
Properties and Functions of Fibroblasts and Myofibroblasts in Myocardial Infarction


Abstract
:1. Introduction
2. The Fibroblasts in Normal Adult Hearts
3. Homeostatic Functions of Fibroblasts in the Adult Mammalian Heart
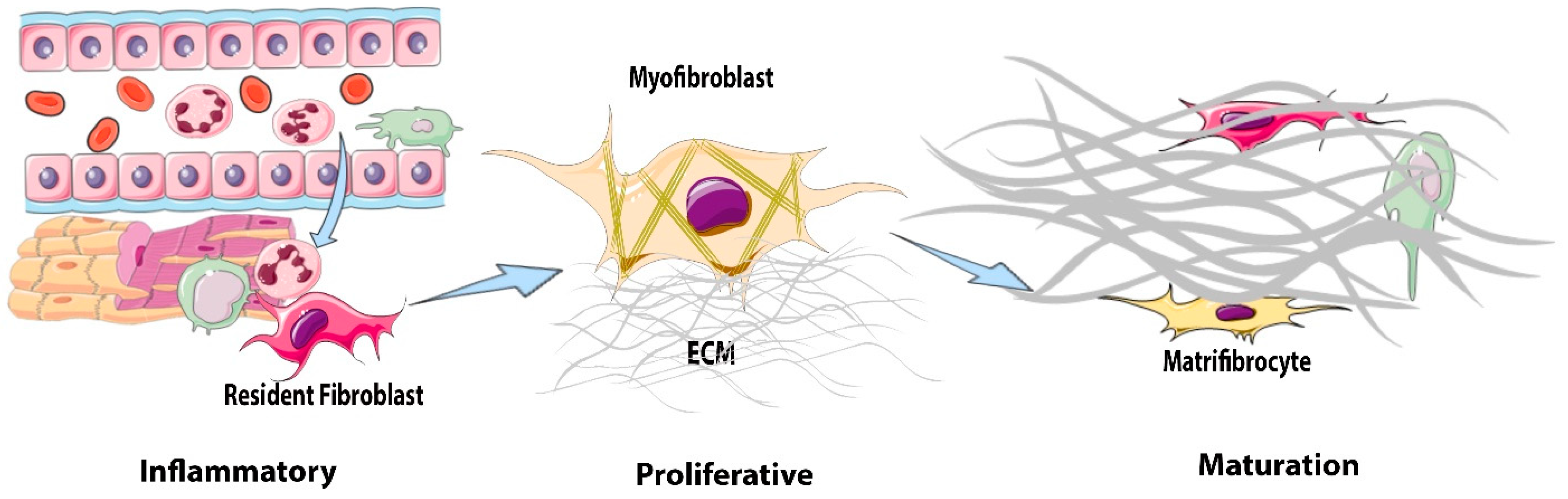
4. The Reparative Response after Myocardial Infarction: From Inflammation to Fibrosis
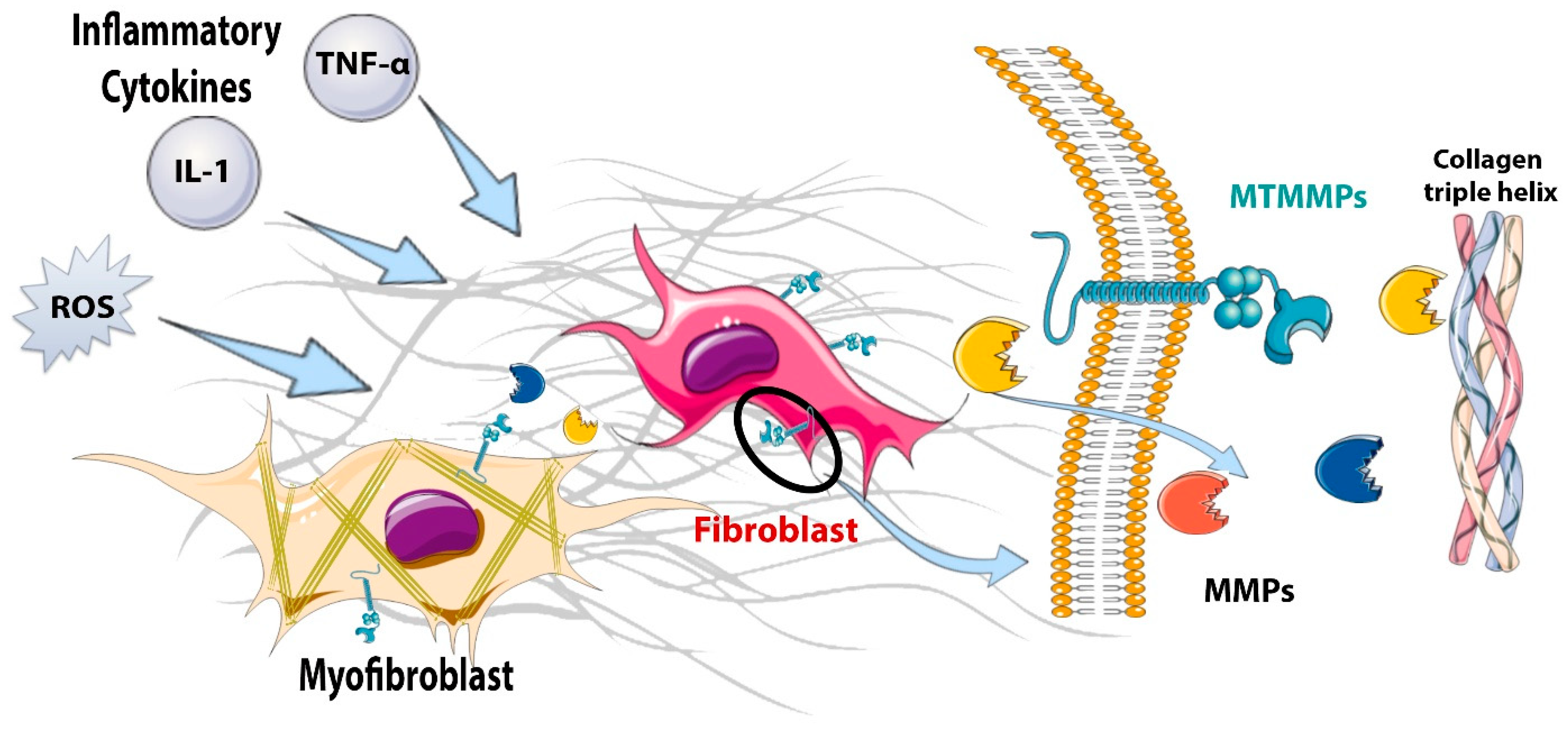
5. Fibroblasts during the Inflammatory Phase: Pro-Inflammatory and Matrix-Degrading Functions
Do Fibroblasts in the Infarct Zone Die?
