
Glucose Starvation or Pyruvate Dehydrogenase Activation Induce a Broad, ERK5-Mediated, Metabolic Remodeling Leading to Fatty Acid Oxidation

 , , ,
, , ,  ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. In Vivo Mouse Experiments
2.3. Cell Lines and Culture Conditions
2.4. Glucose Starvation
2.5. Human Liver Samples and Preparation of PHHs Cultures
2.6. Reagents and Antibodies
2.7. Transient Transfection
2.8. Plasmids
2.9. Counting and Determination of Cell Viability
2.10. RT-QPCR
2.11. Immunoblotting
2.12. Cellular Lipid Uptake
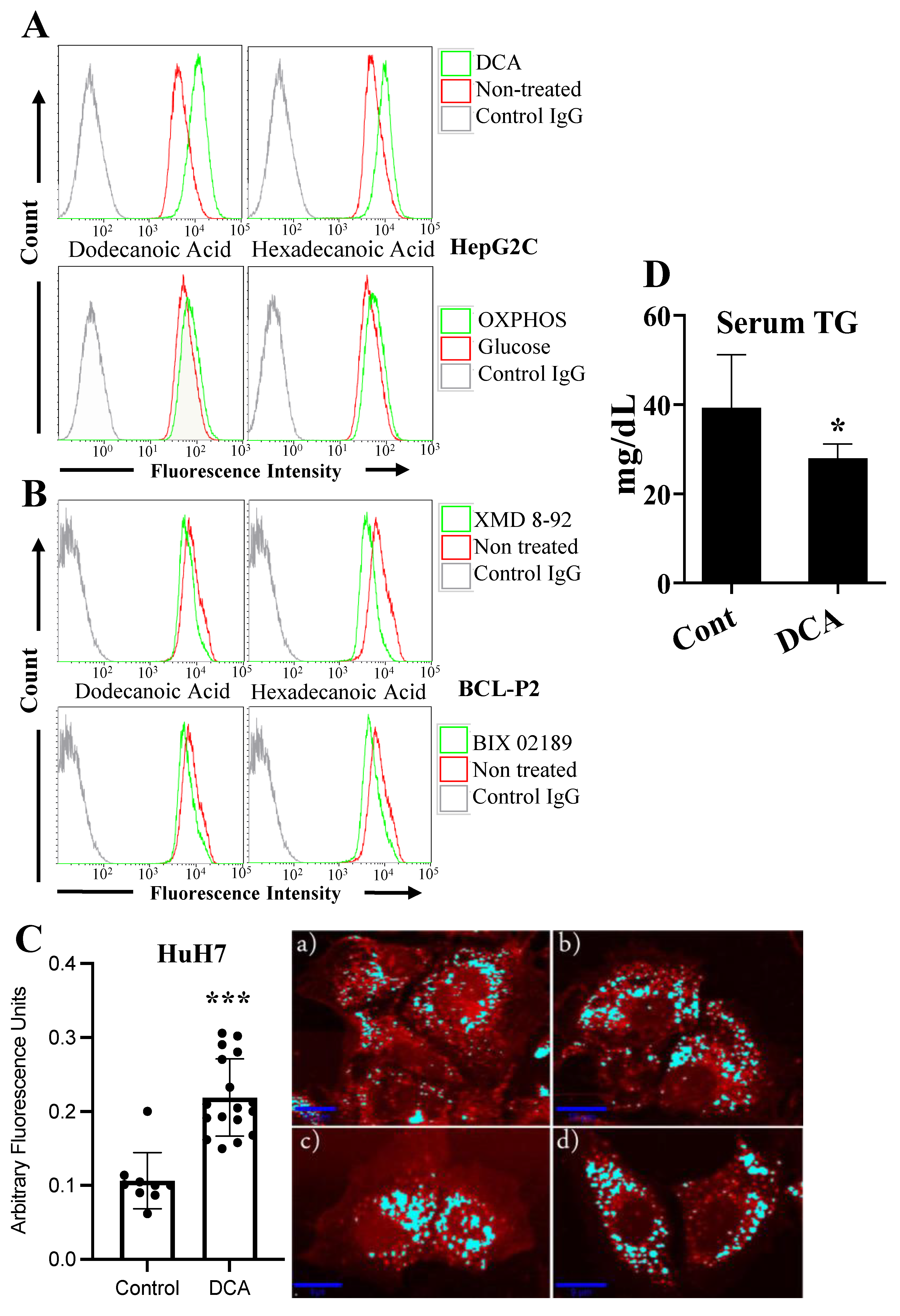
2.13. Serum Triglycerides
2.14. Extracellular Flux Analysis
2.15. Flow Cytometry
2.16. Statistical Analysis
3. Results
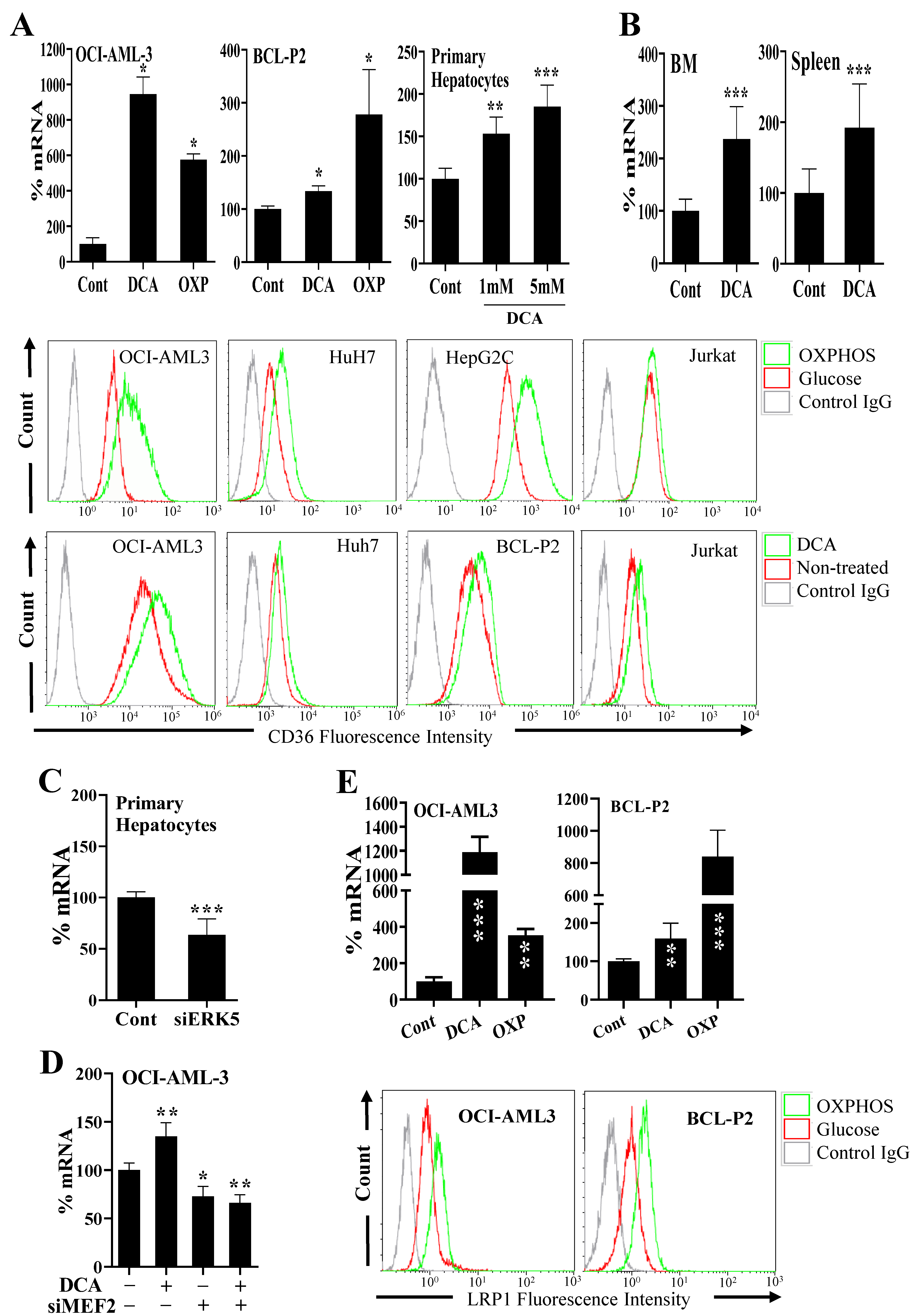
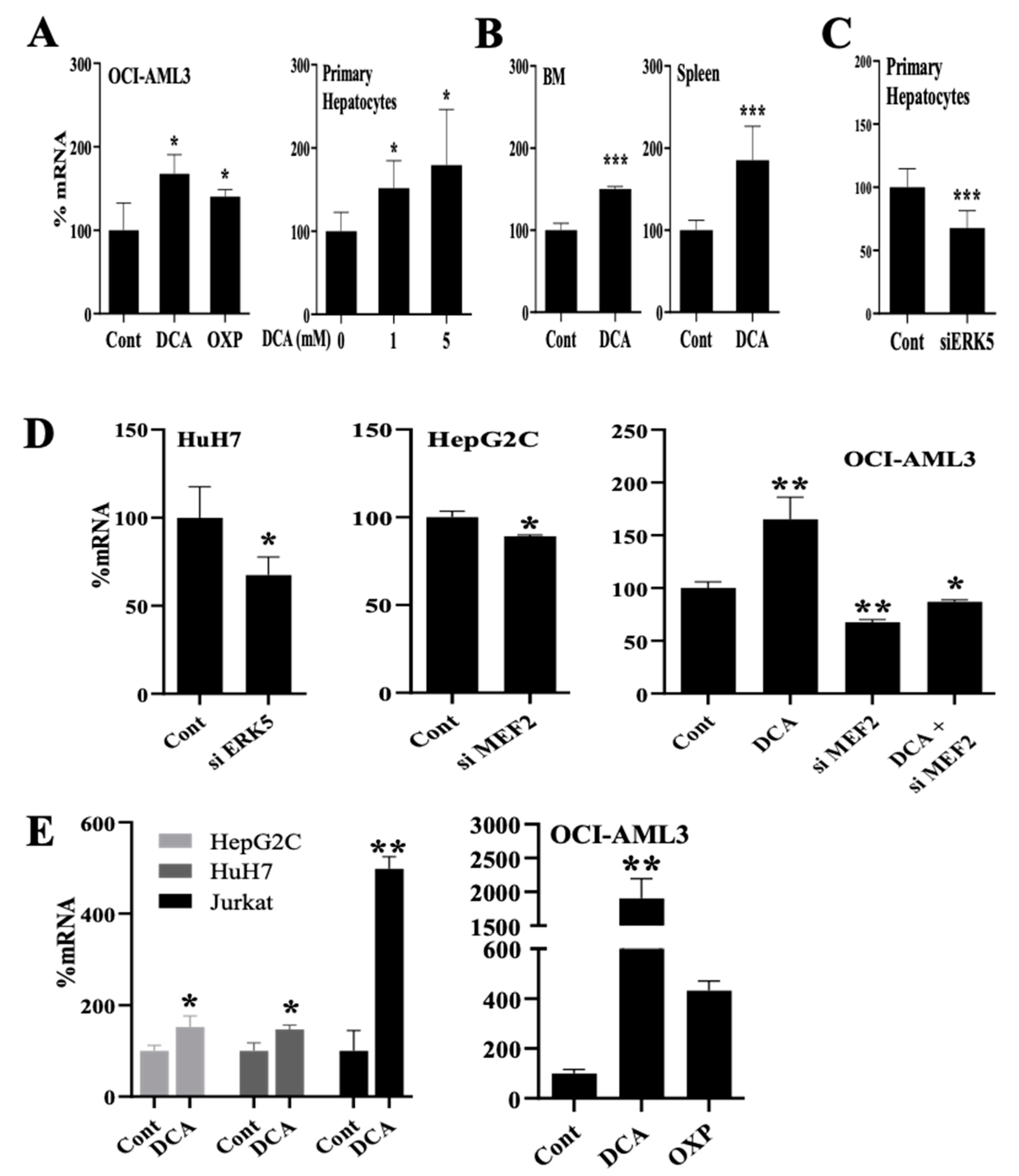
3.1. Changes in Metabolism Regulate Expression of Proteins Involved in Lipid Uptake through the ERK5 Pathway
3.2. Changes in Metabolism Regulate Lipid Uptake through the ERK5 Pathway
3.3. Changes in Metabolism Regulate the Expression of Enzymes Involved in Lipid Esterification through the ERK5 Pathway
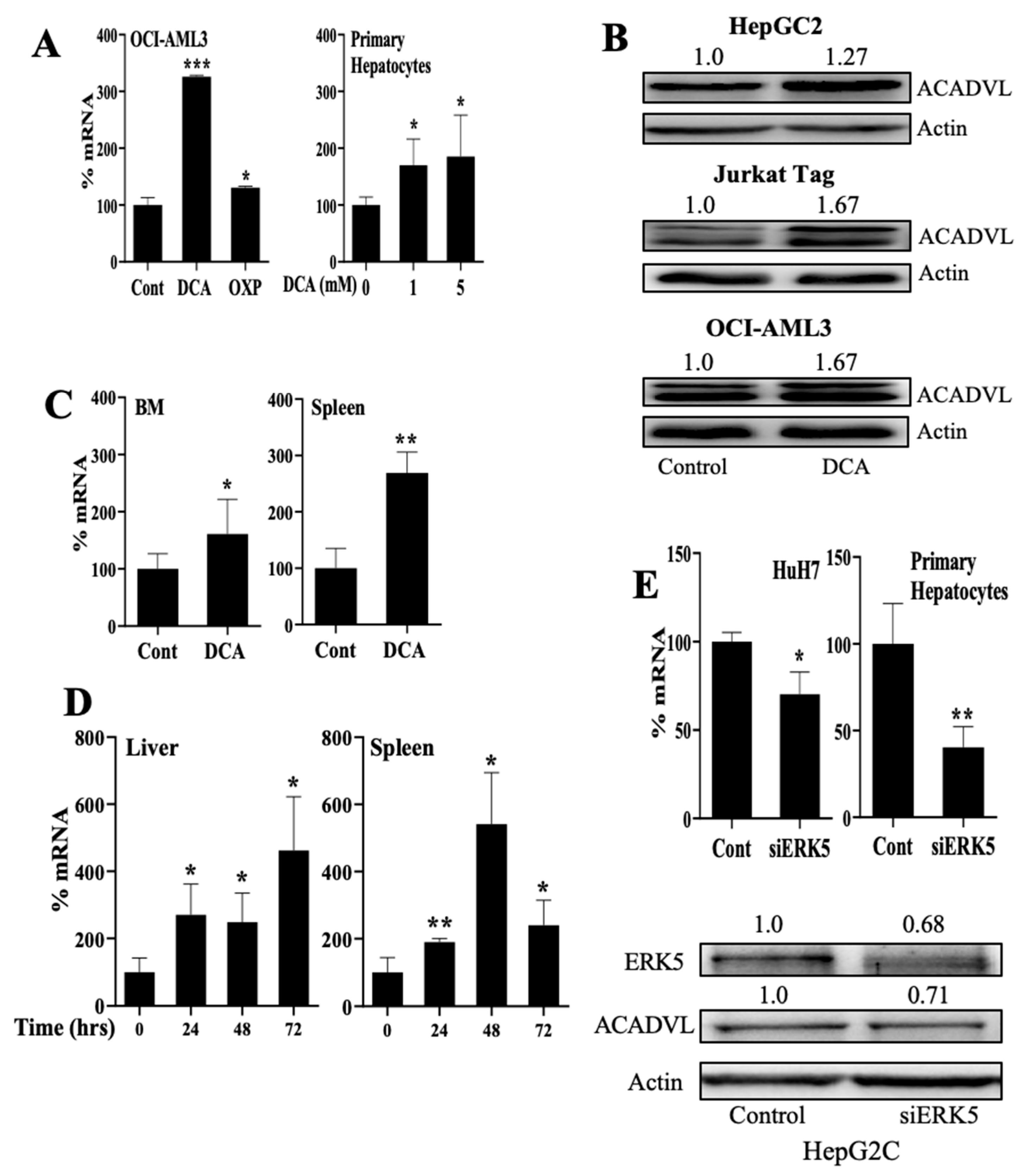
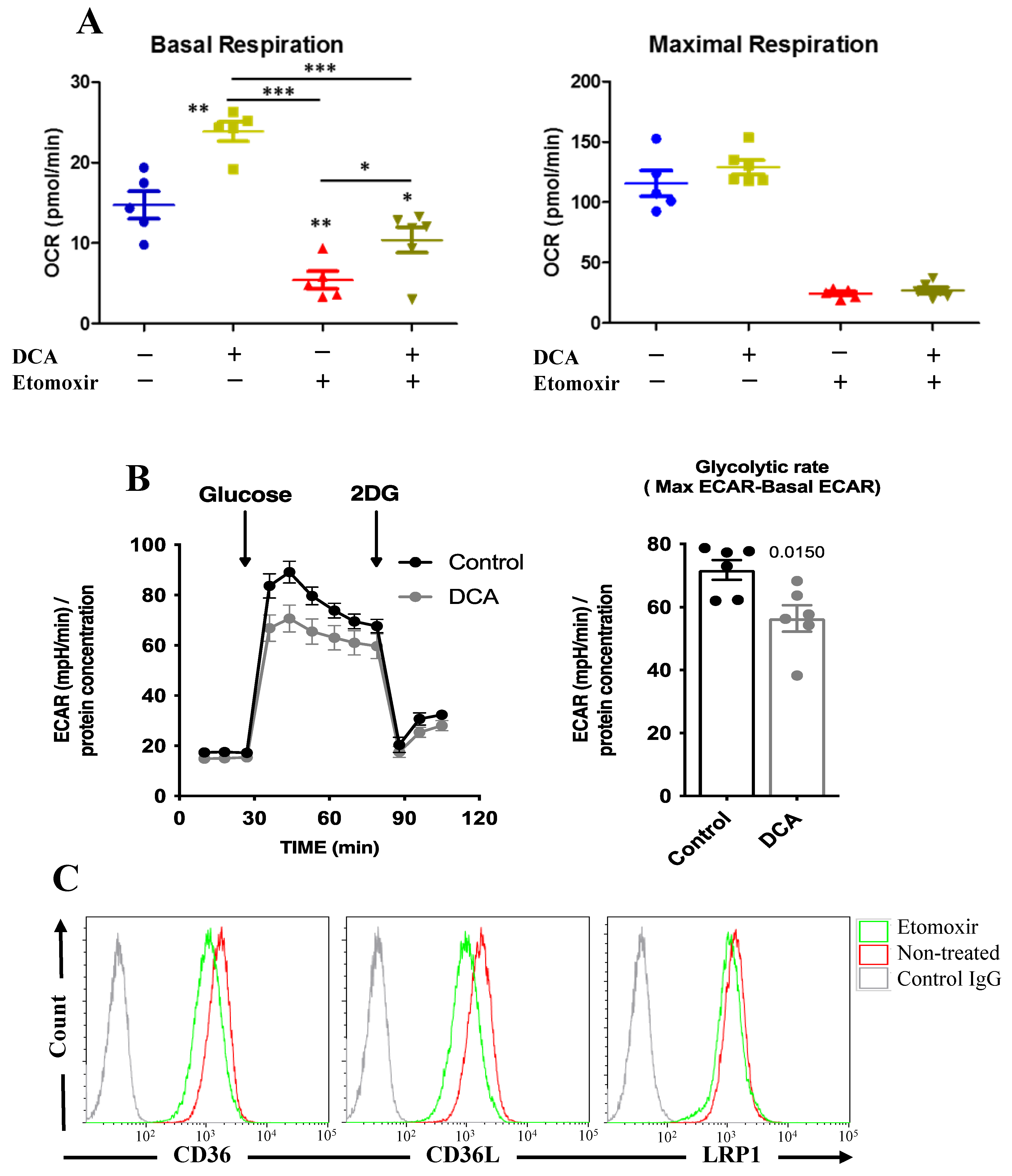
3.4. Changes in Metabolism Regulate FAO through an ERK5-Dependent Pathway
4. Discussion
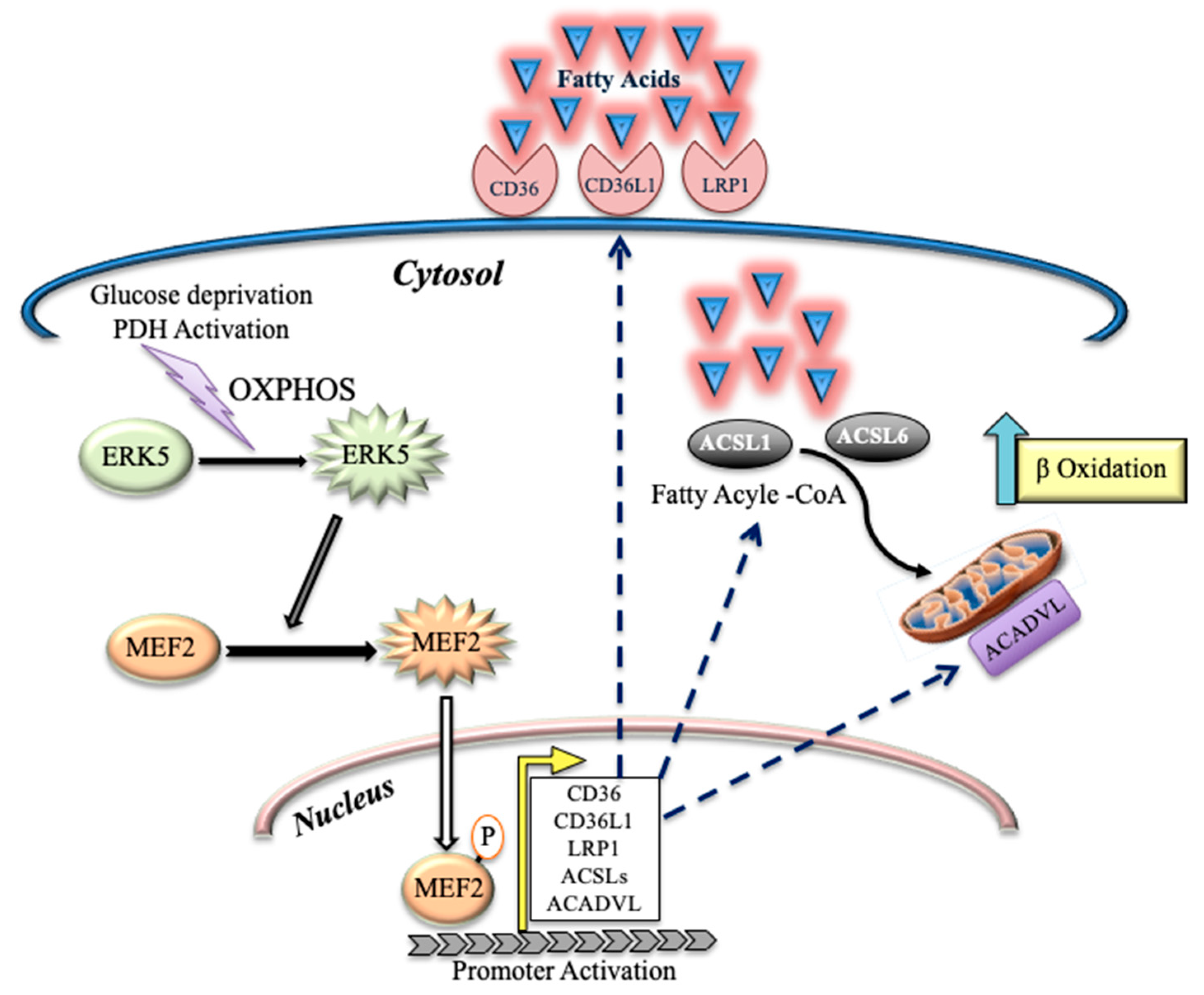
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ho, W.-C.; Zhang, J. Evolutionary Adaptations to New Environments Generally Reverse Plastic Phenotypic Changes. Nat. Commun. 2018, 9, 350. [Google Scholar] [CrossRef] [PubMed]
