
Rabeprazole Promotes Vascular Repair and Resolution of Sepsis-Induced Inflammatory Lung Injury through HIF-1α

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Mouse Model of Endotoxemia Sepsis
2.3. High Throughput Screening for HIF Inducers
2.4. Primary Culture of Human Lung Microvascular ECs (HLMVECs)
2.5. Assessment of Lung Edema and Vascular Permeability
2.6. Measurement of Lung Myeloperoxidase (MPO) Activity
2.7. Molecular Analyses
2.8. Histological Analysis
2.9. Statistical Analysis
3. Results
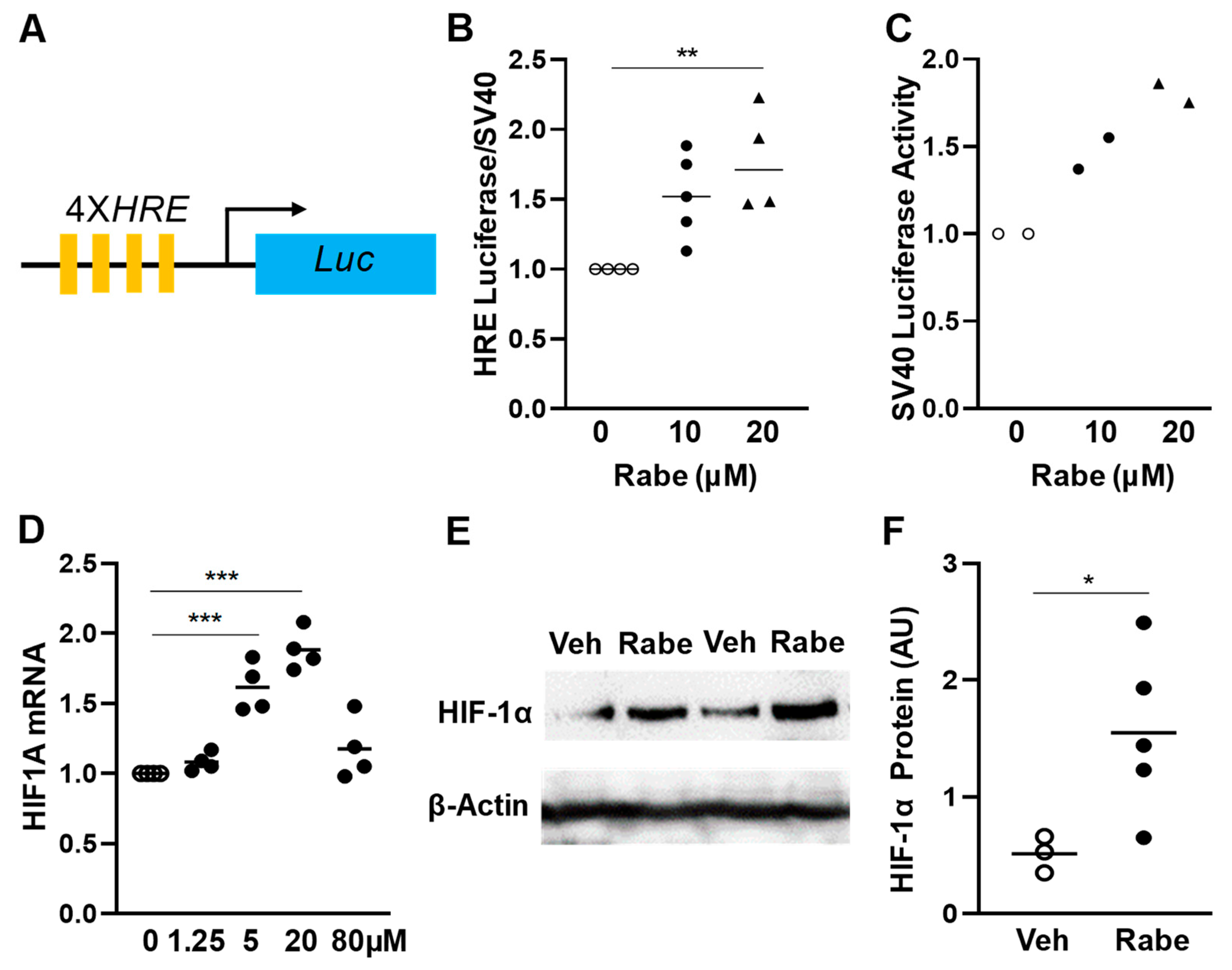
3.1. Rabeprazole Is an HIF-1α Inducer in Lung Endothelial Cells
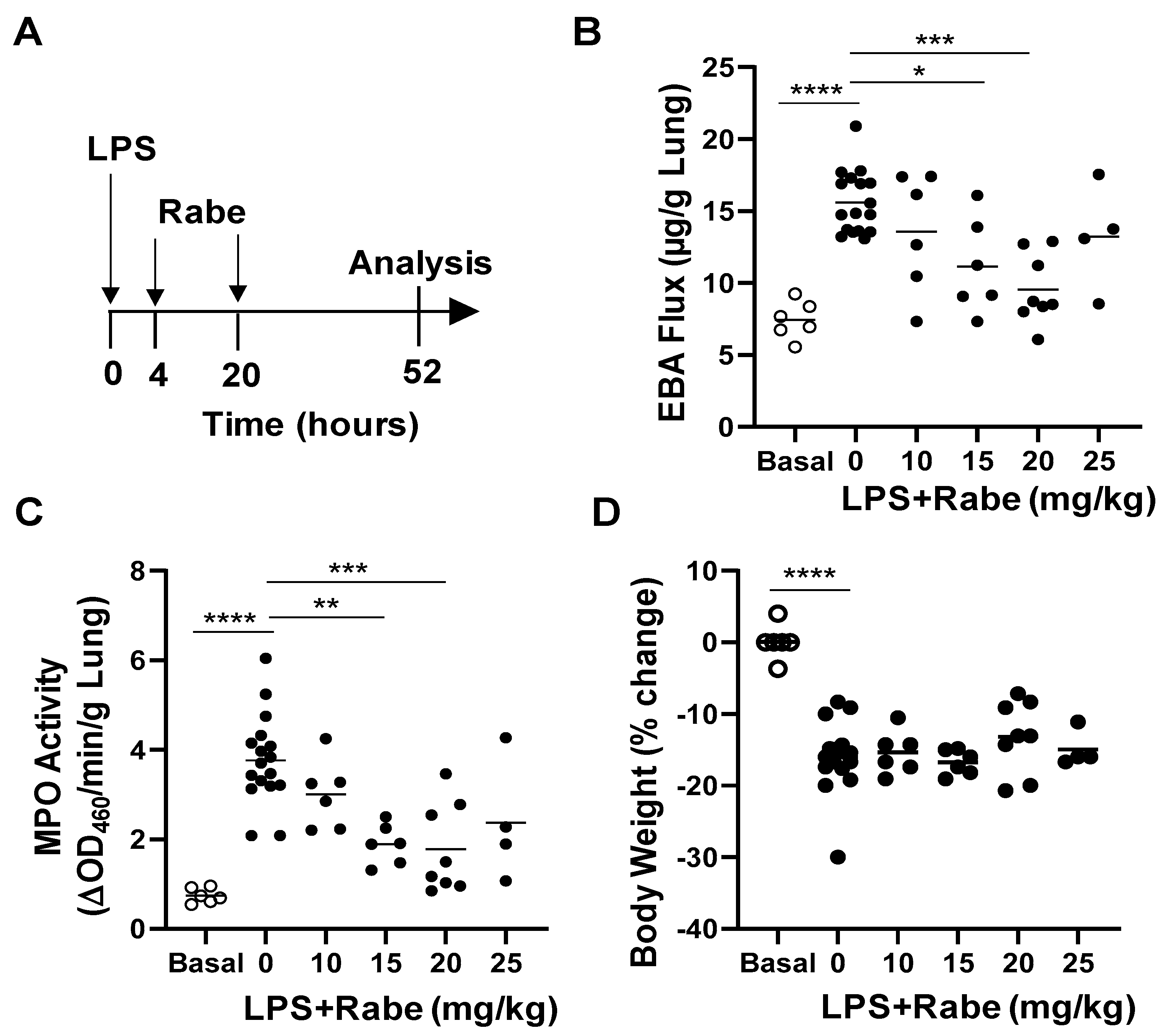
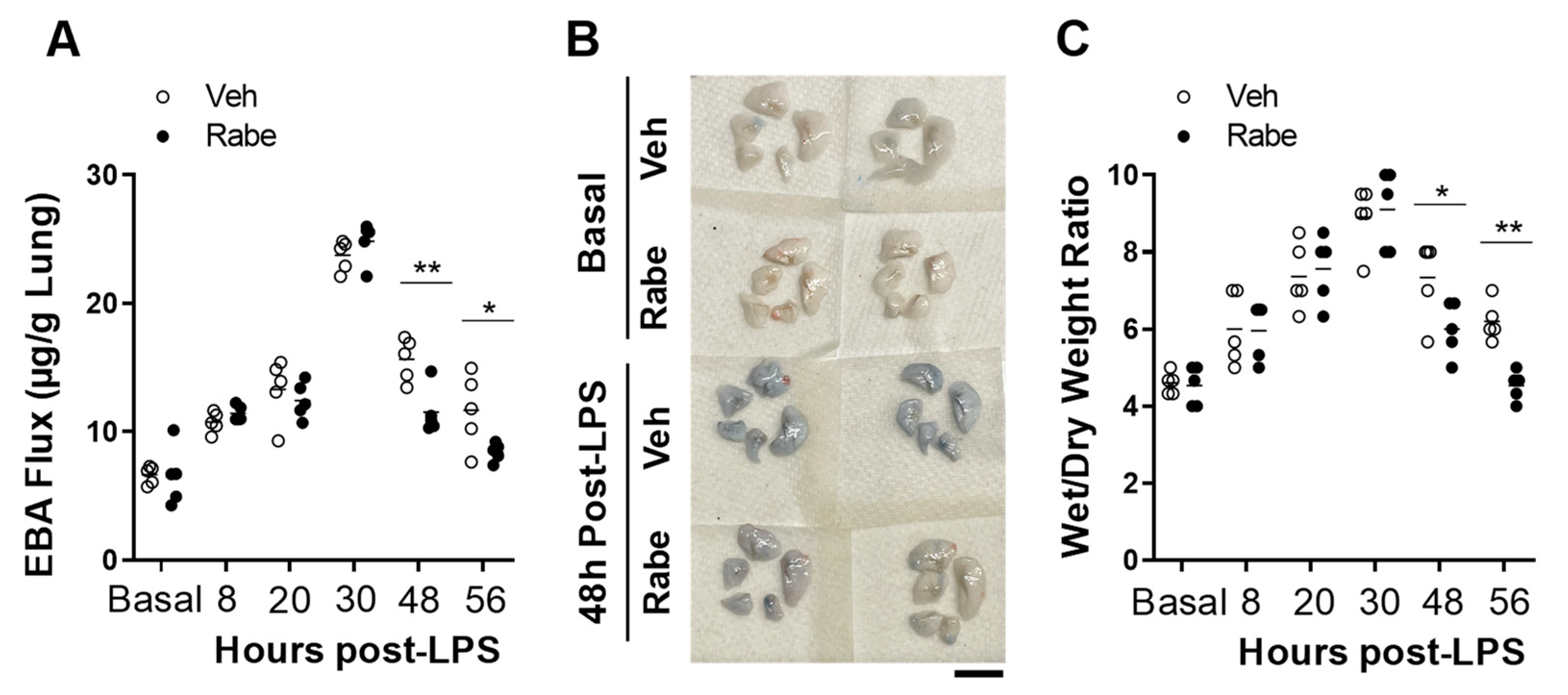
3.2. Rabeprazole Treatment Promotes Lung Vascular Repair Following LPS Challenge
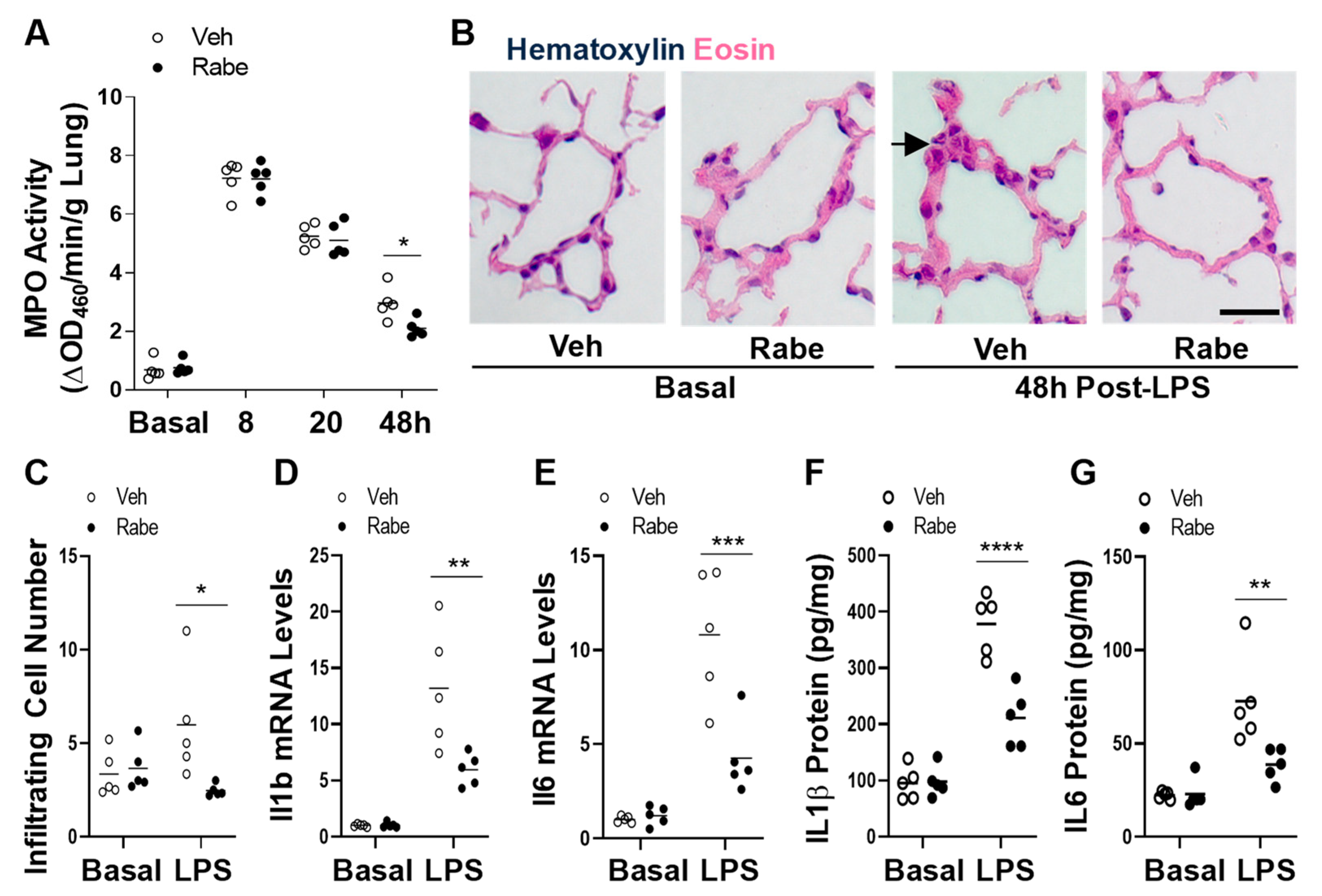
3.3. Rabeprazole Treatment Augments Resolution of Lung Inflammation after LPS Challenge
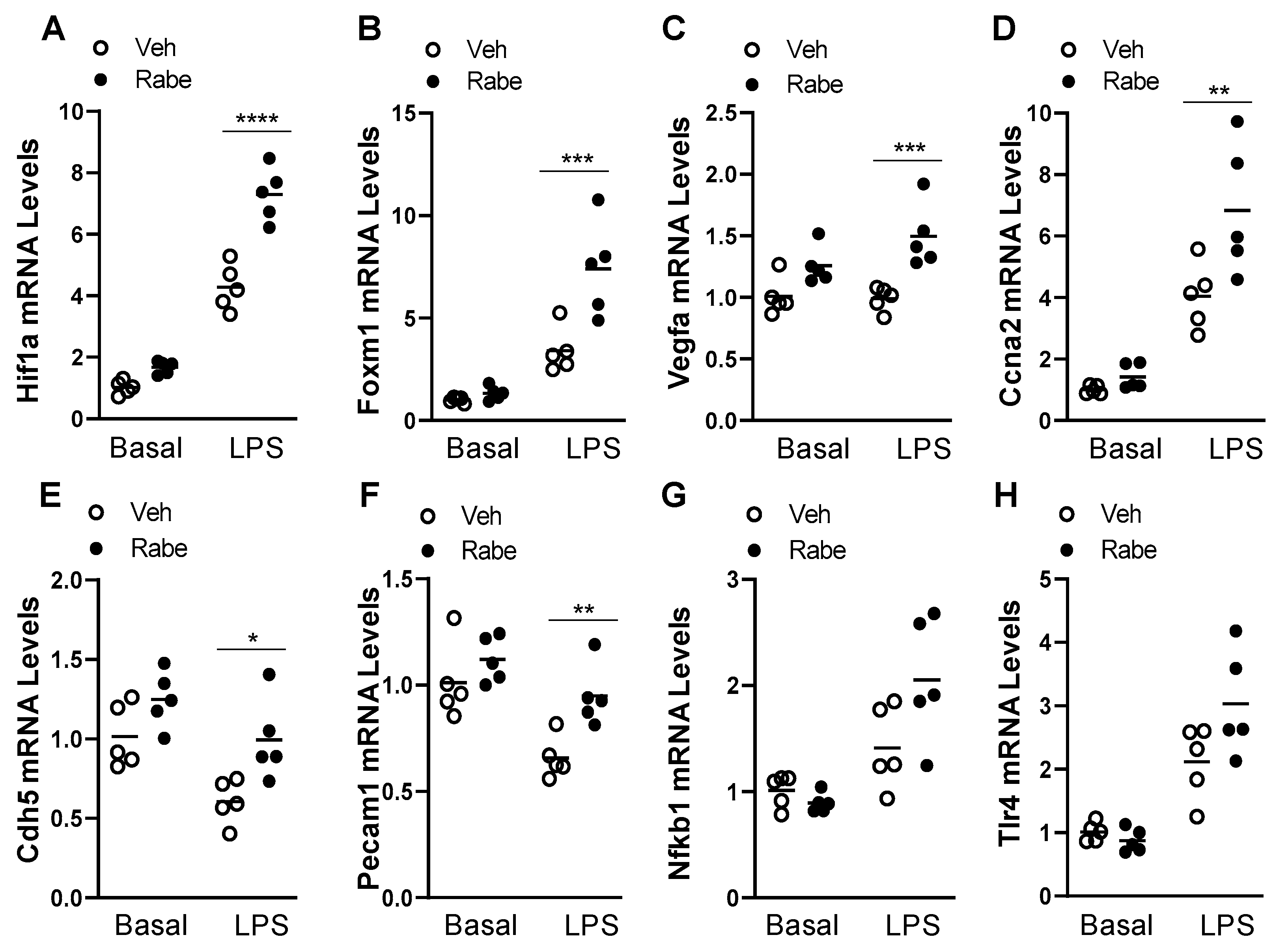
3.4. Rabeprazole Increases HIF-1α/FoxM1 Signaling after LPS Challenge
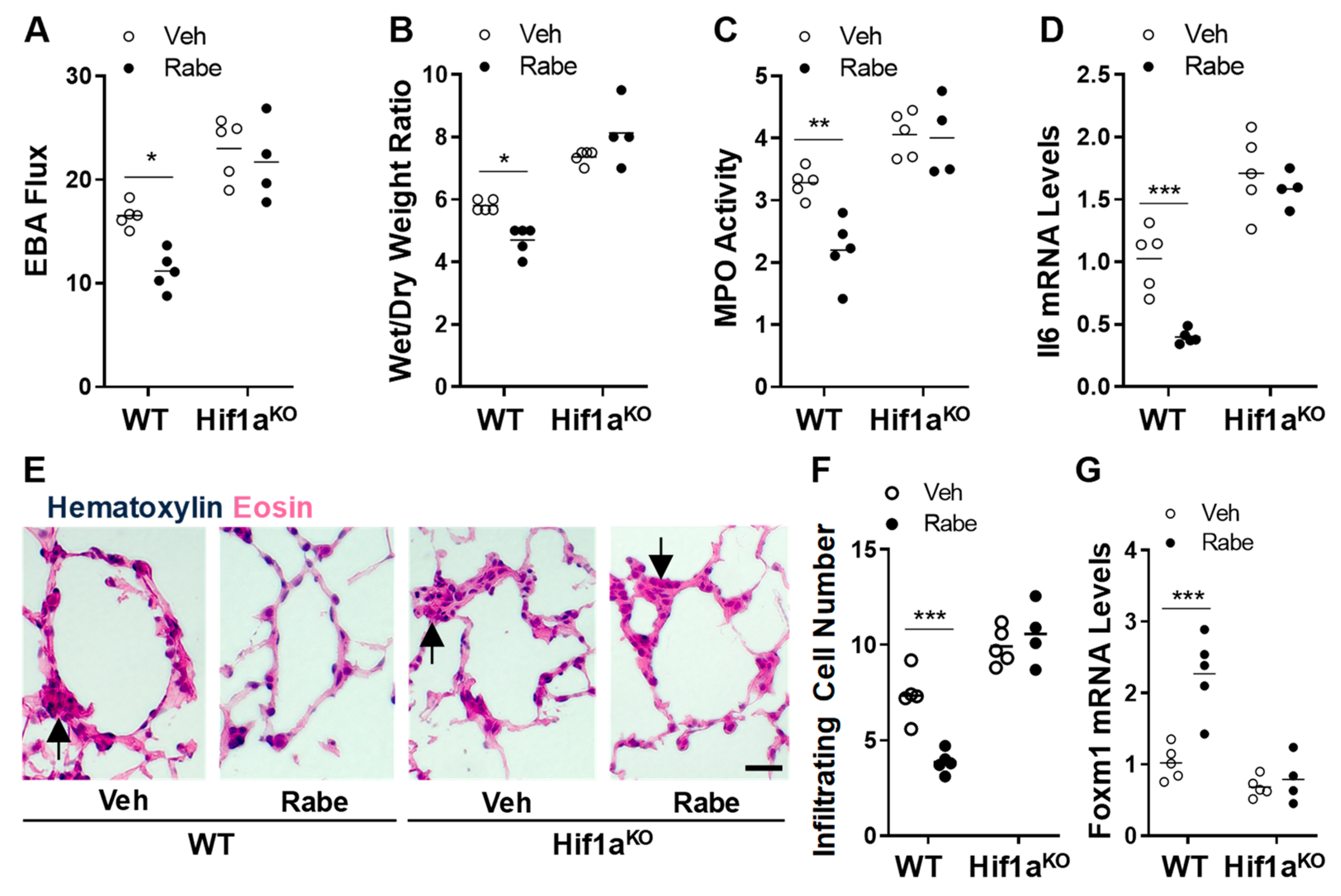
3.5. Rabeprazole Enhances Vascular Repair and Resolution of Sepsis-Induced Lung Inflammation through Endothelial HIF-1α
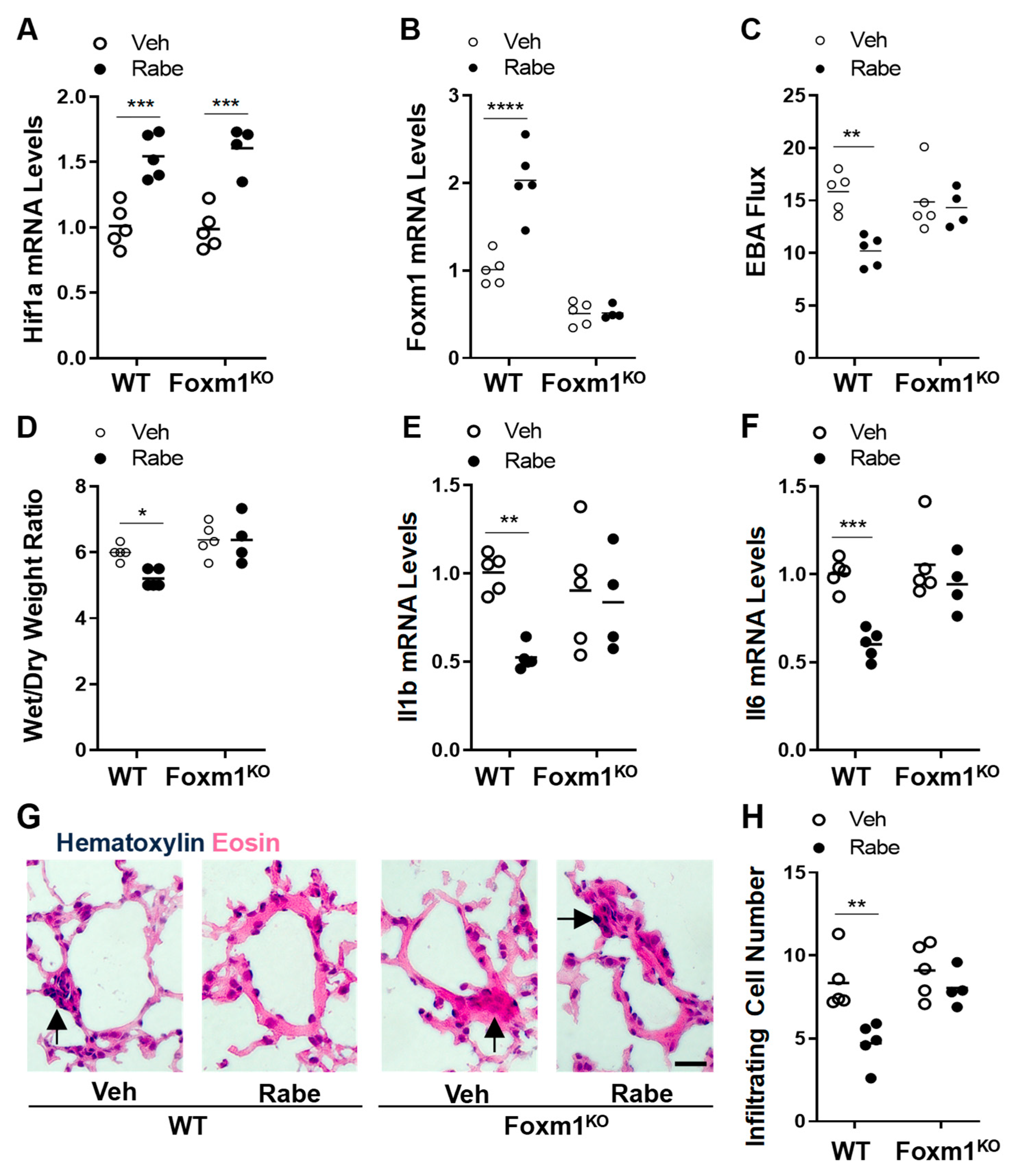
3.6. FoxM1 Is the Downstream Transcriptional Factor of HIF-1α Mediating Rabeprazole-Induced Reparative Effects
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, W.C. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003, 101, 3765–3777. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Gao, X.P.; Zhao, Y.D.; Mirza, M.K.; Frey, R.S.; Kalinichenko, V.V.; Wang, I.C.; Costa, R.H.; Malik, A.B. Endothelial cell-restricted disruption of FoxM1 impairs endothelial repair following LPS-induced vascular injury. J. Clin. Investig. 