
Detection of Cellular Senescence in Human Primary Melanocytes and Malignant Melanoma Cells In Vitro

 ,
,
Abstract
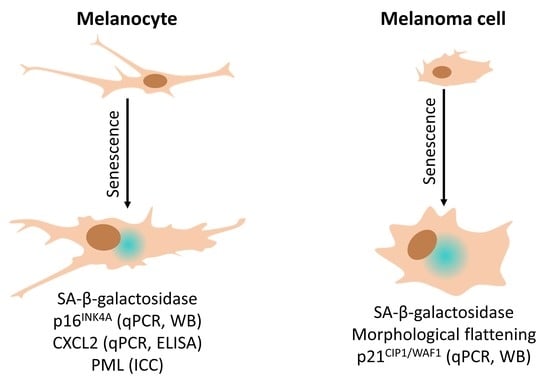
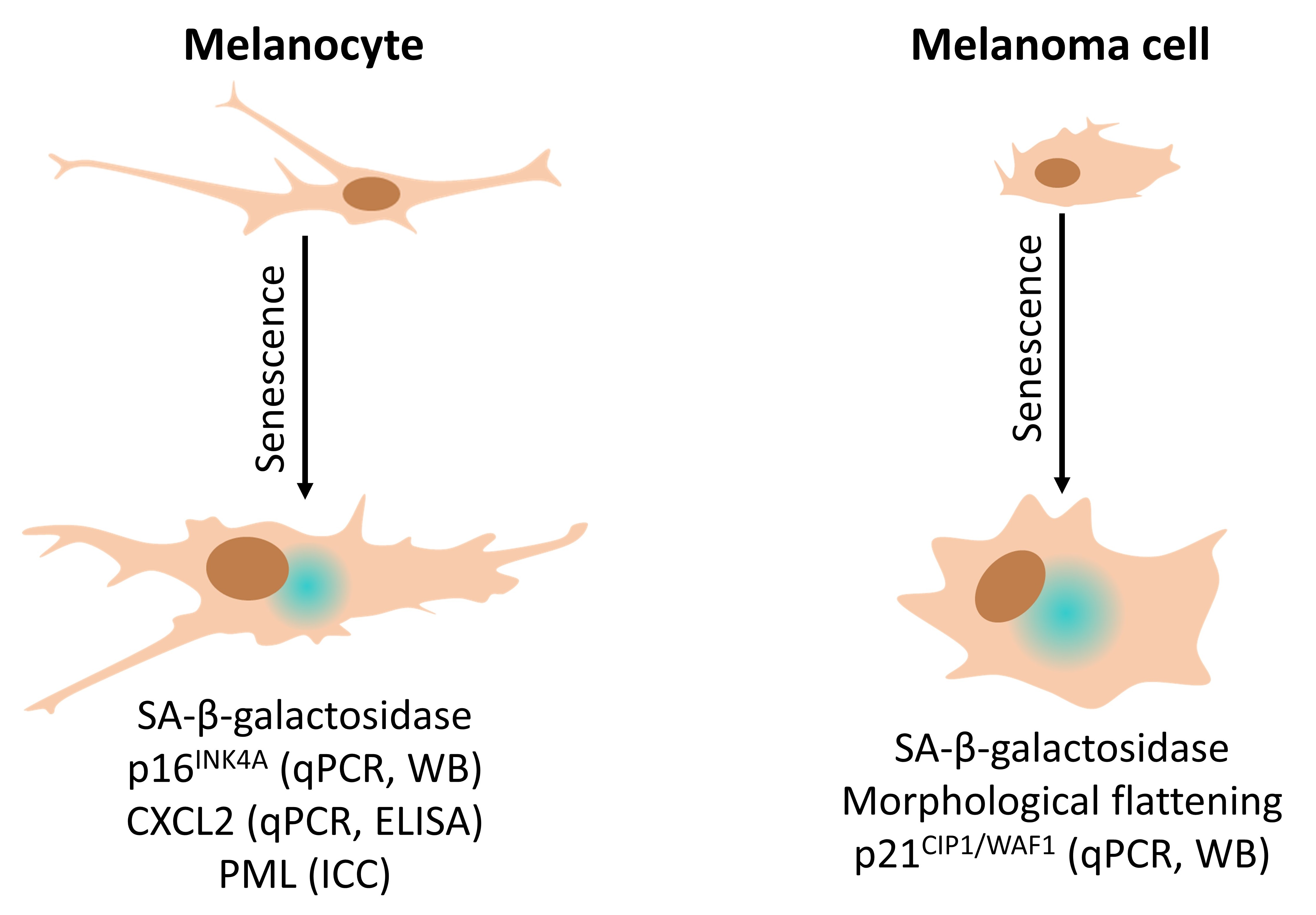
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Lentiviral Transduction of Melanocytes
2.3. Induction of Senescence in Melanoma Cells
2.4. Analysis of mRNA Expression Using Real-Time PCR
2.5. Western Blot Protein Analysis
2.6. Immunofluorescent Stainings
2.7. Real-Time Cell Proliferation Analysis (RTCA)
2.8. XTT Cell Viability Assay
2.9. Staining of Senescence-Associated Beta-Galactosidase Activity
2.10. Flow Cytometry of Fluorescent Beta-Galactosidase Substrates
2.11. Statistical Analysis
3. Results
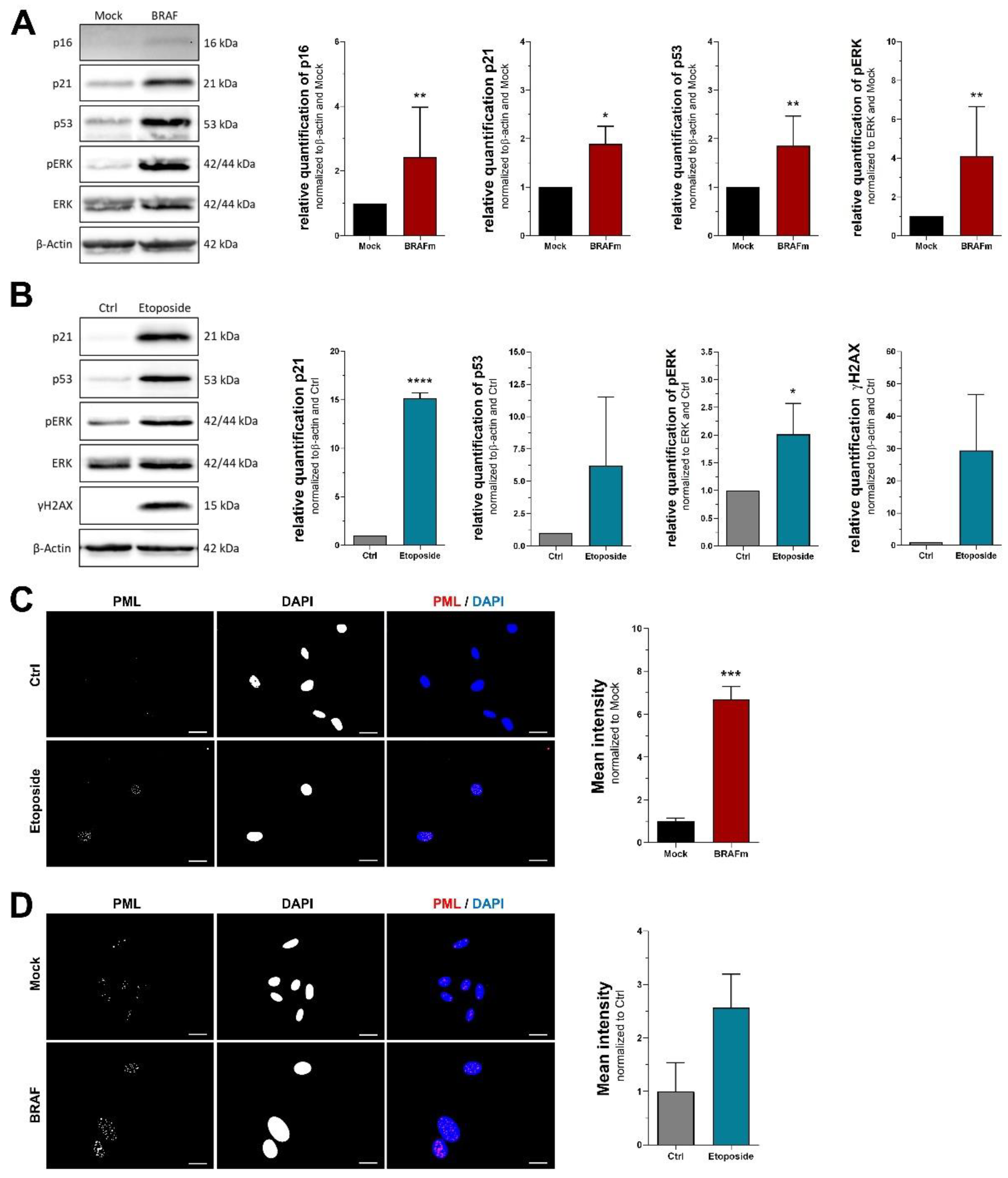
3.1. RNA Markers of Senescence
3.2. Protein Markers of Senescence
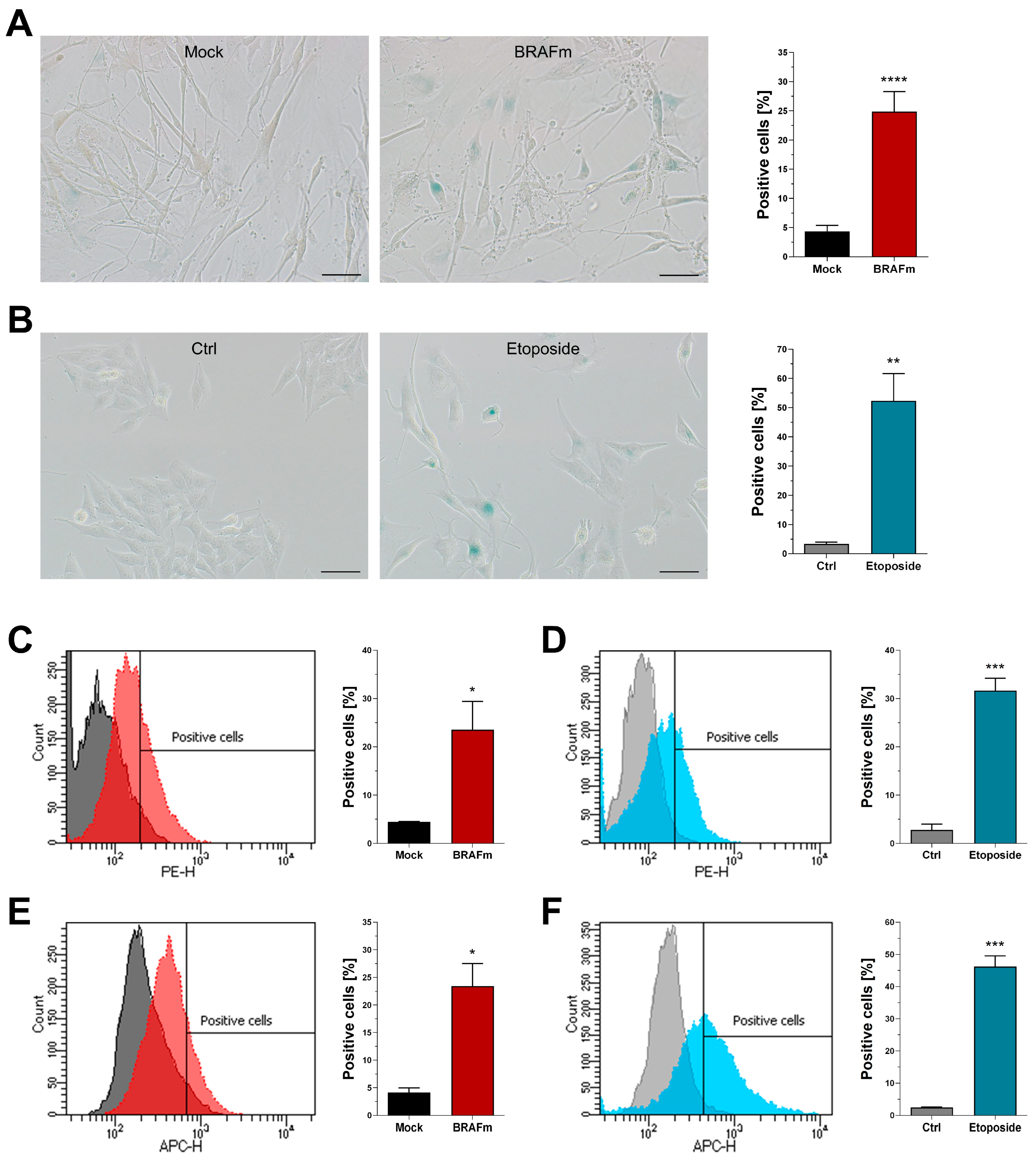
3.3. Functional and Morphological Markers of Senescence
3.4. Quantification of Senescence-Associated Beta-Galactosidase Activity
3.5. Validating Selected Markers for Detection of Cellular Senescence in Melanoma Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Rhinn, M.; Ritschka, B.; Keyes, W.M. Cellular senescence in development, regeneration and disease. Development 2019, 146, dev151837. [Google Scholar] [CrossRef] [Green Version]
- McHugh, D.; Gil, J. Senescence and Aging: Causes, Consequences, and Therapeutic Avenues. J. Cell Biol. 2017, 217, 65–77. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; Van Der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-Associated Senescence-like Cell Cycle Arrest of Human Naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Shain, A.H.; Bastian, B.C. From Melanocytes to Melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Leclerc, J.; Ballotti, R.; Bertolotto, C. Pathways from senescence to melanoma: Focus on MITF sumoylation. Oncogene 2017, 36, 6659–6667. [Google Scholar] [CrossRef]