- Rothman, D.L.; Shulman, R.G. Two Transition States of the Glycogen Shunt and Two Steady States of Gene Expression Support Metabolic Flexibility and the Warburg Effect in Cancer. Neoplasia 2021, 23, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, R.R. Metabolic Interactions between Glucose and Fatty Acids in Humans. Am. J. Clin. Nutr. 1998, 67, 519S–526S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, W.-C.; Sutter, B.M.; Ruess, H.; Barnes, S.D.; Malladi, V.S.; Tu, B.P. Glucose Starvation Induces a Switch in the Histone Acetylome for Activation of Gluconeogenic and Fat Metabolism Genes. Mol. Cell 2022, 82, 60–74. [Google Scholar] [CrossRef]
- Kankotia, S.; Stacpoole, P.W. Dichloroacetate and Cancer: New Home for an Orphan Drug? Biochim. Biophys. Acta 2014, 1846, 617–629. [Google Scholar] [CrossRef]
- Stacpoole, P.W. The Pharmacology of Dichloroacetate. Metabolism 1989, 38, 1124–1144. [Google Scholar] [CrossRef]
- Moore, G.W.; Swift, L.L.; Rabinowitz, D.; Crofford, O.B.; Oates, J.A.; Stacpoole, P.W. Reduction of Serum Cholesterol in Two Patients with Homozygous Familial Hypercholesterolemia by Dichloroacetate. Atherosclerosis 1979, 33, 285–293. [Google Scholar] [CrossRef]
- Khan, A.U.H.; Allende-Vega, N.; Gitenay, D.; Gerbal-Chaloin, S.; Gondeau, C.; Vo, D.N.; Belkahla, S.; Orecchioni, S.; Talarico, G.; Bertolini, F.; et al. The PDK1 Inhibitor Dichloroacetate Controls Cholesterol Homeostasis through the ERK5/MEF2 Pathway. Sci. Rep. 2017, 7, 10654. [Google Scholar] [CrossRef] [Green Version]
- Mainali, R.; Zabalawi, M.; Long, D.; Buechler, N.; Quillen, E.; Key, C.-C.; Zhu, X.; Parks, J.S.; Furdui, C.; Stacpoole, P.W.; et al. Dichloroacetate Reverses Sepsis-Induced Hepatic Metabolic Dysfunction. Elife 2021, 10, e64611. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Moore, G.W.; Kornhauser, D.M. Metabolic Effects of Dichloroacetate in Patients with Diabetes Mellitus and Hyperlipoproteinemia—NEJM. N. Engl. J. Med. 1978, 298, 526–530. [Google Scholar] [CrossRef] [Green Version]
