewicz, M.; Supko, J.G.; Sahani, D.V.; Blaszkowsky, L.S.; Meyerhardt, J.A.; Abrams, T.A.; McCleary, N.J.; Bhargava, P.; Muzikansky, A.; et al. Efficacy, safety, pharmacokinetics, and biomarkers of cediranib monotherapy in advanced hepatocellular carcinoma: A phase ii study. Clin. Cancer Res. 2013, 19, 1557–1566. [Google Scholar] [CrossRef]
- Ningarhari, M.; Caruso, S.; Hirsch, T.Z.; Bayard, Q.; Franconi, A.; Vedie, A.L.; Noblet, B.; Blanc, J.F.; Amaddeo, G.; Ganne, N.; et al. Telomere length is key to hepatocellular carcinoma diversity and telomerase addiction is an actionable therapeutic target. J. Hepatol. 2021, 74, 1155–1166. [Google Scholar] [CrossRef]
- Herz, C.; Hertrampf, A.; Zimmermann, S.; Stetter, N.; Wagner, M.; Kleinhans, C.; Erlacher, M.; Schuler, J.; Platz, S.; Rohn, S.; et al. The isothiocyanate erucin abrogates telomerase in hepatocellular carcinoma cells in vitro and in an orthotopic xenograft tumour model of hcc. J. Cell Mol. Med. 2014, 18, 2393–2403. [Google Scholar] [CrossRef]
- Moon, D.O.; Kang, S.H.; Kim, K.C.; Kim, M.O.; Choi, Y.H.; Kim, G.Y. Sulforaphane decreases viability and telomerase activity in hepatocellular carcinoma hep3b cells through the reactive oxygen species-dependent pathway. Cancer Lett. 2010, 295, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Rentschler, M.; Braumuller, H.; Briquez, P.S.; Wieder, T. Cytokine-induced senescence in the tumor microenvironment and its effects on anti-tumor immune responses. Cancers 2022, 14, 1364. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; MacFawn, I.P. Type i interferons and related pathways in cell senescence. Aging Cell 2020, 19, e13234. [Google Scholar] [CrossRef] [PubMed]
- Braumuller, H.; Wieder, T.; Brenner, E.; Assmann, S.; Hahn, M.; Alkhaled, M.; Schilbach, K.; Essmann, F.; Kneilling, M.; Griessinger, C.; et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013, 494, 361–365. [Google Scholar] [CrossRef] [Green Version]
- Griessinger, C.M.; Schmid, A.M.; Sonanini, D.; Schorg, B.F.; Jarboui, M.A.; Bukala, D.; Mucha, N.; Fehrenbacher, B.; Steinhilber, J.; Martella, M.; et al. The administration route of tumor-antigen-specific t-helper cells differentially modulates the tumor microenvironment and senescence. Carcinogenesis 2019, 40, 289–302. [Google Scholar] [CrossRef]
- Schilbach, K.; Welker, C.; Krickeberg, N.; Kaisser, C.; Schleicher, S.; Hashimoto, H. In the absence of a tcr signal il-2/il-12/18-stimulated gammadelta t cells demonstrate potent anti-tumoral function through direct killing and senescence induction in cancer cells. Cancers 2020, 12, 130. [Google Scholar] [CrossRef] [Green Version]
- Reimann, M.; Lee, S.; Loddenkemper, C.; Dorr, J.R.; Tabor, V.; Aichele, P.; Stein, H.; Dorken, B.; Jenuwein, T.; Schmitt, C.A. Tumor stroma-derived tgf-β limits myc-driven lymphomagenesis via suv39h1-dependent senescence. Cancer Cell 2010, 17, 262–272. [Google Scholar] [CrossRef] [Green Version]
- Di Mitri, D.; Toso, A.; Chen, J.J.; Sarti, M.; Pinton, S.; Jost, T.R.; D’Antuono, R.; Montani, E.; Garcia-Escudero, R.; Guccini, I.; et al. Tumour-infiltrating gr-1+ myeloid cells antagonize senescence in cancer. Nature 2014, 515, 134–137. [Google Scholar] [CrossRef]
- Ahmetlic, F.; Fauser, J.; Riedel, T.; Bauer, V.; Flessner, C.; Homberg, N.; Hennel, R.; Brenner, E.; Lauber, K.; Rocken, M.; et al. Therapy of lymphoma by immune checkpoint inhibitors: The role of t cells, nk cells and cytokine-induced tumor senescence. J. Immunother. Cancer 2021, 9, e001660. [Google Scholar] [CrossRef]