2006, 116, 2333–2343. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Dai, Z.; Cai, L.; Sun, K.; Cho, J.; Albertine, K.H.; Malik, A.B.; Schraufnagel, D.E.; Zhao, Y.Y. Endothelial p110gammaPI3K Mediates Endothelial Regeneration and Vascular Repair After Inflammatory Vascular Injury. Circulation 2016, 133, 1093–1103. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Zhang, X.; Zhao, D.X.; Yin, J.; Hu, G.; Evans, C.E.; Zhao, Y.Y. Endothelial Hypoxia-Inducible Factor-1alpha Is Required for Vascular Repair and Resolution of Inflammatory Lung Injury through Forkhead Box Protein M1. Am. J. Pathol. 2019, 189, 1664–1679. [Google Scholar] [CrossRef]
- De Backer, D.; Creteur, J.; Preiser, J.C.; Dubois, M.J.; Vincent, J.L. Microvascular blood flow is altered in patients with sepsis. Am. J. Respir. Crit. Care Med. 2002, 166, 98–104. [Google Scholar] [CrossRef]
- Trzeciak, S.; Dellinger, R.P.; Parrillo, J.E.; Guglielmi, M.; Bajaj, J.; Abate, N.L.; Arnold, R.C.; Colilla, S.; Zanotti, S.; Hollenberg, S.M.; et al. Early microcirculatory perfusion derangements in patients with severe sepsis and septic shock: Relationship to hemodynamics, oxygen transport, and survival. Ann. Emerg. Med. 2007, 49, 88–98. [Google Scholar] [CrossRef]
- Sakr, Y.; Dubois, M.J.; De Backer, D.; Creteur, J.; Vincent, J.L. Persistent microcirculatory alterations are associated with organ failure and death in patients with septic shock. Crit. Care Med. 2004, 32, 1825–1831. [Google Scholar] [CrossRef]
- Goldenberg, N.M.; Steinberg, B.E.; Slutsky, A.S.; Lee, W.L. Broken barriers: A new take on sepsis pathogenesis. Sci. Transl. Med. 2011, 3, 88ps25. [Google Scholar] [CrossRef] [Green Version]