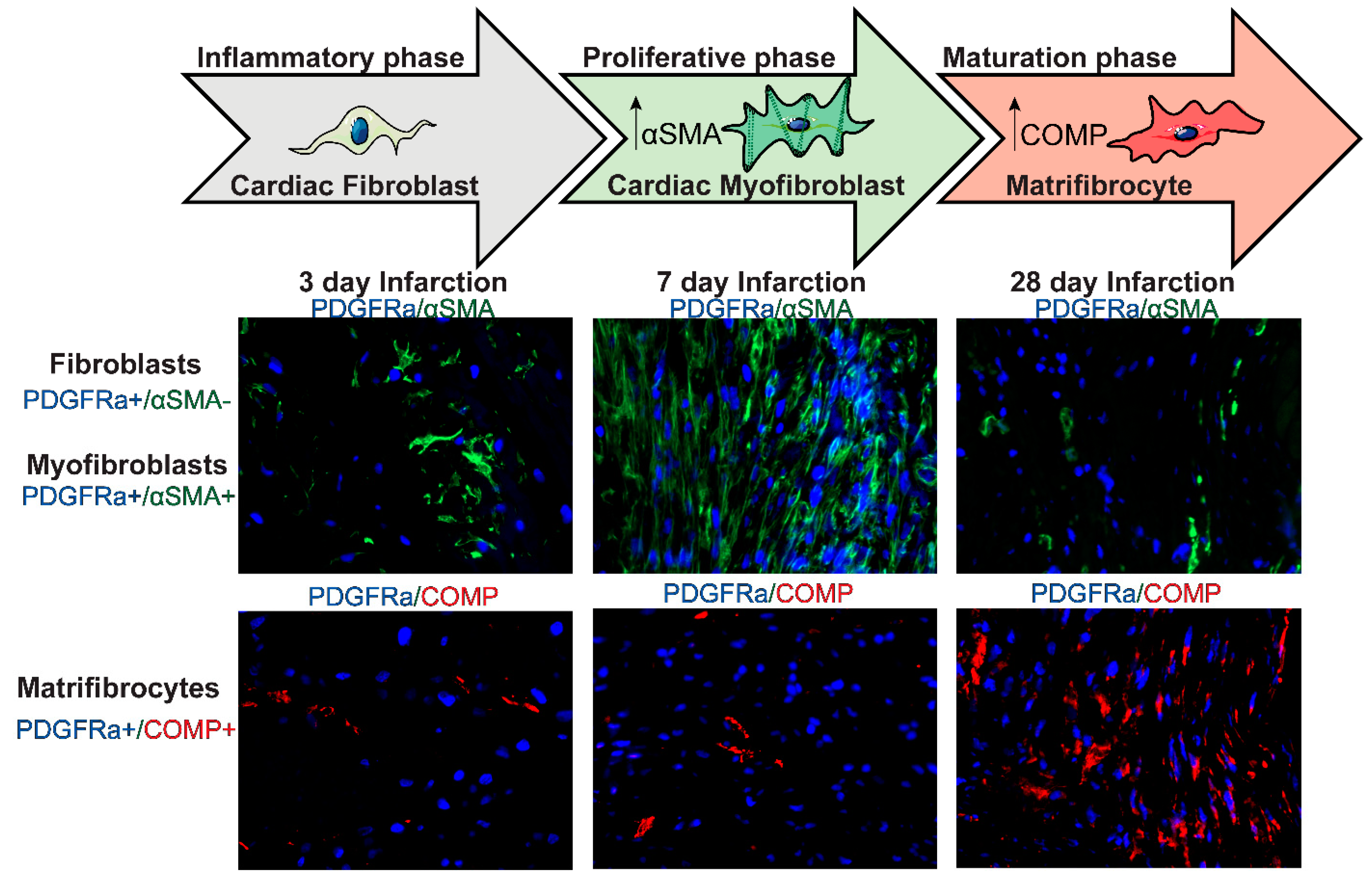
6. Fibroblasts during the Proliferative Phase of Infarct Healing: Myofibroblast Conversion and Acquisition of a Migratory, Proliferative, and Matrix-Synthetic Phenotype
6.1. The Potential Role of Infarct Fibroblasts in Phagocytosis and Suppression of Inflammation
6.2. The Cellular Origins of Fibroblast Expansion in the Infarcted Myocardium
6.3. Myofibroblast Conversion
6.4. Molecular Signals Serving as Activators of Infarct Myofibroblasts
6.4.1. Cytokines
6.4.2. Growth Factors: The Role of TGF-βs, Fibroblast Growth Factors (FGF)s, and PDGFs
6.4.3. Neurohumoral Pathways
6.4.4. Matricellular Proteins in Activation of Infarct Fibroblasts: The ECM as a Signaling Hub
6.4.5. The Intracellular Network of Molecular Cascades Involved in Fibroblast Activation

{kind=link}
{kind=link}
{kind=link}
| Mediator | Fibroblast-Specific Approach | Role in Repair and Remodeling of the Infarcted Heart | Role in Modulation of Fibroblast Phenotype | Proposed Mechanism | Ref. |
|---|---|---|---|---|---|
| IL1-R1 | Tamoxifen-inducible Col1a2 CreERT mice were used for fibroblast- specific deletion of IL1-R1. | Fibroblast-specific IL1-R1 drives adverse cardiac remodeling and promotes ventricular wall thinning and collagen deposition in a model of non-reperfused infarction. | IL1-R1 promotes a pro-fibrotic and matrix-degrading phenotype in fibroblasts. | IL1-α interacts with IL1-R1 to activate p38 and NFκB signaling, leading to increased expression of IL6, MMP3, and MMP9 and secretion of IL-6 and MMP-3. | [85] |
| Smad3 | Myofibroblast-specific deletion used Postn-Cre mice. | Myofibroblast-specific Smad3 protects from cardiac rupture in the non-reperfused infarction model and attenuates chamber dilation. | Smad3 limits fibroblast proliferation and promotes collagen synthesis. It also mediates the formation of organized myofibroblast arrays. | Smad3 regulates fibroblast function via integrin-mediated NOX-2 expression. Moreover, Smad3-dependent activation of the GTPase RhoA dictates fibroblast alignment via the regulation of cell polarity pathways. | [108,110] |
| Smad7 | Postn-Cre mice were used for myofibroblast-specific knockout of Smad7. | Myofibroblast Smad7 protects the infarcted heart from adverse remodeling and from heart failure-related death. Smad7 limits post-infarction fibrosis in the border zone and in the papillary muscles. | Smad7 attenuates myofibroblast activation and the synthesis of structural and matricellular ECM proteins. | Smad7 inhibits the TGFβ response via the inactivation of Smad2/3. Smad7 also binds to ErbB2 and restrains the activation of ErbB1/2 in a TGF-β-independent manner to suppress the expression of fibrogenic genes. | [5] |
| GSK3β | Postn-Cre mice and tamoxifen-inducible Col1a2-Cre mice were used for deletion in myofibroblasts and fibroblasts, respectively. | Myofibroblast-specific GSK3β negatively regulates fibrosis to limit adverse ventricular remodeling in the infarcted heart. | GSK3β functions to suppress myofibroblast activation and pro-fibrotic signaling. | GSK3β inhibits TGF-β-dependent Smad3 transcriptional activation to limit fibrogenic signaling. | [163] |
| P38α | TCF-21 or Postn-MCM mouse models were used to generate fibroblast and myofibroblast-specific p38α KO mice. | P38α-dependent signaling in fibroblasts drives fibrosis to promote adverse post-infarction remodeling. | P38 mediates cardiac fibroblast activation. | P38 transduces mechanical and cytokine signals via serum response factor and calcineurin to promote myofibroblast differentiation. | [140] |
| GRK2 | Myofibroblast-specific GRK2 deletion was achieved using Postn-MCM mice. | GRK2 signaling contributes to pathological cardiac remodeling via promoting fibrosis and infarct expansion, leading to cardiac dysfunction. | GRK2 promotes myofibroblast activation and collagen deposition in vivo. | GRK2 interacts with Gβγ, promoting the downregulation of fibroblast β-adrenergic receptors. This decreases downstream cAMP production, resulting in the activation of pro-fibrotic signals. | [164] |
| Lats1/2 | Tcf21-MCM mice were used for fibroblast-specific Lats1/2 deletion. | Lats1/2 limit pro-fibrotic signaling. | Lats1/2 maintain the resting fibroblast phenotype and prevents activation to myofibroblast. | Lats1/2 act to maintain the quiescent fibroblast cell state via inhibiting the YAP-induced activation of pro-fibrogenic genes. | [31] |
| YAP | Tcf21-MCM and Col1a1 CreERT mice were used for YAP deletion in fibroblasts. | YAP promotes post-infarction fibrosis. | YAP activation in fibroblasts promotes myofibroblast proliferation and differentiation and ECM gene expression. | YAP activation induces MRTF-A expression to facilitate myofibroblast differentiation and profibrotic gene expression. | [162] |
| 5-HT2B | Tcf21-MCM and Postn-MCM were used to delete 5-HT2B in resting and activated fibroblasts, respectively. | 5-HT2B expression directly contributes to excessive scar formation, leading to adverse cardiac remodeling and impaired cardiac function post-MI. | 5-HT2B promotes fibroblast proliferation and migration and the expression of ECM remodeling genes. | 5-HT2B-dependent fibroblast responses are mediated via Dnajb4 expression and Src phosphorylation. | [165] |
| Hsp47 | Postn-MCM mice were used for the generation of myofibroblast-specific Hsp47 KO mice. | Hsp47 expression in myofibroblasts mediates scar formation post-MI. | HSP47 promotes fibroblast proliferation and mediates the expression of collagens without affecting the expression of α-SMA. | HSP47 enhances the expression of Snail and Zeb1 to transcriptionally activate ECM-related genes. It also downregulates the expression of cell cycle inhibitory kinases to facilitate cell proliferation. | [32] |
| Sox9 | Postn-MCM mice were used for myofibroblast-specific KO. | Myofibroblast-specific Sox9 facilitates MI-induced left ventricular dysfunction, inflammation, and tissue scarring. | Sox9 activity promotes fibroblast activation, proliferation, migration, and contractile function. | Sox9 up-regulates ECM-related gene synthesis, inflammation, and proteolysis. | [166] |
| AMPKα1 | Postn-Cre was used for the deletion of AMPKα1 in myofibroblasts. | AMPKα1 activation in myofibroblasts limits adverse post-infarction remodeling post-MI. | Myofibroblast-specific AMPKα1 inhibits fibroblast proliferation in response to injury. | AMPKα1 inhibits fibroblast activation and proliferation via the miR-125b-5p-dependent silencing of connexin-43. | [167] |