- Brenner, E.; Schorg, B.F.; Ahmetlic, F.; Wieder, T.; Hilke, F.J.; Simon, N.; Schroeder, C.; Demidov, G.; Riedel, T.; Fehrenbacher, B.; et al. Cancer immune control needs senescence induction by interferon-dependent cell cycle regulator pathways in tumours. Nat. Commun. 2020, 11, 1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, M.; Ikeda, H.; Sato, Y.; Nakanuma, Y. Proinflammatory cytokine-induced cellular senescence of biliary epithelial cells is mediated via oxidative stress and activation of atm pathway: A culture study. Free Radic. Res. 2008, 42, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Leaver, D.J.; Hermans, S.J.; Kelly, G.L.; Brennan, M.S.; Downer, N.L.; Nguyen, N.; Wichmann, J.; McRae, H.M.; Yang, Y.; et al. Inhibitors of histone acetyltransferases kat6a/b induce senescence and arrest tumour growth. Nature 2018, 560, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Augello, G.; Puleio, R.; Emma, M.R.; Cusimano, A.; Loria, G.R.; McCubrey, J.A.; Montalto, G.; Cervello, M. A pten inhibitor displays preclinical activity against hepatocarcinoma cells. Cell Cycle 2016, 15, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; Deursen, J.V.; Goronzy, J.; et al. Therapy-induced senescence: Opportunities to improve anticancer therapy. J. Natl. Cancer Inst. 2021, 113, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Borghesan, M.; Fusilli, C.; Rappa, F.; Panebianco, C.; Rizzo, G.; Oben, J.A.; Mazzoccoli, G.; Faulkes, C.; Pata, I.; Agodi, A.; et al. DNA hypomethylation and histone variant macroh2a1 synergistically attenuate chemotherapy-induced senescence to promote hepatocellular carcinoma progression. Cancer Res. 2016, 76, 594–606. [Google Scholar] [CrossRef] [Green Version]
- Anerillas, C.; Herman, A.B.; Munk, R.; Garrido, A.; Lam, K.G.; Payea, M.J.; Rossi, M.; Tsitsipatis, D.; Martindale, J.L.; Piao, Y.; et al. A bdnf-trkb autocrine loop enhances senescent cell viability. Nat. Commun. 2022, 13, 6228. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of bcl-w and bcl-xl. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Leung, C.O.N.; Tong, M.; Chung, K.P.S.; Zhou, L.; Che, N.; Tang, K.H.; Ding, J.; Lau, E.Y.T.; Ng, I.O.L.; Ma, S.; et al. Overriding adaptive resistance to sorafenib through combination therapy with src homology 2 domain-containing phosphatase 2 blockade in hepatocellular carcinoma. Hepatology 2020, 72, 155–168. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut microbiota promotes obesity-associated liver cancer through pge2-mediated suppression of antitumor immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghesan, M.; Hoogaars, W.M.H.; Varela-Eirin, M.; Talma, N.; Demaria, M. A senescence-centric view of aging: Implications for longevity and disease. Trends Cell Biol. 2020, 30, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Huangyang, P.; Burrows, M.; Guo, K.; Riscal, R.; Godfrey, J.; Lee, K.E.; Lin, N.; Lee, P.; Blair, I.A.; et al. Fbp1 loss disrupts liver metabolism and promotes tumorigenesis through a hepatic stellate cell senescence secretome. Nat. Cell Biol. 2020, 22, 728–739. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef] [Green Version]
- Thadathil, N.; Selvarani, R.; Mohammed, S.; Nicklas, E.H.; Tran, A.L.; Kamal, M.; Luo, W.; Brown, J.L.; Lawrence, M.M.; Borowik, A.K.; et al. Senolytic treatment reduces cell senescence and necroptosis in sod1 knockout mice that is associated with reduced inflammation and hepatocellular carcinoma. Aging Cell 2022, 21, e13676. [Google Scholar] [CrossRef]