- Saleh, T.; Carpenter, V.J. Potential Use of Senolytics for Pharmacological Targeting of Precancerous Lesions. Mol. Pharmacol. 2021, 100, 580–587. [Google Scholar] [CrossRef]
- L’Hôte, V.; Courbeyrette, R.; Pinna, G.; Cintrat, J.C.; Le Pavec, G.; Delaunay-Moisan, A.; Mann, C.; Thuret, J.Y. Ouabain and Chloroquine Trigger Senolysis of BRAF-V600E-Induced Senescent Cells by Targeting Autophagy. Aging Cell 2021, 20, e13447. [Google Scholar] [CrossRef]
- Hosoya, N.; Miyagawa, K. Targeting DNA Damage Response in Cancer Therapy. Cancer Sci. 2014, 105, 370–388. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.R.; Nelson, P.S. Cellular senescence and cancer chemotherapy resistance. Drug Resist. Updat. 2012, 15, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Guillon, J.; Petit, C.; Toutain, B.; Guette, C.; Lelievre, E.; Coqueret, O. Chemotherapy-induced senescence, an adaptive mechanism driving resistance and tumor heterogeneity. Cell Cycle 2019, 18, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Matt, S.; Hofmann, T.G. The DNA damage-induced cell death response: A roadmap to kill cancer cells. Experientia 2016, 73, 2829–2850. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-Associated Beta-Galactosidase Is Lysosomal Beta-Galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- Itahana, K.; Campisi, J.; Dimri, G.P. Methods to Detect Biomarkers of Cellular Senescence: The Senescence-Associated Beta-Galactosidase Assay. Methods Mol. Biol. 2007, 371, 21–31. [Google Scholar] [CrossRef]
- Feuerer, L.; Lamm, S.; Henz, I.; Kappelmann-Fenzl, M.; Haferkamp, S.; Meierjohann, S.; Hellerbrand, C.; Kuphal, S.; Bosserhoff, A.K. Role of Melanoma Inhibitory Activity in Melanocyte Senescence. Pigment. Cell Melanoma Res. 2019, 32, 777–791. [Google Scholar] [CrossRef]