- Stacpoole, P.W.; Wright, E.C.; Baumgartner, T.G.; Bersin, R.M.; Buchalter, S.; Curry, S.H.; Duncan, C.A.; Harman, E.M.; Henderson, G.N.; Jenkinson, S. A Controlled Clinical Trial of Dichloroacetate for Treatment of Lactic Acidosis in Adults. The Dichloroacetate-Lactic Acidosis Study Group. N. Engl. J. Med. 1992, 327, 1564–1569. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, P.; Engelstad, K.; Wei, Y.; Jhung, S.; Sano, M.C.; Shungu, D.C.; Millar, W.S.; Hong, X.; Gooch, C.L.; Mao, X.; et al. Dichloroacetate Causes Toxic Neuropathy in MELAS: A Randomized, Controlled Clinical Trial. Neurology 2006, 66, 324–330. [Google Scholar] [CrossRef]
- Felitsyn, N.; Stacpoole, P.W.; Notterpek, L. Dichloroacetate Causes Reversible Demyelination in Vitro: Potential Mechanism for Its Neuropathic Effect. J. Neurochem. 2007, 100, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U.H.; Allende-Vega, N.; Gitenay, D.; Garaude, J.; Vo, D.N.; Belkhala, S.; Gerbal-Chaloin, S.; Gondeau, C.; Daujat-Chavanieu, M.; Delettre, C.; et al. Mitochondrial Complex I Activity Signals Antioxidant Response through ERK5. Sci. Rep. 2018, 8, 7420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkahla, S.; Haq Khan, A.U.; Gitenay, D.; Alexia, C.; Gondeau, C.; Vo, D.N.; Orecchioni, S.; Talarico, G.; Bertolini, F.; Cartron, G.; et al. Changes in Metabolism Affect Expression of ABC Transporters through ERK5 and Depending on P53 Status. Oncotarget 2018, 9, 1114–1129. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.U.H.; Rathore, M.G.; Allende-Vega, N.; Vo, D.N.; Belkhala, S.; Orecchioni, S.; Talarico, G.; Bertolini, F.; Cartron, G.; Lecellier, C.H.; et al. Human Leukemic Cells Performing Oxidative Phosphorylation (OXPHOS) Generate an Antioxidant Response Independently of Reactive Oxygen Species (ROS) Production. EBioMedcine 2016, 3, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Barsyte-Lovejoy, D.; Galanis, A.; Clancy, A.; Sharrocks, A.D. ERK5 Is Targeted to Myocyte Enhancer Factor 2A (MEF2A) through a MAPK Docking Motif. Biochem. J. 2004, 381, 693–699. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Kravchenko, V.V.; Tapping, R.I.; Han, J.; Ulevitch, R.J.; Lee, J.D. BMK1/ERK5 Regulates Serum-Induced Early Gene Expression through Transcription Factor MEF2C. EMBO J. 1997, 16, 7054–7066. [Google Scholar] [CrossRef] [Green Version]
- Young, A.; Wu, W.; Sun, W.; Benjamin Larman, H.; Wang, N.; Li, Y.S.; Shyy, J.Y.; Chien, S.; Garcia-Cardena, G. Flow Activation of AMP-Activated Protein Kinase in Vascular Endothelium Leads to Kruppel-like Factor 2 Expression. Arter. Thromb. Vasc. Biol. 2009, 29, 1902–1908. [Google Scholar] [CrossRef] [Green Version]
- Monti, M.; Celli, J.; Missale, F.; Cersosimo, F.; Russo, M.; Belloni, E.; Di Matteo, A.; Lonardi, S.; Vermi, W.; Ghigna, C.; et al. Clinical Significance and Regulation of ERK5 Expression and Function in Cancer. Cancers 2022, 14, 348. [Google Scholar] [CrossRef]