- Kovacovicova, K.; Skolnaja, M.; Heinmaa, M.; Mistrik, M.; Pata, P.; Pata, I.; Bartek, J.; Vinciguerra, M. Senolytic cocktail dasatinib+quercetin (d+q) does not enhance the efficacy of senescence-inducing chemotherapy in liver cancer. Front. Oncol. 2018, 8, 459. [Google Scholar] [CrossRef]
- Missiaen, R.; Anderson, N.M.; Kim, L.C.; Nance, B.; Burrows, M.; Skuli, N.; Carens, M.; Riscal, R.; Steensels, A.; Li, F.; et al. Gcn2 inhibition sensitizes arginine-deprived hepatocellular carcinoma cells to senolytic treatment. Cell Metab. 2022, 34, 1151–1167.e1157. [Google Scholar] [CrossRef]
- Cai, Y.; Zhou, H.; Zhu, Y.; Sun, Q.; Ji, Y.; Xue, A.; Wang, Y.; Chen, W.; Yu, X.; Wang, L.; et al. Elimination of senescent cells by β-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Res. 2020, 30, 574–589. [Google Scholar] [CrossRef]
- Pi, L.; Rooprai, J.; Allan, D.S.; Atkins, H.; Bredeson, C.; Fulcher, A.J.; Ito, C.; Ramsay, T.; Shorr; Stanford, W.L.; et al. Evaluating dose-limiting toxicities of mdm2 inhibitors in patients with solid organ and hematologic malignancies: A systematic review of the literature. Leuk. Res. 2019, 86, 106222. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. Mtor regulates mapkapk2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. Mtor regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting il1a translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Alimbetov, D.; Davis, T.; Brook, A.J.; Cox, L.S.; Faragher, R.G.; Nurgozhin, T.; Zhumadilov, Z.; Kipling, D. Suppression of the senescence-associated secretory phenotype (sasp) in human fibroblasts using small molecule inhibitors of p38 map kinase and mk2. Biogerontology 2016, 17, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.; Kohno, S.; Okada, N.; Okahashi, N.; Teranishi, K.; Matsuda, F.; Shimizu, H.; Linn, P.; Nagatani, N.; Yamamura, M.; et al. Treatment of retinoblastoma 1-intact hepatocellular carcinoma with cyclin-dependent kinase 4/6 inhibitor combination therapy. Hepatology 2021, 74, 1971–1993. [Google Scholar] [CrossRef]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. Jak inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.Y.; Chiu, D.K.; Yuen, V.W.; Law, C.T.; Wong, B.P.; Thu, K.L.; Cescon, D.W.; Soria-Bretones, I.; Cheu, J.W.; Lee, D.; et al. Cfi-402257, a ttk inhibitor, effectively suppresses hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2022, 119, e2119514119. [Google Scholar] [CrossRef]
- Chen, H.A.; Ho, Y.J.; Mezzadra, R.; Adrover, J.M.; Smolkin, R.; Zhu, C.; Woess, K.; Bernstein, N.; Schmitt, G.; Fong, L.; et al. Senescence rewires microenvironment sensing to facilitate anti-tumor immunity. Cancer Discov. 2022. ahead of print. [Google Scholar] [CrossRef]
- Reimann, M.; Schrezenmeier, J.; Richter-Pechanska, P.; Dolnik, A.; Hick, T.P.; Schleich, K.; Cai, X.; Fan, D.N.Y.; Lohneis, P.; Masswig, S.; et al. Adaptive t-cell immunity controls senescence-prone myd88- or card11-mutant b-cell lymphomas. Blood 2021, 137, 2785–2799. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin d-cdk4 kinase destabilizes pd-l1 via cullin 3-spop to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Lelliott, E.J.; Kong, I.Y.; Zethoven, M.; Ramsbottom, K.M.; Martelotto, L.G.; Meyran, D.; Zhu, J.J.; Costacurta, M.; Kirby, L.; Sandow, J.J.; et al. Cdk4/6 inhibition promotes antitumor immunity through the induction of t-cell memory. Cancer Discov. 