- Matthay, M.A.; McAuley, D.F.; Ware, L.B. Clinical trials in acute respiratory distress syndrome: Challenges and opportunities. Lancet Respir. Med. 2017, 5, 524–534. [Google Scholar] [CrossRef]
- Yunt, Z.X.; Mohning, M.P.; Barthel, L.; Kearns, M.T.; Tuder, R.M.; Hyde, D.M.; Henson, P.M.; Janssen, W.J. Kinetics of the angiogenic response in lung endothelium following acute inflammatory injury with bleomycin. Exp. Lung Res. 2014, 40, 415–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimaldi, D.; Vincent, J.L. Clinical trial research in focus: Rethinking trials in sepsis. Lancet Respir. Med. 2017, 5, 610–611. [Google Scholar] [CrossRef]
- Bolte, C.; Kalin, T.V.; Kalinichenko, V.V. Molecular, cellular, and bioengineering approaches to stimulate lung regeneration after injury. Semin. Cell Dev. Biol. 2020, 100, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Komuro, I. Regeneration of the endothelium as a novel therapeutic strategy for acute lung injury. J. Clin. Investig. 2006, 116, 2316–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Transcriptional regulation by hypoxia-inducible factor 1 molecular mechanisms of oxygen homeostasis. Trends Cardiovasc. Med. 1996, 6, 151–157. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 2001, 13, 167–171. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci. STKE 2007, 2007, cm8. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Minamishima, Y.A.; Kaelin, W.G., Jr. Reactivation of hepatic EPO synthesis in mice after PHD loss. Science 2010, 329, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Duan, C. Hypoxia-inducible factor 3 biology: Complexities and emerging themes. Am. J. Physiol. Cell Physiol. 2016, 310, C260–C269. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Rha, J.; Unger, T.L.; Wu, C.H.; Shutt, H.P.