| Muscleblind-like1 (MBNL1) | Tcf21-MCM was used for the fibroblast-specific deletion and overexpression of MBNL1. Postn-MCM was used for the deletion of MBNL1 in activated fibroblasts. | Myofibroblast-specific MBNL1 facilitates the acute wound-healing response post-MI and promotes tissue fibrosis. | MBNL1 promotes myofibroblast transition and contractile function in fibroblasts. | The RNA-binding protein MBNL1 binds to and stabilizes mRNA encoding CnAβ and SRF, promoting myofibroblast differentiation and profibrotic gene expression. | [168] |
| Fibronectin | Tcf21-MCM was used for the ablation of fibronectin in fibroblasts. | Fibronectin polymerization facilitates adverse cardiac remodeling and fibrosis post I/R injury. | Polymerized FN promotes fibroblast proliferation and migration and collagen matrix deposition. | Fibronectin activates c-myc signaling, leading to integrin β1 activation and the downstream phosphorylation of FAK. | [169] |
6.5. Angiogenic Effects of Infarct Fibroblasts
7. Fibroblasts during the Maturation Phase of Infarct Healing
Matrifibrocyte Transition and Negative Regulation of the Fibrotic Response
8. Fibroblast Heterogeneity in Infarcted Hearts
9. Chronic Activation of Fibroblasts in the Remodeling of Non-Infarcted Myocardium
10. Fibroblasts and Myofibroblasts as Therapeutic Targets in Myocardial Infarction
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Frangogiannis, N.G. Pathophysiology of Myocardial Infarction. Compr. Physiol. 2015, 5, 1841–1875. [Google Scholar] [CrossRef]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.M.; Burgos Villar, K.N.; Small, E.M. Fibroblast contributions to ischemic cardiac remodeling. Cell Signal. 2021, 77, 109824. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Shinde, A.V.; Frangogiannis, N.G. Validation of diagnostic criteria and histopathological characterization of cardiac rupture in the mouse model of nonreperfused myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H948–H964. [Google Scholar] [CrossRef] [PubMed]
- Humeres, C.; Shinde, A.V.; Hanna, A.; Alex, L.; Hernandez, S.C.; Li, R.; Chen, B.; Conway, S.J.; Frangogiannis, N.G. Smad7 effects on TGF-beta and ErbB2 restrain myofibroblast activation and protect from postinfarction heart failure. J. Clin. Investig. 2022, 132, e146926. [Google Scholar] [CrossRef]
- Ivey, M.J.; Tallquist, M.D. Defining the Cardiac Fibroblast. Circ. J. 2016, 80, 2269–2276. [Google Scholar] [CrossRef] [Green Version]
- Buechler, M.B.; Pradhan, R.N.; Krishnamurty, A.T.; Cox, C.; Calviello, A.K.; Wang, A.W.; Yang, Y.A.; Tam, L.; Caothien, R.; Roose-Girma, M.; et al. Cross-tissue organization of the fibroblast lineage. Nature 2021, 593, 575–579. [Google Scholar] [CrossRef]
- Muhl, L.; Genove, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef]
- Tallquist, M.D. Cardiac Fibroblast Diversity. Annu. Rev. Physiol. 2020, 82, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Zak, R. Development and proliferative capacity of cardiac muscle cells. Circ. Res. 1974, 35 (Suppl. II), 17–26. [Google Scholar]
- Nag, A.C. Study of non-muscle cells of the adult mammalian heart: A fine structural analysis and distribution. Cytobios 1980, 28, 41–61. [Google Scholar] [PubMed]
- Banerjee, I.; Fuseler, J.W.; Price, R.L.; Borg, T.K.; Baudino, T.A. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1883–H1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Camelliti, P.; Green, C.R.; LeGrice, I.; Kohl, P. Fibroblast network in rabbit sinoatrial node: Structural and functional identification of homogeneous and heterogeneous cell coupling. Circ. Res. 2004, 94, 828–835. [Google Scholar] [CrossRef] [Green Version]
- Franke, W.W.; Schmid, E.; Osborn, M.; Weber, K. Intermediate-sized filaments of human endothelial cells. J. Cell Biol. 1979, 81, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Gabbiani, G.; Schmid, E.; Winter, S.; Chaponnier, C.; de Ckhastonay, C.; Vandekerckhove, J.; Weber, K.; Franke, W.W. Vascular smooth muscle cells differ from other smooth muscle cells: Predominance of vimentin filaments and a specific alpha-type actin. Proc. Natl. Acad. Sci. USA 1981, 78, 298–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmouliere, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Dixon, I.M.C.; Landry, N.M.; Rattan, S.G. Periostin Reexpression in Heart Disease Contributes to Cardiac Interstitial Remodeling by Supporting the Cardiac Myofibroblast Phenotype. Adv. Exp. Med. Biol. 2019, 1132, 35–41. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Saxena, A.; Su, Y.; Frangogiannis, N.G. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1363–H1372. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Kostin, S.; Strøm, C.C.; Aplin, M.; Lyngbaek, S.; Theilade, J.; Grigorian, M.; Andersen, C.B.; Lukanidin, E.; Lerche Hansen, J.; et al. S100A4 is upregulated in injured myocardium and promotes growth and survival of cardiac myocytes. Cardiovasc. Res. 2007, 75, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Acciani, T.; Le Cras, T.; Lutzko, C.; Perl, A.K. Dynamic regulation of platelet-derived growth factor receptor alpha expression in alveolar fibroblasts during realveolarization. Am. J. Respir. Cell Mol. Biol. 2012, 47, 517–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alex, L.; Tuleta, I.; Harikrishnan, V.; Frangogiannis, N.G. Validation of Specific and Reliable Genetic Tools to Identify, Label, and Target Cardiac Pericytes in Mice. J. Am. Heart Assoc. 2022, 11, e023171. [Google Scholar] [CrossRef] [PubMed]
- Ieda, M.; Tsuchihashi, T.; Ivey, K.N.; Ross, R.S.; Hong, T.T.; Shaw, R.M.; Srivastava, D. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev. Cell 2009, 16, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivey, M.J.; Kuwabara, J.T.; Riggsbee, K.L.; Tallquist, M.D. Platelet-derived growth factor receptor-alpha is essential for cardiac fibroblast survival. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H330–H344. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chen, B.; Humeres, C.; Alex, L.; Hanna, A.; Frangogiannis, N.G. The role of Smad2 and Smad3 in regulating homeostatic functions of fibroblasts in vitro and in adult mice. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118703. [Google Scholar] [CrossRef] [PubMed]
- Nicin, L.; Wagner, J.U.G.; Luxan, G.; Dimmeler, S. Fibroblast-mediated intercellular crosstalk in the healthy and diseased heart. FEBS Lett. 2021, 10, e019338. [Google Scholar] [CrossRef]
- Weber, K.T. Cardiac interstitium in health and disease: The fibrillar collagen network. J. Am. Coll. Cardiol. 1989, 13, 1637–1652. [Google Scholar] [CrossRef] [Green Version]
- Kohl, P.; Camelliti, P.; Burton, F.L.; Smith, G.L. Electrical coupling of fibroblasts and myocytes: Relevance for cardiac propagation. J. Electrocardiol. 2005, 38, 45–50. [Google Scholar] [CrossRef]