2021, 11, 2582–2601. [Google Scholar] [CrossRef] [PubMed]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A cancer cell program promotes t cell exclusion and resistance to checkpoint blockade. Cell 2018, 175, 984–997.e924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. Cdk4/6 inhibition augments antitumor immunity by enhancing t-cell activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.W.; Johmura, Y.; Suzuki, N.; Omori, S.; Migita, T.; Yamaguchi, K.; Hatakeyama, S.; Yamazaki, S.; Shimizu, E.; Imoto, S.; et al. Blocking pd-l1-pd-1 improves senescence surveillance and ageing phenotypes. Nature 2022, 611, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent cells evade immune clearance via hla-e-mediated nk and cd8(+) t cell inhibition. Nat. Commun. 2019, 10, 2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Blery, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-nkg2a mab is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both t and nk cells. Cell 2018, 175, 1731–1743.e1713. [Google Scholar] [CrossRef] [Green Version]
- Kohga, K.; Takehara, T.; Tatsumi, T.; Ohkawa, K.; Miyagi, T.; Hiramatsu, N.; Kanto, T.; Kasugai, T.; Katayama, K.; Kato, M.; et al. Serum levels of soluble major histocompatibility complex (mhc) class i-related chain a in patients with chronic liver diseases and changes during transcatheter arterial embolization for hepatocellular carcinoma. Cancer Sci. 2008, 99, 1643–1649. [Google Scholar] [CrossRef]
- Munoz, D.P.; Yannone, S.M.; Daemen, A.; Sun, Y.; Vakar-Lopez, F.; Kawahara, M.; Freund, A.M.; Rodier, F.; Wu, J.D.; Desprez, P.Y.; et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight 2019, 4, e124716. [Google Scholar] [CrossRef]
- de Andrade, L.F.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F., Jr.; Harvey, C.J.; et al. Antibody-mediated inhibition of mica and micb shedding promotes nk cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef] [Green Version]
- da Silva, P.H.A.; Xing, S.; Kotini, A.G.; Papapetrou, E.P.; Song, X.; Wucherpfennig, K.W.; Mascarenhas, J.; de Andrade, L.F. Mica/b antibody induces macrophage-mediated immunity against acute myeloid leukemia. Blood 2022, 139, 205–216. [Google Scholar] [CrossRef]
- Kim, K.M.; Noh, J.H.; Bodogai, M.; Martindale, J.L.; Yang, X.; Indig, F.E.; Basu, S.K.; Ohnuma, K.; Morimoto, C.; Johnson, P.F.; et al. Identification of senescent cell surface targetable protein dpp4. Genes Dev. 2017, 31, 1529–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishina, S.; Hino, K. Cd26/dpp4 as a therapeutic target in nonalcoholic steatohepatitis associated hepatocellular carcinoma. Cancers 2022, 14, 454. [Google Scholar] [CrossRef]
- Dietrich, P.; Wormser, L.; Fritz, V.; Seitz, T.; De Maria, M.; Schambony, A.; Kremer, A.E.; Gunther, C.; Itzel, T.; Thasler, W.E.; et al. Molecular crosstalk between y5 receptor and neuropeptide y drives liver cancer. J. Clin. Investig. 2020, 130, 2509–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishina, S.; Yamauchi, A.; Kawaguchi, T.; Kaku, K.; Goto, M.; Sasaki, K.; Hara, Y.; Tomiyama, Y.; Kuribayashi, F.; Torimura, T.; et al. Dipeptidyl peptidase 4 inhibitors reduce hepatocellular carcinoma by activating lymphocyte chemotaxis in mice. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 115–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, W.H.; Sue, S.P.; Liang, H.L.; Tseng, C.W.; Lin, H.C.; Wen, W.L.; Lee, M.Y. Dipeptidyl peptidase 4 inhibitors decrease the risk of hepatocellular carcinoma in patients with chronic hepatitis c infection and type 2 diabetes mellitus: A nationwide study in taiwan. Front. Public Health 2021, 9, 711723. [Google Scholar] [CrossRef] [PubMed]