- Kasler, H.G.; Victoria, J.; Duramad, O.; Winoto, A. ERK5 Is a Novel Type of Mitogen-Activated Protein Kinase Containing a Transcriptional Activation Domain. Mol. Cell Biol. 2000, 20, 8382–8389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecca, B.; Rovida, E. Impact of ERK5 on the Hallmarks of Cancer. Int. J. Mol. Sci. 2019, 20, 1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Cooley, B.C.; Li, W.; Chen, Y.; Vasquez-Vivar, J.; Scoggins, N.O.; Cameron, S.J.; Morrell, C.N.; Silverstein, R.L. Platelet CD36 Promotes Thrombosis by Activating Redox Sensor ERK5 in Hyperlipidemic Conditions. Blood 2017, 129, 2917–2927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristea, S.; Coles, G.L.; Hornburg, D.; Gershkovitz, M.; Arand, J.; Cao, S.; Sen, T.; Williamson, S.C.; Kim, J.W.; Drainas, A.P.; et al. The MEK5-ERK5 Kinase Axis Controls Lipid Metabolism in Small-Cell Lung Cancer. Cancer Res. 2020, 80, 1293–1303. [Google Scholar] [CrossRef] [Green Version]
- Nigro, P.; Abe, J.; Berk, B.C. Flow Shear Stress and Atherosclerosis: A Matter of Site Specificity. Antioxid. Redox Signal. 2011, 15, 1405–1414. [Google Scholar] [CrossRef]
- Kim, M.; Kim, S.; Lim, J.H.; Lee, C.; Choi, H.C.; Woo, C.H. Laminar Flow Activation of ERK5 Protein in Vascular Endothelium Leads to Atheroprotective Effect via NF-E2-Related Factor 2 (Nrf2) Activation. J. Biol. Chem. 2012, 287, 40722–40731. [Google Scholar] [CrossRef] [Green Version]
- Allende-Vega, N.; Marco Brualla, J.; Falvo, P.; Alexia, C.; Constantinides, M.; de Maudave, A.F.; Coenon, L.; Gitenay, D.; Mitola, G.; Massa, P.; et al. Metformin Sensitizes Leukemic Cells to Cytotoxic Lymphocytes by Increasing Expression of Intercellular Adhesion Molecule-1 (ICAM-1). Sci. Rep. 2022, 12, 1341. [Google Scholar] [CrossRef]
- Pichard, L.; Raulet, E.; Fabre, G.; Ferrini, J.B.; Ourlin, J.C.; Maurel, P. Human Hepatocyte Culture. Methods Mol. Biol. 2006, 320, 283–293. [Google Scholar] [CrossRef]