- Zimmermann, T.; Gebhardt, L.; Kreiss, L.; Schneider, C.; Arndt, S.; Karrer, S.; Friedrich, O.; Fischer, M.; Bosserhoff, A.-K. Acidified Nitrite Contributes to the Antitumor Effect of Cold Atmospheric Plasma on Melanoma Cells. Int. J. Mol. Sci. 2021, 22, 3757. [Google Scholar] [CrossRef]
- Schiffner, S.; Braunger, B.M.; de Jel, M.M.; Coupland, S.E.; Tamm, E.R.; Bosserhoff, A.K. Tg(Grm1) Transgenic Mice: A Murine Model That Mimics Spontaneous Uveal Melanoma in Humans? Exp. Eye Res. 2014, 127, 59–68. [Google Scholar] [CrossRef]
- Schreyer, L.; Mittermeier, C.; Franz, M.J.; Meier, M.A.; Martin, D.E.; Maier, K.C.; Huebner, K.; Schneider-Stock, R.; Singer, S.; Holzer, K.; et al. Tetraspanin 5 (TSPAN5), a Novel Gatekeeper of the Tumor Suppressor DLC1 and Myocardin-Related Transcription Factors (MRTFs), Controls HCC Growth and Senescence. Cancers 2021, 13, 5373. [Google Scholar] [CrossRef]
- Bosserhoff, A.K.; Ellmann, L.; Kuphal, S. Melanoblasts in Culture as an in Vitro System to Determine Molecular Changes in Melanoma. Exp. Dermatol. 2011, 20, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.S.; Dreesen, O. Biomarkers of Cellular Senescence and Skin Aging. Front. Genet. 2018, 9, 247. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Lei, T.; Guo, P.; Yu, J.; Xu, Q.; Luo, Y.; Ke, R.; Huang, D. Mechanisms Shaping the Role of ERK1/2 in Cellular Senescence. Mol. Med. Rep. 2018, 19, 759–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What Do All the Mutations Mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef]