- Yen, F.S.; Wei, J.C.; Yip, H.T.; Hwu, C.M.; Hou, M.C.; Hsu, C.C. Dipeptidyl peptidase-4 inhibitors may accelerate cirrhosis decompensation in patients with diabetes and liver cirrhosis: A nationwide population-based cohort study in taiwan. Hepatol. Int. 2021, 15, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic car t cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Jurtz, V.I.; Jessen, L.E.; Bentzen, A.K.; Jespersen, M.C.; Mahajan, S.; Vita, R.; Jensen, K.K.; Marcatili, P.; Hadrup, S.R.; Peters, B.; et al. Nettcr: Sequence-based prediction of tcr binding to peptide-mhc complexes using convolutional neural networks. bioRxiv 2018. preprint. [Google Scholar] [CrossRef]
- Sbierski-Kind, J.; Grenkowitz, S.; Schlickeiser, S.; Sandforth, A.; Friedrich, M.; Kunkel, D.; Glauben, R.; Brachs, S.; Mai, K.; Thurmer, A.; et al. Effects of caloric restriction on the gut microbiome are linked with immune senescence. Microbiome 2022, 10, 57. [Google Scholar] [CrossRef]
- Gao, W.; Jia, Z.; Tian, Y.; Yang, P.; Sun, H.; Wang, C.; Ding, Y.; Zhang, M.; Zhang, Y.; Yang, D.; et al. Hbx protein contributes to liver carcinogenesis by h3k4me3 modification through stabilizing wd repeat domain 5 protein. Hepatology 2020, 71, 1678–1695. [Google Scholar] [CrossRef]
- Idrissi, M.E.; Hachem, H.; Koering, C.; Merle, P.; Thenoz, M.; Mortreux, F.; Wattel, E. Hbx triggers either cellular senescence or cell proliferation depending on cellular phenotype. J. Viral Hepat 2016, 23, 130–138. [Google Scholar] [CrossRef]
- Kim, Y.J.; Jung, J.K.; Lee, S.Y.; Jang, K.L. Hepatitis b virus x protein overcomes stress-induced premature senescence by repressing p16(ink4a) expression via DNA methylation. Cancer Lett. 2010, 288, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Ou, D.P.; Tao, Y.M.; Tang, F.Q.; Yang, L.Y. The hepatitis b virus x protein promotes hepatocellular carcinoma metastasis by upregulation of matrix metalloproteinases. Int. J. Cancer 2007, 120, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Hao, J.; Cui, S.; Worthley, D.L.; Zhang, S.; Wang, Z.; Shi, J.; Liu, L.; Wang, X.; Ke, A.; et al. Diverse modes of clonal evolution in hbv-related hepatocellular carcinoma revealed by single-cell genome sequencing. Cell Res. 2018, 28, 359–373. [Google Scholar] [CrossRef]
- Wandrer, F.; Han, B.; Liebig, S.; Schlue, J.; Manns, M.P.; Schulze-Osthoff, K.; Bantel, H. Senescence mirrors the extent of liver fibrosis in chronic hepatitis c virus infection. Aliment. Pharmacol. Ther. 2018, 48, 270–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.S.; Park, S.H.; Jang, K.L. Hepatitis c virus core protein overcomes stress-induced premature senescence by down-regulating p16 expression via DNA methylation. Cancer Lett. 2012, 321, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B.; D’Angelica, M.I.; Abbott, D.E.; Anaya, D.A.; Anders, R.; Are, C.; Bachini, M.; Borad, M.; Brown, D.; Burgoyne, A.; et al. Hepatobiliary cancers, version 2.2021, nccn clinical practice guidelines in oncology. J. Natl. Compr. Canc. Netw. 2021, 19, 541–565. [Google Scholar] [CrossRef]