- Garaude, J.; Cherni, S.; Kaminski, S.; Delepine, E.; Chable-Bessia, C.; Benkirane, M.; Borges, J.; Pandiella, A.; Iniguez, M.A.; Fresno, M.; et al. ERK5 Activates NF-KappaB in Leukemic T Cells and Is Essential for Their Growth In Vivo. J. Immunol. 2006, 177, 7607–7617. [Google Scholar] [CrossRef]
- Charni, S.; de Bettignies, G.; Rathore, M.G.; Aguilo, J.I.; van den Elsen, P.J.; Haouzi, D.; Hipskind, R.A.; Enriquez, J.A.; Sanchez-Beato, M.; Pardo, J.; et al. Oxidative Phosphorylation Induces de Novo Expression of the MHC Class I in Tumor Cells through the ERK5 Pathway. J. Immunol. 2010, 185, 3498–3503. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-Driven Oxidative Phosphorylation Is a Major ATP Source in Transformed Mammalian Cells in Both Normoxia and Hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Allende-Vega, N.; Krzywinska, E.; Orecchioni, S.; Lopez-Royuela, N.; Reggiani, F.; Talarico, G.; Rossi, J.F.; Rossignol, R.; Hicheri, Y.; Cartron, G.; et al. The Presence of Wild Type P53 in Hematological Cancers Improves the Efficacy of Combinational Therapy Targeting Metabolism. Oncotarget 2015, 6, 19228–19245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of Scavenger Receptor SR-BI as a High Density Lipoprotein Receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C. Lipoprotein Receptor Signalling in Atherosclerosis. Cardiovasc. Res. 2020, 116, 1254–1274. [Google Scholar] [CrossRef]
- Nilsson-Ehle, P.; Garfinkel, A.S.; Schotz, M.C. Lipolytic Enzymes and Plasma Lipoprotein Metabolism. Annu. Rev. Biochem. 1980, 49, 667–693. [Google Scholar] [CrossRef]