- Gaudesius, G.; Miragoli, M.; Thomas, S.P.; Rohr, S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ. Res. 2003, 93, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Hill, M.C.; Li, L.; Deshmukh, V.; Martin, T.J.; Wang, J.; Martin, J.F. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 2019, 33, 1491–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, H.; Kanisicak, O.; Vagnozzi, R.J.; Johansen, A.K.; Maliken, B.D.; Prasad, V.; Boyer, J.G.; Brody, M.J.; Schips, T.; Kilian, K.K.; et al. Cell-specific ablation of Hsp47 defines the collagen-producing cells in the injured heart. JCI Insight 2019, 4, e128722. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, I.M.; Robeson, K.Z.; Regnier, M.; Davis, J. Controlling cardiac fibrosis through fibroblast state space modulation. Cell. Signal. 2021, 79, 109888. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell. Cardiol. 2014, 70C, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Saxena, A.; Chen, W.; Su, Y.; Rai, V.; Uche, O.U.; Li, N.; Frangogiannis, N.G. IL-1 Induces Proinflammatory Leukocyte Infiltration and Regulates Fibroblast Phenotype in the Infarcted Myocardium. J. Immunol. 2013, 191, 4838–4848. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Michael, L.H.; Entman, M.L. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb). Cardiovasc. Res. 2000, 48, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Cleutjens, J.P.; Verluyten, M.J.; Smiths, J.F.; Daemen, M.J. Collagen remodeling after myocardial infarction in the rat heart. Am. J. Pathol. 1995, 147, 325–338. [Google Scholar]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.A.; Das, A.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Interleukin-1alpha stimulates proinflammatory cytokine expression in human cardiac myofibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1117–H1127. [Google Scholar] [CrossRef]
- Mitchell, M.D.; Laird, R.E.; Brown, R.D.; Long, C.S. IL-1beta stimulates rat cardiac fibroblast migration via MAP kinase pathways. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1139–H1147. [Google Scholar] [CrossRef]
- Richter, K.; Kietzmann, T. Reactive oxygen species and fibrosis: Further evidence of a significant liaison. Cell Tissue Res. 2016, 365, 591–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boza, P.; Ayala, P.; Vivar, R.; Humeres, C.; Caceres, F.T.; Munoz, C.; Garcia, L.; Hermoso, M.A.; Diaz-Araya, G. Expression and function of toll-like receptor 4 and inflammasomes in cardiac fibroblasts and myofibroblasts: IL-1beta synthesis, secretion, and degradation. Mol. Immunol. 2016, 74, 96–105. [Google Scholar] [CrossRef]
- Lugrin, J.; Parapanov, R.; Rosenblatt-Velin, N.; Rignault-Clerc, S.; Feihl, F.; Waeber, B.; Muller, O.; Vergely, C.; Zeller, M.; Tardivel, A.; et al. Cutting Edge: IL-1alpha Is a Crucial Danger Signal Triggering Acute Myocardial Inflammation during Myocardial Infarction. J. Immunol. 2015, 194, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Lindsey, M.L.; Michael, L.H.; Youker, K.A.; Bressler, R.B.; Mendoza, L.H.; Spengler, R.N.; Smith, C.W.; Entman, M.L. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 1998, 98, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.G.; Ballantyne, C.M.; Michael, L.H.; Kukielka, G.L.; Youker, K.A.; Lindsey, M.L.; Hawkins, H.K.; Birdsall, H.H.; MacKay, C.R.; LaRosa, G.J.; et al. Induction of monocyte chemoattractant protein-1 in the small veins of the ischemic and reperfused canine myocardium. Circulation 1997, 95, 693–700. [Google Scholar] [CrossRef]
- Gwechenberger, M.; Mendoza, L.H.; Youker, K.A.; Frangogiannis, N.G.; Smith, C.W.; Michael, L.H.; Entman, M.L. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation 1999, 99, 546–551. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Sandanger, O.; Ranheim, T.; Vinge, L.E.; Bliksoen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef] [Green Version]
- Anzai, A.; Choi, J.L.; He, S.; Fenn, A.M.; Nairz, M.; Rattik, S.; McAlpine, C.S.; Mindur, J.E.; Chan, C.T.; Iwamoto, Y.; et al. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J. Exp. Med. 2017, 214, 3293–3310. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Guo, M.; Zeng, W.; Wang, Y.; Yang, L.; Pang, X.; Li, H.; Suo, Y.; Jiang, X.; Yu, C. Matrix metalloproteinase 9 secreted by hypoxia cardiac fibroblasts triggers cardiac stem cell migration in vitro. Stem. Cells Int. 2015, 2015, 836390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, A.E.; Kandalam, V.; Chakrabarti, S.; Wang, X.; Penninger, J.M.; Davidge, S.T.; Oudit, G.Y.; Kassiri, Z. Tumor necrosis factor induces matrix metalloproteinases in cardiomyocytes and cardiofibroblasts differentially via superoxide production in a PI3Kgamma-dependent manner. Am. J. Physiol. Cell Physiol. 2010, 298, C679–C692. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Shinde, A.V.; Li, R.; Alex, L.; Humeres, C.; Balasubramanian, P.; Frangogiannis, N.G. Collagen denaturation in the infarcted myocardium involves temporally distinct effects of MT1-MMP-dependent proteolysis and mechanical tension. Matrix Biol. 2021, 99, 18–42. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of cell death in heart disease. Arter. Thromb. Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, R.B.; Murry, C.E.; Steenbergen, C., Jr.; Reimer, K.A. Development of cell injury in sustained acute ischemia. Circulation 1990, 82, II2–II12. [Google Scholar] [PubMed]
- Reimer, K.A.; Lowe, J.E.; Rasmussen, M.M.; Jennings, R.B. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation 1977, 56, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Mayorga, M.; Bahi, N.; Ballester, M.; Comella, J.X.; Sanchis, D. Bcl-2 is a key factor for cardiac fibroblast resistance to programmed cell death. J. Biol. Chem. 2004, 279, 34882–34889. [Google Scholar] [CrossRef] [Green Version]
- Abrial, M.; Da Silva, C.C.; Pillot, B.; Augeul, L.; Ivanes, F.; Teixeira, G.; Cartier, R.; Angoulvant, D.; Ovize, M.; Ferrera, R. Cardiac fibroblasts protect cardiomyocytes against lethal ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2014, 68, 56–65. [Google Scholar] [CrossRef]
- Wan, E.; Yeap, X.Y.; Dehn, S.; Terry, R.; Novak, M.; Zhang, S.; Iwata, S.; Han, X.; Homma, S.; Drosatos, K.; et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 2013, 113, 1004–1012. [Google Scholar] [CrossRef]
- Chen, B.; Huang, S.; Su, Y.; Wu, Y.J.; Hanna, A.; Brickshawana, A.; Graff, J.; Frangogiannis, N.G. Macrophage Smad3 Protects the Infarcted Heart, Stimulating Phagocytosis and Regulating Inflammation. Circ. Res. 2019, 125, 55–70. [Google Scholar] [CrossRef]