- Böhme, I.; Bosserhoff, A. Extracellular Acidosis Triggers a Senescence-like Phenotype in Human Melanoma Cells. Pigment. Cell Melanoma Res. 2019, 33, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Jochems, F.; Thijssen, B.; De Conti, G.; Jansen, R.; Pogacar, Z.; Groot, K.; Wang, L.; Schepers, A.; Wang, C.; Jin, H.; et al. The Cancer SENESCopedia: A Delineation of Cancer Cell Senescence. Cell Rep. 2021, 36, 109441. [Google Scholar] [CrossRef]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular Senescence and Tumor Suppressor Gene P16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 485. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of P53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Larsson, L.G. Oncogene- and Tumor Suppressor Gene-Mediated Suppression of Cellular Senescence. Semin. Cancer Biol. 2011, 21, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors P21 and P16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Te Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA Damage Is Able to Induce Senescence in Tumor Cells In Vitro and In Vivo 1. Cancer Res. 2002, 62, 1876–1883. [Google Scholar] [PubMed]
- Brien, G.L.; Bracken, A.P. The PCL1-p53 axis promotes cellular quiescence. Cell Cycle 2016, 15, 305–306. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Bhatia, R. Stem Cell Quiescence. Clin. Cancer Res. 2011, 17, 4936–4941. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Lee, S.; Park, J.Y.; Lee, S.Y.; Kim, H.S. Induction of P53-Dependent Apoptosis by Prostaglandin A2. Biomolecules 2020, 10, 492. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Simpson, E.R.; Brown, K.A. P53: Protection against Tumor Growth beyond Effects on Cell Cycle and Apoptosis. Cancer Res. 2015, 75, 5001–5007. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Wang, L.; Tang, J.; Cao, P.; Luo, Z.; Sun, J.; Kiflu, A.; Sai, B.; Zhang, M.; Wang, F.; et al. Activation of Anaphase-Promoting Complex by P53 Induces a State of Dormancy in Cancer Cells against Chemotherapeutic Stress. Oncotarget 2016, 7, 25478–25492. [Google