- Tang, Y.; Zhou, J.; Hooi, S.C.; Jiang, Y.-M.; Lu, G.-D. Fatty Acid Activation in Carcinogenesis and Cancer Development: Essential Roles of Long-Chain Acyl-CoA Synthetases. Oncol. Lett. 2018, 16, 1390–1396. [Google Scholar] [CrossRef] [Green Version]
- Wajner, M.; Amaral, A.U. Mitochondrial Dysfunction in Fatty Acid Oxidation Disorders: Insights from Human and Animal Studies. Biosci. Rep. 2015, 36, e00281. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Taketani, T. Management and Diagnosis of Mitochondrial Fatty Acid Oxidation Disorders: Focus on Very-Long-Chain Acyl-CoA Dehydrogenase Deficiency. J. Hum. Genet. 2019, 64, 73–85. [Google Scholar] [CrossRef]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty Acid Oxidation: An Emerging Facet of Metabolic Transformation in Cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef]
- Matsuhashi, T.; Hishiki, T.; Zhou, H.; Ono, T.; Kaneda, R.; Iso, T.; Yamaguchi, A.; Endo, J.; Katsumata, Y.; Atsushi, A.; et al. Activation of Pyruvate Dehydrogenase by Dichloroacetate Has the Potential to Induce Epigenetic Remodeling in the Heart. J. Mol. Cell. Cardiol. 2015, 82, 116–124. [Google Scholar] [CrossRef]
- Hayashi, M.; Kim, S.W.; Imanaka-Yoshida, K.; Yoshida, T.; Abel, E.D.; Eliceiri, B.; Yang, Y.; Ulevitch, R.J.; Lee, J.D. Targeted Deletion of BMK1/ERK5 in Adult Mice Perturbs Vascular Integrity and Leads to Endothelial Failure. J. Clin. Investig. 2004, 113, 1138–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Ruiz-Velasco, A.; Wang, S.; Khan, S.; Zi, M.; Jungmann, A.; Dolores Camacho-Munoz, M.; Guo, J.; Du, G.; Xie, L.; et al. Metabolic Stress-Induced Cardiomyopathy Is Caused by Mitochondrial Dysfunction Due to Attenuated Erk5 Signaling. Nat Commun. 2017, 8, 494. [Google Scholar] [CrossRef] [PubMed]