- Hofmann, U.; Frantz, S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ. Res. 2015, 116, 354–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobaczewski, M.; Xia, Y.; Bujak, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am. J. Pathol. 2010, 176, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Watari, K.; Tajima, M.; Nakaya, T.; Matsuda, S.; Ohara, H.; Nishihara, H.; Yamaguchi, H.; Hashimoto, A.; Nishida, M.; et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J. Clin. Investig. 2017, 127, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Willems, I.E.; Havenith, M.G.; De Mey, J.G.; Daemen, M.J. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am. J. Pathol. 1994, 145, 868–875. [Google Scholar] [PubMed]
- Aisagbonhi, O.; Rai, M.; Ryzhov, S.; Atria, N.; Feoktistov, I.; Hatzopoulos, A.K. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Model Mech. 2011, 4, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, N.; Bosca, L.; Zandbergen, H.R.; Kovacic, J.C.; Narula, N.; Gonzalez-Ramos, S.; Fernandez-Velasco, M.; Agrawal, S.; Paz-Garcia, M.; Gupta, S.; et al. Transition of Macrophages to Fibroblast-Like Cells in Healing Myocardial Infarction. J. Am. Coll. Cardiol. 2019, 74, 3124–3135. [Google Scholar] [CrossRef]
- Mollmann, H.; Nef, H.M.; Kostin, S.; von Kalle, C.; Pilz, I.; Weber, M.; Schaper, J.; Hamm, C.W.; Elsasser, A. Bone marrow-derived cells contribute to infarct remodelling. Cardiovasc. Res. 2006, 71, 661–671. [Google Scholar] [CrossRef]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; Lin, S.-C.J.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef] [Green Version]
- Moore-Morris, T.; Cattaneo, P.; Guimaraes-Camboa, N.; Bogomolovas, J.; Cedenilla, M.; Banerjee, I.; Ricote, M.; Kisseleva, T.; Zhang, L.; Gu, Y.; et al. Infarct Fibroblasts Do Not Derive From Bone Marrow Lineages. Circ. Res. 2018, 122, 583–590. [Google Scholar] [CrossRef]
- Blankesteijn, W.M.; Essers-Janssen, Y.P.; Verluyten, M.J.; Daemen, M.J.; Smits, J.F. A homologue of Drosophila tissue polarity gene frizzled is expressed in migrating myofibroblasts in the infarcted rat heart. Nat. Med. 1997, 3, 541–544. [Google Scholar] [CrossRef]
- Shinde, A.V.; Humeres, C.; Frangogiannis, N.G. The role of alpha-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta 2017, 1863, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Detillieux, K.A.; Sheikh, F.; Kardami, E.; Cattini, P.A. Biological activities of fibroblast growth factor-2 in the adult myocardium. Cardiovasc. Res. 2003, 57, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Stawowy, P.; Margeta, C.; Kallisch, H.; Seidah, N.G.; Chrétien, M.; Fleck, E.; Graf, K. Regulation of matrix metalloproteinase MT1-MMP/MMP-2 in cardiac fibroblasts by TGF-beta1 involves furin-convertase. Cardiovasc. Res. 2004, 63, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Freed, D.H.; Chilton, L.; Li, Y.; Dangerfield, A.L.; Raizman, J.E.; Rattan, S.G.; Visen, N.; Hryshko, L.V.; Dixon, I.M. Role of myosin light chain kinase in cardiotrophin-1-induced cardiac myofibroblast cell migration. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H514–H522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manso, A.M.; Kang, S.M.; Ross, R.S. Integrins, focal adhesions, and cardiac fibroblasts. J. Investig. Med. 2009, 57, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Murphy-Ullrich, J.E. The de-adhesive activity of matricellular proteins: Is intermediate cell adhesion an adaptive state? J. Clin. Investig. 2001, 107, 785–790. [Google Scholar] [CrossRef] [Green Version]
- Ghersi, G.; Dong, H.; Goldstein, L.A.; Yeh, Y.; Hakkinen, L.; Larjava, H.S.; Chen, W.T. Regulation of fibroblast migration on collagenous matrix by a cell surface peptidase complex. J. Biol. Chem. 2002, 277, 29231–29241. [Google Scholar] [CrossRef] [Green Version]
- Shinde, A.V.; Frangogiannis, N.G. Mechanisms of Fibroblast Activation in the Remodeling Myocardium. Curr. Pathobiol. Rep. 2017, 5, 145–152. [Google Scholar] [CrossRef]
- Gibb, A.A.; Lazaropoulos, M.P.; Elrod, J.W. Myofibroblasts and Fibrosis: Mitochondrial and Metabolic Control of Cellular Differentiation. Circ. Res. 2020, 127, 427–447. [Google Scholar] [CrossRef]
- Ma, F.; Li, Y.; Jia, L.; Han, Y.; Cheng, J.; Li, H.; Qi, Y.; Du, J. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS ONE 2012, 7, e35144. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Kim, H.; Liu, X.; Sugiura, H.; Kohyama, T.; Fang, Q.; Wen, F.Q.; Abe, S.; Wang, X.; Atkinson, J.J.; et al. Matrix metalloproteinase-9 activates TGF-beta and stimulates fibroblast contraction of collagen gels. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L1006–L1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurantz, D.; Cowling, R.T.; Villarreal, F.J.; Greenberg, B.H. Tumor necrosis factor-alpha upregulates angiotensin II type 1 receptors on cardiac fibroblasts. Circ. Res. 1999, 85, 272–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maqbool, A.; Hemmings, K.E.; O’Regan, D.J.; Ball, S.G.; Porter, K.E.; Turner, N.A. Interleukin-1 has opposing effects on connective tissue growth factor and tenascin-C expression in human cardiac fibroblasts. Matrix Biol. 2013, 32, 208–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamaoki, M.; Imanaka-Yoshida, K.; Yokoyama, K.; Nishioka, T.; Inada, H.; Hiroe, M.; Sakakura, T.; Yoshida, T. Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am. J. Pathol. 2005, 167, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Bageghni, S.A.; Hemmings, K.E.; Yuldasheva, N.Y.; Maqbool, A.; Gamboa-Esteves, F.O.; Humphreys, N.E.; Jackson, M.S.; Denton, C.P.; Francis, S.; Porter, K.E.; et al. Fibroblast-specific deletion of interleukin-1 receptor-1 reduces adverse cardiac remodeling following myocardial infarction. JCI Insight 2019, 5, e125074. [Google Scholar] [CrossRef] [Green Version]
- Melendez, G.C.; McLarty, J.L.; Levick, S.P.; Du, Y.; Janicki, J.S.; Brower, G.L. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 2010, 56, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Muller, J.; Gorressen, S.; Grandoch, M.; Feldmann, K.; Kretschmer, I.; Lehr, S.; Ding, Z.; Schmitt, J.P.; Schrader, J.; Garbers, C.; et al. Interleukin-6-dependent phenotypic modulation of cardiac fibroblasts after acute myocardial infarction. Basic Res. Cardiol. 2014, 109, 440. [Google Scholar] [CrossRef]
- Wang, J.H.; Zhao, L.; Pan, X.; Chen, N.N.; Chen, J.; Gong, Q.L.; Su, F.; Yan, J.; Zhang, Y.; Zhang, S.H. Hypoxia-stimulated cardiac fibroblast production of IL-6 promotes myocardial fibrosis via the TGF-β1 signaling pathway. Lab. Investig. 2016, 96, 839–852. [Google Scholar] [CrossRef]
- Chou, C.H.; Hung, C.S.; Liao, C.W.; Wei, L.H.; Chen, C.W.; Shun, C.T.; Wen, W.F.; Wan, C.H.; Wu, X.M.; Chang, Y.Y.; et al. IL-6 trans-signalling contributes to aldosterone-induced cardiac fibrosis. Cardiovasc. Res. 2018, 114, 690–702. [Google Scholar] [CrossRef]