Scholar] [CrossRef]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Birch, J.; Gil, J. Senescence and the SASP: Many Therapeutic Avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, C.R.; Ritchie, S.; Pereira, B.A.; Timpson, P. Overcoming the Senescence-associated Secretory Phenotype (SASP): A Complex Mechanism of Resistance in the Treatment of Cancer. Mol. Oncol. 2021, 15, 3242–3255. [Google Scholar] [CrossRef]
- Tang, W.; Wang, W.; Zhang, Y.; Liu, S.; Liu, Y.; Zheng, D. Tumour Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL)-Induced Chemokine Release in Both TRAIL-Resistant and TRAIL-Sensitive Cells via Nuclear Factor Kappa B. FEBS J. 2008, 276, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.H.; Hales, C.N.; Ozanne, S.E. DNA Damage, Cellular Senescence and Organismal Ageing: Causal or Correlative? Nucleic Acids Res. 2007, 35, 7417–7428. [Google Scholar] [CrossRef]
- Kuo, L.J.; Yang, L.-X. γ-H2AX-A Novel Biomarker for DNA Double-Strand Breaks. In Vivo 2008, 22, 305–309. [Google Scholar] [PubMed]
- Previati, M.; Missiroli, S.; Perrone, M.; Caroccia, N.; Paliotto, F.; Milani, D.; Giorgi, C. Functions and dys-functions of promyelocytic leukemia protein PML. Rend. Lince 2018, 29, 411–420. [Google Scholar] [CrossRef]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef]
- Boucher, M.-J.; Jean, D.; Vézina, A.; Rivard, N. Dual role of MEK/ERK signaling in senescence and transformation of intestinal epithelial cells. Am. J. Physiol. Liver Physiol. 2004, 286, G736–G746. [Google Scholar] [CrossRef]
- Anerillas, C.; Abdelmohsen, K.; Gorospe, M. Regulation of senescence traits by MAPKs. GeroScience 2020, 42, 397–408. [Google Scholar] [CrossRef]