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor-2 regulates vascular tumorigenesis in mice. Oncogene 2008, 27, 5354–5358. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.K.; Sun, Y.; Zhao, Y.D.; Potula, H.H.; Frey, R.S.; Vogel, S.M.; Malik, A.B.; Zhao, Y.Y. FoxM1 regulates re-annealing of endothelial adherens junctions through transcriptional control of beta-catenin expression. J. Exp. Med. 2010, 207, 1675–1685. [Google Scholar] [CrossRef]
- Wu, C.; Evans, C.E.; Dai, Z.; Huang, X.; Zhang, X.; Jin, H.; Hu, G.; Song, Y.; Zhao, Y.Y. Lipopolysaccharide-induced endotoxemia in corn oil-preloaded mice causes an extended course of lung injury and repair and pulmonary fibrosis: A translational mouse model of acute respiratory distress syndrome. PLoS ONE 2017, 12, e0174327. [Google Scholar] [CrossRef]
- Mirza, M.K.; Yuan, J.; Gao, X.P.; Garrean, S.; Brovkovych, V.; Malik, A.B.; Tiruppathi, C.; Zhao, Y.Y. Caveolin-1 deficiency dampens Toll-like receptor 4 signaling through eNOS activation. Am. J. Pathol. 2010, 176, 2344–2351. [Google Scholar] [CrossRef]
- Tanigawa, T.; Watanabe, T.; Higuchi, K.; Machida, H.; Okazaki, H.; Yamagami, H.; Watanabe, K.; Tominaga, K.; Fujiwara, Y.; Oshitani, N.; et al. Lansoprazole, a Proton Pump Inhibitor, Suppresses Production of Tumor Necrosis Factor-alpha and Interleukin-1beta Induced by Lipopolysaccharide and Helicobacter Pylori Bacterial Components in Human Monocytic Cells via Inhibition of Activation of Nuclear Factor-kappaB and Extracellular Signal-Regulated Kinase. J. Clin. Biochem. Nutr. 2009, 45, 86–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsuka, M.; Hashimoto, Y.; Nakatake, R.; Okuyama, T.; Hatta, M.; Yoshida, T.; Okumura, T.; Nishizawa, M.; Kaibori, M.; Sekimoto, M. Omeprazole