- Fredj, S.; Bescond, J.; Louault, C.; Potreau, D. Interactions between cardiac cells enhance cardiomyocyte hypertrophy and increase fibroblast proliferation. J. Cell. Physiol. 2005, 202, 891–899. [Google Scholar] [CrossRef]
- Banerjee, I.; Fuseler, J.W.; Intwala, A.R.; Baudino, T.A. IL-6 loss causes ventricular dysfunction, fibrosis, reduced capillary density, and dramatically alters the cell populations of the developing and adult heart. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1694–H1704. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.D.; Jones, G.M.; Laird, R.E.; Hudson, P.; Long, C.S. Cytokines regulate matrix metalloproteinases and migration in cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2007, 362, 200–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siwik, D.A.; Chang, D.L.; Colucci, W.S. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ. Res. 2000, 86, 1259–1265. [Google Scholar] [CrossRef] [Green Version]
- Bujak, M.; Dobaczewski, M.; Chatila, K.; Mendoza, L.H.; Li, N.; Reddy, A.; Frangogiannis, N.G. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am. J. Pathol. 2008, 173, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurantz, D.; Cowling, R.T.; Varki, N.; Frikovsky, E.; Moore, C.D.; Greenberg, B.H. IL-1beta and TNF-alpha upregulate angiotensin II type 1 (AT1) receptors on cardiac fibroblasts and are associated with increased AT1 density in the post-MI heart. J. Mol. Cell. Cardiol. 2005, 38, 505–515. [Google Scholar] [CrossRef]
- Turner, N.A.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Modulatory effect of interleukin-1α on expression of structural matrix proteins, MMPs and TIMPs in human cardiac myofibroblasts: Role of p38 MAP kinase. Matrix Biol. 2010, 29, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Das, A.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Human cardiac fibroblasts express ICAM-1, E-selectin and CXC chemokines in response to proinflammatory cytokine stimulation. Int. J. Biochem. Cell Biol. 2011, 43, 1450–1458. [Google Scholar] [CrossRef]
- Bräuninger, H.; Thottakara, T.; Schön, J.; Voss, S.; Dhople, V.; Warnke, S.; Scherschel, K.; Schrage, B.; Kirchhof, P.; Blankenberg, S.; et al. Cytokine-Mediated Alterations of Human Cardiac Fibroblast’s Secretome. Int. J. Mol. Sci. 2021, 22, 12262. [Google Scholar] [CrossRef]
- Shi, S.; Yi, J.L. S100A8/A9 promotes MMP-9 expression in the fibroblasts from cardiac rupture after myocardial infarction by inducing macrophages secreting TNFα. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3925–3935. [Google Scholar] [CrossRef]
- He, Y.H.; Zhang, H.N.; Zhang, G.P.; Hou, N.; Xiao, Q.; Huang, Y.; Wu, J.H.; Luo, M.S.; Zhang, G.S.; Yi, Q.; et al. A physiological concentration of glucocorticoid inhibits the pro-inflammatory cytokine-induced proliferation of adult rat cardiac fibroblasts: Roles of extracellular signal-regulated kinase 1/2 and nuclear factor-κB. Clin. Exp. Pharmacol. Physiol. 2011, 38, 739–746. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Transforming growth factor-beta in myocardial disease. Nat. Rev. Cardiol. 2022. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Dewald, O.; Ren, G.; Duerr, G.D.; Zoerlein, M.; Klemm, C.; Gersch, C.; Tincey, S.; Michael, L.H.; Entman, M.L.; Frangogiannis, N.G. Of mice and dogs: Species-specific differences in the inflammatory response following myocardial infarction. Am. J. Pathol. 2004, 164, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G.; Ren, G.; Dewald, O.; Zymek, P.; Haudek, S.; Koerting, A.; Winkelmann, K.; Michael, L.H.; Lawler, J.; Entman, M.L. The critical role of endogenous Thrombospondin (TSP)-1 in preventing expansion of healing myocardial infarcts. Circulation 2005, 111, 2935–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Sarrazy, V.; Koehler, A.; Chow, M.L.; Zimina, E.; Li, C.X.; Kato, H.; Caldarone, C.A.; Hinz, B. Integrins alphavbeta5 and alphavbeta3 promote latent TGF-beta1 activation by human cardiac fibroblast contraction. Cardiovasc. Res. 2014, 102, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. Transforming Growth Factor (TGF)-beta in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Bujak, M.; Li, N.; Gonzalez-Quesada, C.; Mendoza, L.H.; Wang, X.F.; Frangogiannis, N.G. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ. Res. 2010, 107, 418–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, P.; Shinde, A.V.; Su, Y.; Russo, I.; Chen, B.; Saxena, A.; Conway, S.J.; Graff, J.M.; Frangogiannis, N.G. Opposing Actions of Fibroblast and Cardiomyocyte Smad3 Signaling in the Infarcted Myocardium. Circulation 2018, 137, 707–724. [Google Scholar] [CrossRef]
- Bugg, D.; Bretherton, R.; Kim, P.; Olszewski, E.; Nagle, A.; Schumacher, A.E.; Chu, N.; Gunaje, J.; DeForest, C.A.; Stevens, K.; et al. Infarct Collagen Topography Regulates Fibroblast Fate via p38-Yes-Associated Protein Transcriptional Enhanced Associate Domain Signals. Circ. Res. 2020, 127, 1306–1322. [Google Scholar] [CrossRef]
- Huang, S.; Chen, B.; Su, Y.; Alex, L.; Humeres, C.; Shinde, A.V.; Conway, S.J.; Frangogiannis, N.G. Distinct roles of myofibroblast-specific Smad2 and Smad3 signaling in repair and remodeling of the infarcted heart. J. Mol. Cell. Cardiol. 2019, 132, 84–97. [Google Scholar] [CrossRef]
- Hanna, A.; Frangogiannis, N.G. The Role of the TGF-beta Superfamily in Myocardial Infarction. Front. Cardiovasc. Med. 2019, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- House, S.L.; Branch, K.; Newman, G.; Doetschman, T.; Schultz, J.l.J. Cardioprotection induced by cardiac-specific overexpression of fibroblast growth factor-2 is mediated by the MAPK cascade. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2167–H2175. [Google Scholar] [CrossRef] [PubMed]
- Virag, J.A.; Rolle, M.L.; Reece, J.; Hardouin, S.; Feigl, E.O.; Murry, C.E. Fibroblast growth factor-2 regulates myocardial infarct repair: Effects on cell proliferation, scar contraction, and ventricular function. Am. J. Pathol. 2007, 171, 1431–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zymek, P.; Bujak, M.; Chatila, K.; Cieslak, A.; Thakker, G.; Entman, M.L.; Frangogiannis, N.G. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J. Am. Coll. Cardiol. 2006, 48, 2315–2323. [Google Scholar] [CrossRef] [Green Version]
- Simm, A.; Nestler, M.; Hoppe, V. Mitogenic effect of PDGF-AA on cardiac fibroblasts. Basic Res. Cardiol. 1998, 93 (Suppl. 3), 40–43. [Google Scholar] [CrossRef] [PubMed]
- Saraswati, S.; Lietman, C.D.; Li, B.; Mathew, S.; Zent, R.; Young, P.P. Small proline-rich repeat 3 is a novel coordinator of PDGFRbeta and integrin beta1 crosstalk to augment proliferation and matrix synthesis by cardiac fibroblasts. FASEB J. 2020, 34, 7885–7904. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.T.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2012, 10, 15–26. [Google Scholar] [CrossRef]