- Inamdar, G.S.; Madhunapantula, S.V.; Robertson, G.P. Targeting the MAPK pathway in melanoma: Why some approaches succeed and other fail. Biochem. Pharmacol. 2010, 80, 624–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial Dysfunction and Cell Senescence: Deciphering a Complex Relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Macedo, J.C.; Vaz, S.; Bakker, B.; Ribeiro, R.; Bakker, P.L.; Escandell, J.M.; Ferreira, M.G.; Medema, R.; Foijer, F.; Logarinho, E. FoxM1 repression during human aging leads to mitotic decline and aneuploidy-driven full senescence. Nat. Commun. 2018, 9, 2834. [Google Scholar] [CrossRef] [Green Version]
- Flor, A.C.; Wolfgeher, D.; Wu, D.; Kron, S.J. A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov. 2017, 3, 17075. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| CDKN2A | GGAGCAGCATGGAGCCTTCGGC | CCACCAGCGTGTCCAGGAAGC |
| CDKN1A | CGAGGCACCGAGGCACTCAGAGG | CCTGCCTCCTCCCAACTCATCCC |
| TP53 | AAGTCTAGAGCCACCGTCCA | AGTCTGGCTGCCAATCCA |
| CXCL2 | ATCAATGTGACGGCAGGGAAA | CGAAACCTCTCTGCTCTAACAC |
| CCL8 | CCCAGGTGCAGTGTGACATTA | GGGAGGACCCCACAACACTA |
| 18s | TCTGTGATGCCCTTAGATGTCC | CCATCCAATCGGTAGTAGCG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimmermann, T.; Pommer, M.; Kluge, V.; Chiheb, C.; Muehlich, S.; Bosserhoff, A.-K. Detection of Cellular Senescence in Human Primary Melanocytes and Malignant Melanoma Cells In Vitro. Cells 2022, 11, 1489. https://doi.org/10.3390/cells11091489
Zimmermann T, Pommer M, Kluge V, Chiheb C, Muehlich S, Bosserhoff A-K. Detection of Cellular Senescence in Human Primary Melanocytes and Malignant Melanoma Cells In Vitro. Cells. 2022; 11(9):1489. https://doi.org/10.3390/cells11091489
Chicago/Turabian StyleZimmermann, Tom, Michaela Pommer, Viola Kluge, Chafia Chiheb, Susanne Muehlich, and Anja-Katrin Bosserhoff. 2022. "Detection of Cellular Senescence in Human Primary Melanocytes and Malignant Melanoma Cells In Vitro" Cells 11, no. 9: 1489. https://doi.org/10.3390/cells11091489