Increases Survival Through the Inhibition of Inflammatory Mediaters in Two Rat Sepsis Models. Shock 2022, 57, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Nakatake, R.; Hishikawa, H.; Kotsuka, M.; Ishizaki, M.; Matsui, K.; Nishizawa, M.; Yoshizawa, K.; Kaibori, M.; Okumura, T. The Proton Pump Inhibitor Lansoprazole Has Hepatoprotective Effects in In Vitro and In Vivo Rat Models of Acute Liver Injury. Dig. Dis. Sci. 2019, 64, 2854–2866. [Google Scholar] [CrossRef] [PubMed]
- Geeviman, K.; Babu, D.; Prakash Babu, P. Pantoprazole Induces Mitochondrial Apoptosis and Attenuates NF-κB Signaling in Glioma Cells. Cell Mol. Neurobiol. 2018, 38, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Handa, O.; Yoshida, N.; Fujita, N.; Tanaka, Y.; Ueda, M.; Takagi, T.; Kokura, S.; Naito, Y.; Okanoue, T.; Yoshikawa, T. Molecular mechanisms involved in anti-inflammatory effects of proton pump inhibitors. Inflamm. Res. 2006, 55, 476–480. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, Y.Y. Transgenic expression of FoxM1 promotes endothelial repair following lung injury induced by polymicrobial sepsis in mice. PLoS ONE 2012, 7, e50094. [Google Scholar] [CrossRef] [Green Version]
- Langtry, H.D.; Markham, A. Rabeprazole: A review of its use in acid-related gastrointestinal disorders. Drugs 1999, 58, 725–742. [Google Scholar] [CrossRef]
- Pace, F.; Pallotta, S.; Casalini, S.; Porro, G.B. A review of rabeprazole in the treatment of acid-related diseases. Ther. Clin. Risk Manag. 2007, 3, 363–379. [Google Scholar]