- Regan, C.P.; Li, W.; Boucher, D.M.; Spatz, S.; Su, M.S.; Kuida, K. Erk5 Null Mice Display Multiple Extraembryonic Vascular and Embryonic Cardiovascular Defects. Proc. Natl. Acad. Sci. USA 2002, 99, 9248–9253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, S.J.; Sarvis, B.K.; Cado, D.; Winoto, A. ERK5 MAPK Regulates Embryonic Angiogenesis and Acts as a Hypoxia-Sensitive Repressor of Vascular Endothelial Growth Factor Expression. J. Biol. Chem. 2002, 277, 43344–43351. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, A.U.H.; Salehi, H.; Alexia, C.; Valdivielso, J.M.; Bozic, M.; Lopez-Mejia, I.C.; Fajas, L.; Gerbal-Chaloin, S.; Daujat-Chavanieu, M.; Gitenay, D.; et al. Glucose Starvation or Pyruvate Dehydrogenase Activation Induce a Broad, ERK5-Mediated, Metabolic Remodeling Leading to Fatty Acid Oxidation. Cells 2022, 11, 1392. https://doi.org/10.3390/cells11091392
Khan AUH, Salehi H, Alexia C, Valdivielso JM, Bozic M, Lopez-Mejia IC, Fajas L, Gerbal-Chaloin S, Daujat-Chavanieu M, Gitenay D, et al. Glucose Starvation or Pyruvate Dehydrogenase Activation Induce a Broad, ERK5-Mediated, Metabolic Remodeling Leading to Fatty Acid Oxidation. Cells. 2022; 11(9):1392. https://doi.org/10.3390/cells11091392
Chicago/Turabian StyleKhan, Abrar Ul Haq, Hamideh Salehi, Catherine Alexia, Jose M. Valdivielso, Milica Bozic, Isabel C. Lopez-Mejia, Lluis Fajas, Sabine Gerbal-Chaloin, Martine Daujat-Chavanieu, Delphine Gitenay, and et al. 2022. "Glucose Starvation or Pyruvate Dehydrogenase Activation Induce a Broad, ERK5-Mediated, Metabolic Remodeling Leading to Fatty Acid Oxidation" Cells 11, no. 9: 1392. https://doi.org/10.3390/cells11091392