- Casak, S.J.; Donoghue, M.; Fashoyin-Aje, L.; Jiang, X.; Rodriguez, L.; Shen, Y.L.; Xu, Y.; Jiang, X.; Liu, J.; Zhao, H.; et al. Fda approval summary: Atezolizumab plus bevacizumab for the treatment of patients with advanced unresectable or metastatic hepatocellular carcinoma. Clin. Cancer Res. 2021, 27, 1836–1841. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: The checkmate 040 randomized clinical trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in keynote-240: A randomized, double-blind, phase iii trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Kelley, R.K.; Sangro, B.; Harris, W.; Ikeda, M.; Okusaka, T.; Kang, Y.K.; Qin, S.; Tai, D.W.; Lim, H.Y.; Yau, T.; et al. Safety, efficacy, and pharmacodynamics of tremelimumab plus durvalumab for patients with unresectable hepatocellular carcinoma: Randomized expansion of a phase i/ii study. J. Clin. Oncol. 2021, 39, 2991–3001. [Google Scholar] [CrossRef]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Senescence Inducer | ||||||
|---|---|---|---|---|---|---|
| Category | Compound | Regimens | Phase | Population | Results | Study Number |
| CDK4/6 Inhibitor | Palbociclib (PD0332991) | Singular treatment | II | 23 patients with advanced HCC | OS: 40 (24–96) wk, 11/23 (47.83%) with severe side effects | NCT01356628 |
| Abemaciclib | Combined with nivolumab | II | 7 patients with inoperable HCC | 1/7 (14.3% ORR), PFS: 2.1 (1.6 to 5.6) mon, OS: 7.9 (2.6 to 22.7) mon, 3/7 (42.86%) severe side effects | NCT03781960 | |
| Ribociclib (LEE011) | Combined with chemoembolization | Ib/II | 5 patients with advanced HCC | 1/5 (20.00%) severe side effects | NCT02524119 | |
| SHR6390 | Combined with anti-PD-1 Inhibitor SHR-1210 | Ib/II | 41 advanced colorectal cancer, non-small-cell lung cancer and HCC | Ongoing | NCT03601598 | |
| VEGFR-2 tyrosine kinase inhibitor | AZD2171 (Cediranib maleate) | Singular treatment | II | 17 patients with locally advanced unresectable or metastatic HCC | OS: 11.7 (7.5 to 13.6) mon, 5/17 (29.41%) severe side effects | NCT00427973 |
| Senescence eliminator | ||||||
| Senolytics | Navitoclax | Combined with Sorafenib (Nexavar) | I | 44 patients with relapsed or refractory solid organ tumors including HCC | Ongoing | NCT02143401 |
| Dasatinib (BMS-354825) | Singular treatment | I | 80 patients with unresectable or metastatic solid tumors including HCC | Completed | NCT00608361 | |
| Dasatinib (BMS-354825) | Singular treatment | II | 25 patients with unresectable advanced HCC | PFS: 3.7 (1.8 to 6.4) mon, OS: 7.5 (2.9 to 13.6) mon, 13/25 (52.00%) serious side effects | NCT00459108 | |
| mTOR inhibitor | Temsirolimus | Combined with Sorafenib | I/II | Advanced HCC | Cancelled | NCT01335074 |
| Combined with pegylated liposomal doxorubicin (Doxil®) | II | Advanced HCC | Cancelled | NCT01281943 | ||
| ABI-009 | Combined with anti-PD1 inhibitor nivolumab | I/II | 26 patients with advanced sarcoma and certain cancers including HCC (with genetic mutations sensitive to mTOR inhibitors) | 4/26 (15%) serious side effects | NCT03190174 | |
| Anti-DPP4 inhibitor | Sitagliptin | Singular treatment before HCC resection | I | 14 HCC patients undergoing liver resection | No severe adverse event | NCT02650427 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, X.; Guillot, A.; Liu, H. Cellular Senescence in Hepatocellular Carcinoma: The Passenger or the Driver? Cells 2023, 12, 132. https://doi.org/10.3390/cells12010132
Cai X, Guillot A, Liu H. Cellular Senescence in Hepatocellular Carcinoma: The Passenger or the Driver? Cells. 2023; 12(1):132. https://doi.org/10.3390/cells12010132
Chicago/Turabian StyleCai, Xiurong, Adrien Guillot, and Hanyang Liu. 2023. "Cellular Senescence in Hepatocellular Carcinoma: The Passenger or the Driver?" Cells 12, no. 1: 132. https://doi.org/10.3390/cells12010132