- Carswell, C.I.; Goa, K.L. Rabeprazole: An update of its use in acid-related disorders. Drugs 2001, 61, 2327–2356. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, L.; Marsboom, G.; Jambusaria, A.; Xiong, S.; Toth, P.T.; Benevolenskaya, E.V.; Rehman, J.; Malik, A.B. Sox17 is required for endothelial regeneration following inflammation-induced vascular injury. Nat. Commun. 2019, 10, 2126. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Malkovskiy, A.V.; Tian, W.; Sung, Y.K.; Sun, W.; Hsu, J.L.; Manickam, S.; Wagh, D.; Joubert, L.M.; Semenza, G.L.; et al. Promotion of airway anastomotic microvascular regeneration and alleviation of airway ischemia by deferoxamine nanoparticles. Biomaterials 2014, 35, 803–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Hsu, J.L.; Tian, W.; Yuan, K.; Olcholski, M.; Perez Vde, J.; Semenza, G.L.; Nicolls, M.R. Tie2-dependent VHL knockdown promotes airway microvascular regeneration and attenuates invasive growth of Aspergillus fumigatus. J. Mol. Med. 2013, 91, 1081–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClendon, J.; Jansing, N.L.; Redente, E.F.; Gandjeva, A.; Ito, Y.; Colgan, S.P.; Ahmad, A.; Riches, D.W.H.; Chapman, H.A.; Mason, R.J.; et al. Hypoxia-Inducible Factor 1alpha Signaling Promotes Repair of the Alveolar Epithelium after Acute Lung Injury. Am. J. Pathol. 2017, 187, 1772–1786. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Bishop, T.; Ratcliffe, P.J.; Yeger, H.; Cutz, E. Hyperplasia and hypertrophy of pulmonary neuroepithelial bodies, presumed airway hypoxia sensors, in hypoxia-inducible factor prolyl hydroxylase-deficient mice. Hypoxia 2016, 4, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19)? Eur. Respir. J. 2020, 56, 2001634. [Google Scholar] [CrossRef]