- Zeigler, A.C.; Nelson, A.R.; Chandrabhatla, A.S.; Brazhkina, O.; Holmes, J.W.; Saucerman, J.J. Computational model predicts paracrine and intracellular drivers of fibroblast phenotype after myocardial infarction. Matrix Biol. 2020, 91–92, 136–151. [Google Scholar] [CrossRef]
- Kagami, S.; Border, W.A.; Miller, D.E.; Noble, N.A. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-beta expression in rat glomerular mesangial cells. J. Clin. Investig. 1994, 93, 2431–2437. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.E.; Katwa, L.C. Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J. Mol. Cell. Cardiol. 1997, 29, 1947–1958. [Google Scholar] [CrossRef]
- Schorb, W.; Booz, G.W.; Dostal, D.E.; Conrad, K.M.; Chang, K.C.; Baker, K.M. Angiotensin II is mitogenic in neonatal rat cardiac fibroblasts. Circ. Res. 1993, 72, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Sadoshima, J.; Izumo, S. Molecular characterization of angiotensin II--induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ. Res. 1993, 73, 413–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabos, M.; Roth, M.; Hahn, A.W.; Erne, P. Characterization of angiotensin II receptors in cultured adult rat cardiac fibroblasts. Coupling to signaling systems and gene expression. J. Clin. Investig. 1994, 93, 2372–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibault, G.; Lacombe, M.J.; Schnapp, L.M.; Lacasse, A.; Bouzeghrane, F.; Lapalme, G. Upregulation of alpha(8)beta(1)-integrin in cardiac fibroblast by angiotensin II and transforming growth factor-beta1. Am. J. Physiol. Cell Physiol. 2001, 281, C1457–C1467. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, P.; Papparella, I.; Petrov, V.; Semplicini, A.; Fagard, R. Angiotensin II-stimulated collagen production in cardiac fibroblasts is mediated by reactive oxygen species. J. Hypertens. 2006, 24, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, N.; Matsubara, H.; Nozawa, Y.; Mori, Y.; Murasawa, S.; Kijima, K.; Maruyama, K.; Masaki, H.; Tsutumi, Y.; Shibazaki, Y.; et al. Angiotensin type 2 receptors are reexpressed by cardiac fibroblasts from failing myopathic hamster hearts and inhibit cell growth and fibrillar collagen metabolism. Circulation 1997, 96, 3954–3962. [Google Scholar] [CrossRef]
- Kurisu, S.; Ozono, R.; Oshima, T.; Kambe, M.; Ishida, T.; Sugino, H.; Matsuura, H.; Chayama, K.; Teranishi, Y.; Iba, O.; et al. Cardiac angiotensin II type 2 receptor activates the kinin/NO system and inhibits fibrosis. Hypertension 2003, 41, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Schieffer, B.; Wirger, A.; Meybrunn, M.; Seitz, S.; Holtz, J.; Riede, U.N.; Drexler, H. Comparative effects of chronic angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade on cardiac remodeling after myocardial infarction in the rat. Circulation 1994, 89, 2273–2282. [Google Scholar] [CrossRef] [Green Version]
- Lijnen, P.; Petrov, V. Induction of cardiac fibrosis by aldosterone. J. Mol. Cell. Cardiol. 2000, 32, 865–879. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Bonnard, B.; Jaisser, F. Roles of Mineralocorticoid Receptors in Cardiovascular and Cardiorenal Diseases. Annu. Rev. Physiol. 2022, 84, 585–610. [Google Scholar] [CrossRef]
- Neumann, S.; Huse, K.; Semrau, R.; Diegeler, A.; Gebhardt, R.; Buniatian, G.H.; Scholz, G.H. Aldosterone and D-glucose stimulate the proliferation of human cardiac myofibroblasts in vitro. Hypertension 2002, 39, 756–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brilla, C.G.; Zhou, G.; Matsubara, L.; Weber, K.T. Collagen metabolism in cultured adult rat cardiac fibroblasts: Response to angiotensin II and aldosterone. J. Mol. Cell. Cardiol. 1994, 26, 809–820. [Google Scholar] [CrossRef]
- Rickard, A.J.; Morgan, J.; Tesch, G.; Funder, J.W.; Fuller, P.J.; Young, M.J. Deletion of mineralocorticoid receptors from macrophages protects against deoxycorticosterone/salt-induced cardiac fibrosis and increased blood pressure. Hypertension 2009, 54, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sun, X.N.; Zeng, M.R.; Zheng, X.J.; Zhang, Y.Y.; Wan, Q.; Zhang, W.C.; Shi, C.; Du, L.J.; Ai, T.J.; et al. Mineralocorticoid Receptor Deficiency in T Cells Attenuates Pressure Overload-Induced Cardiac Hypertrophy and Dysfunction Through Modulating T-Cell Activation. Hypertension 2017, 70, 137–147. [Google Scholar] [CrossRef]
- Rickard, A.J.; Morgan, J.; Bienvenu, L.A.; Fletcher, E.K.; Cranston, G.A.; Shen, J.Z.; Reichelt, M.E.; Delbridge, L.M.; Young, M.J. Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt-mediated inflammation and cardiac fibrosis. Hypertension 2012, 60, 1443–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Borne, S.W.; Isobe, S.; Zandbergen, H.R.; Li, P.; Petrov, A.; Wong, N.D.; Fujimoto, S.; Fujimoto, A.; Lovhaug, D.; Smits, J.F.; et al. Molecular imaging for efficacy of pharmacologic intervention in myocardial remodeling. JACC Cardiovasc. Imaging 2009, 2, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.; Tsutamoto, T.; Wada, A.; Tsutsui, T.; Ishii, C.; Ohno, K.; Fujii, M.; Taniguchi, A.; Hamatani, T.; Nozato, Y.; et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents post-infarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003, 107, 2559–2565. [Google Scholar]
- Kim, J.; Eckhart, A.D.; Eguchi, S.; Koch, W.J. Beta-adrenergic receptor-mediated DNA synthesis in cardiac fibroblasts is dependent on transactivation of the epidermal growth factor receptor and subsequent activation of extracellular signal-regulated kinases. J. Biol. Chem. 2002, 277, 32116–32123. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.A.; Porter, K.E.; Smith, W.H.; White, H.L.; Ball, S.G.; Balmforth, A.J. Chronic beta2-adrenergic receptor stimulation increases proliferation of human cardiac fibroblasts via an autocrine mechanism. Cardiovasc. Res. 2003, 57, 784–792. [Google Scholar] [CrossRef] [Green Version]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017, 136, 549–561. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Kovacic, J.C. Extracellular Matrix in Ischemic Heart Disease, Part 4/4: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 2219–2235. [Google Scholar] [CrossRef] [PubMed]
- Dugina, V.; Fontao, L.; Chaponnier, C.; Vasiliev, J.; Gabbiani, G. Focal adhesion features during myofibroblastic differentiation are controlled by intracellular and extracellular factors. J. Cell Sci. 2001, 114, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Kohan, M.; Muro, A.F.; White, E.S.; Berkman, N. EDA-containing cellular fibronectin induces fibroblast differentiation through binding to alpha4beta7 integrin receptor and MAPK/Erk 1/2-dependent signaling. FASEB J. 2010, 24, 4503–4512. [Google Scholar] [CrossRef] [PubMed]