- Pons, S.; Fodil, S.; Azoulay, E.; Zafrani, L. The vascular endothelium: The cornerstone of organ dysfunction in severe SARS-CoV-2 infection. Crit. Care 2020, 24, 353. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Hariri, L.; Hardin, C.C. Covid-19, Angiogenesis, and ARDS Endotypes. N. Engl. J. Med. 2020, 383, 182–183. [Google Scholar] [CrossRef]
- Mangalmurti, N.S.; Reilly, J.P.; Cines, D.B.; Meyer, N.J.; Hunter, C.A.; Vaughan, A.E. COVID-19-associated Acute Respiratory Distress Syndrome Clarified: A Vascular Endotype? Am. J. Respir. Crit. Care Med. 2020, 202, 750–753. [Google Scholar] [CrossRef]
- Marini, J.J.; Gattinoni, L. Management of COVID-19 Respiratory Distress. JAMA 2020, 323, 2329–2330. [Google Scholar] [CrossRef] [PubMed]
- Chertow, G.M.; Pergola, P.E.; Farag, Y.M.K.; Agarwal, R.; Arnold, S.; Bako, G.; Block, G.A.; Burke, S.; Castillo, F.P.; Jardine, A.G.; et al. Vadadustat in Patients with Anemia and Non-Dialysis-Dependent CKD. N. Engl. J. Med. 2021, 384, 1589–1600. [Google Scholar] [CrossRef]
- Hardt, C.; Beber, M.E.; Rasche, A.; Kamburov, A.; Hebels, D.G.; Kleinjans, J.C.; Herwig, R. ToxDB: Pathway-level interpretation of drug-treatment data. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, X.W.; Zhao, F.; Wang, J.Y.; Wang, H.Y.; Ge, S.H.; Wang, X.; Zhang, L.; Liu, R.; Ba, Y.; Li, H.L.; et al. Tumor microenvironment interruption: A novel anti-cancer mechanism of Proton-pump inhibitor in gastric cancer by suppressing the release of microRNA-carrying exosomes. Am. J. Cancer Res. 2017, 7, 1913–1925. [Google Scholar] [PubMed]
- Chen, M.; Huang, S.L.; Zhang, X.Q.; Zhang, B.; Zhu, H.; Yang, V.W.; Zou, X.P. Reversal effects of pantoprazole on multidrug resistance in human gastric adenocarcinoma cells by down-regulating the V-ATPases/mTOR/HIF-1α/P-gp and MRP1 signaling pathway in vitro and in vivo. J. Cell Biochem. 2012, 113, 2474–2487. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef]
- Semenza, G.L. A compendium of proteins that interact with HIF-1alpha. Exp. Cell Res. 2017, 356, 128–135. [Google Scholar] [CrossRef]
- Simon, M.C. The Hypoxia Response Pathways—Hats Off! N. Engl. J. Med. 2016, 375, 1687–1689. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.G.; Robbins, P.A.; Ratcliffe, P.J. The human side of hypoxia-inducible factor. Br. J. Haematol. 2008, 141, 325–334. [Google Scholar] [CrossRef]
- Evans, C.E. Hypoxia-Inducible Factor Signaling in Inflammatory Lung Injury and Repair. Cells 2022, 11, 183. [Google Scholar] [CrossRef]
- Gong, H.; Rehman, J.; Tang, H.; Wary, K.; Mittal, M.; Chaturvedi, P.; Zhao, Y.Y.; Komarova, Y.A.; Vogel, S.M.; Malik, A.B. HIF2alpha signaling inhibits adherens junctional disruption in acute lung injury. J. Clin. Investig. 2015, 125, 652–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N. Engl. J. Med. 2019, 381, 1001–1010. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evans, C.E.; Peng, Y.; Zhu, M.M.; Dai, Z.; Zhang, X.; Zhao, Y.-Y. Rabeprazole Promotes Vascular Repair and Resolution of Sepsis-Induced Inflammatory Lung Injury through HIF-1α. Cells 2022, 11, 1425. https://doi.org/10.3390/cells11091425
Evans CE, Peng Y, Zhu MM, Dai Z, Zhang X, Zhao Y-Y. Rabeprazole Promotes Vascular Repair and Resolution of Sepsis-Induced Inflammatory Lung Injury through HIF-1α. Cells. 2022; 11(9):1425. https://doi.org/10.3390/cells11091425
Chicago/Turabian StyleEvans, Colin E., Yi Peng, Maggie M. Zhu, Zhiyu Dai, Xianming Zhang, and You-Yang Zhao. 2022. "Rabeprazole Promotes Vascular Repair and Resolution of Sepsis-Induced Inflammatory Lung Injury through HIF-1α" Cells 11, no. 9: 1425. https://doi.org/10.3390/cells11091425