- Arslan, F.; Smeets, M.B.; Riem Vis, P.W.; Karper, J.C.; Quax, P.H.; Bongartz, L.G.; Peters, J.H.; Hoefer, I.E.; Doevendans, P.A.; Pasterkamp, G.; et al. Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction. Circ. Res. 2011, 108, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Lenga, Y.; Koh, A.; Perera, A.S.; McCulloch, C.A.; Sodek, J.; Zohar, R. Osteopontin expression is required for myofibroblast differentiation. Circ. Res. 2008, 102, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Schellings, M.W.; Vanhoutte, D.; Swinnen, M.; Cleutjens, J.P.; Debets, J.; van Leeuwen, R.E.; d’Hooge, J.; Van de Werf, F.; Carmeliet, P.; Pinto, Y.M.; et al. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J. Exp. Med. 2009, 206, 113–123. [Google Scholar] [CrossRef]
- Oka, T.; Xu, J.; Kaiser, R.A.; Melendez, J.; Hambleton, M.; Sargent, M.A.; Lorts, A.; Brunskill, E.W.; Dorn, G.W., 2nd; Conway, S.J.; et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 2007, 101, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Van Aelst, L.N.; Voss, S.; Carai, P.; Van Leeuwen, R.; Vanhoutte, D.; Sanders-van Wijk, S.; Eurlings, L.; Swinnen, M.; Verheyen, F.K.; Verbeken, E.; et al. Osteoglycin prevents cardiac dilatation and dysfunction after myocardial infarction through infarct collagen strengthening. Circ. Res. 2015, 116, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.J.; He, Y.; Li, Y.; Shen, H.; Lin, L.; Zhu, M.; Wang, Z.; Luo, X.; Hill, J.A.; Cao, D.; et al. Matricellular Protein Cilp1 Promotes Myocardial Fibrosis in Response to Myocardial Infarction. Circ. Res. 2021, 129, 1021–1035. [Google Scholar] [CrossRef]
- Murphy-Ullrich, J.E.; Suto, M.J. Thrombospondin-1 regulation of latent TGF-beta activation: A therapeutic target for fibrotic disease. Matrix Biol. 2018, 68–69, 28–43. [Google Scholar] [CrossRef]
- Shimazaki, M.; Nakamura, K.; Kii, I.; Kashima, T.; Amizuka, N.; Li, M.; Saito, M.; Fukuda, K.; Nishiyama, T.; Kitajima, S.; et al. Periostin is essential for cardiac healing after acute myocardial infarction. J. Exp. Med. 2008, 205, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Bein, K.; Simons, M. Thrombospondin type 1 repeats interact with matrix metalloproteinase 2. Regulation of metalloproteinase activity. J. Biol. Chem. 2000, 275, 32167–32173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, P.L.; Filomeno, K.L.; Bagchi, R.A.; Czubryt, M.P. Intracellular signaling of cardiac fibroblasts. Compr. Physiol. 2015, 5, 721–760. [Google Scholar] [CrossRef]
- Sinfield, J.K.; Das, A.; O’Regan, D.J.; Ball, S.G.; Porter, K.E.; Turner, N.A. p38 MAPK alpha mediates cytokine-induced IL-6 and MMP-3 expression in human cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2013, 430, 419–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bageghni, S.A.; Hemmings, K.E.; Zava, N.; Denton, C.P.; Porter, K.E.; Ainscough, J.F.X.; Drinkhill, M.J.; Turner, N.A. Cardiac fibroblast-specific p38alpha MAP kinase promotes cardiac hypertrophy via a putative paracrine interleukin-6 signaling mechanism. FASEB J. 2018, 32, 4941–4954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasek, J.J.; McRae, J.; Owens, G.K.; Haaksma, C.J. Regulation of alpha-smooth muscle actin expression in granulation tissue myofibroblasts is dependent on the intronic CArG element and the transforming growth factor-beta1 control element. Am. J. Pathol. 2005, 166, 1343–1351. [Google Scholar] [CrossRef]
- Lighthouse, J.K.; Small, E.M. Transcriptional control of cardiac fibroblast plasticity. J. Mol. Cell. Cardiol. 2016, 91, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Loirand, G.; Sauzeau, V.; Pacaud, P. Small G proteins in the cardiovascular system: Physiological and pathological aspects. Physiol. Rev. 2013, 93, 1659–1720. [Google Scholar] [CrossRef] [Green Version]
- Phrommintikul, A.; Tran, L.; Kompa, A.; Wang, B.; Adrahtas, A.; Cantwell, D.; Kelly, D.J.; Krum, H. Effects of a Rho kinase inhibitor on pressure overload induced cardiac hypertrophy and associated diastolic dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1804–H1814. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Narang, N.; Chen, P.; Yu, B.; Knapp, M.; Janardanan, J.; Blair, J.; Liao, J.K. Fibroblast deletion of ROCK2 attenuates cardiac hypertrophy, fibrosis, and diastolic dysfunction. JCI Insight 2017, 2, e93187. [Google Scholar] [CrossRef]
- Mia, M.M.; Cibi, D.M.; Binte Abdul Ghani, S.A.; Singh, A.; Tee, N.; Sivakumar, V.; Bogireddi, H.; Cook, S.A.; Mao, J.; Singh, M.K. Loss of Yap/taz in cardiac fibroblasts attenuates adverse remodeling and improves cardiac function. Cardiovasc. Res. 2021, cvab205. [Google Scholar] [CrossRef]
- Francisco, J.; Zhang, Y.; Jeong, J.I.; Mizushima, W.; Ikeda, S.; Ivessa, A.; Oka, S.; Zhai, P.; Tallquist, M.D.; Del Re, D.P. Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition. JACC Basic Transl. Sci. 2020, 5, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Lal, H.; Ahmad, F.; Zhou, J.; Yu, J.E.; Vagnozzi, R.J.; Guo, Y.; Yu, D.; Tsai, E.J.; Woodgett, J.; Gao, E.; et al. Cardiac fibroblast glycogen synthase kinase-3β regulates ventricular remodeling and dysfunction in ischemic heart. Circulation 2014, 130, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Valiente-Alandi, I.; Nieman, M.L.; Sargent, M.A.; Lorenz, J.N.; Molkentin, J.D.; Blaxall, B.C. Pharmacological and Activated Fibroblast Targeting of Gβγ-GRK2 After Myocardial Ischemia Attenuates Heart Failure Progression. J. Am. Coll. Cardiol. 2017, 70, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Snider, J.C.; Riley, L.A.; Mallory, N.T.; Bersi, M.R.; Umbarkar, P.; Gautam, R.; Zhang, Q.; Mahadevan-Jansen, A.; Hatzopoulos, A.K.; Maroteaux, L.; et al. Targeting 5-HT(2B) Receptor Signaling Prevents Border Zone Expansion and Improves Microstructural Remodeling After Myocardial Infarction. Circulation 2021, 143, 1317–1330. [Google Scholar] [CrossRef]
- Scharf, G.M.; Kilian, K.; Cordero, J.; Wang, Y.; Grund, A.; Hofmann, M.; Froese, N.; Wang, X.; Kispert, A.; Kist, R.; et al. Inactivation of Sox9 in fibroblasts reduces cardiac fibrosis and inflammation. JCI Insight 2019, 5, e126721. [Google Scholar] [CrossRef]
- Dufeys, C.; Daskalopoulos, E.P.; Castanares-Zapatero, D.; Conway, S.J.; Ginion, A.; Bouzin, C.; Ambroise, J.; Bearzatto, B.; Gala, J.L.; Heymans, S.; et al. AMPKalpha1 deletion in myofibroblasts exacerbates post-myocardial infarction fibrosis by a connexin 43 mechanism. Basic Res. Cardiol. 2021, 116, 10. [Google Scholar] [CrossRef]
- Davis, J.; Salomonis, N.; Ghearing, N.; Lin, S.C.; Kwong, J.Q.; Mohan, A.; Swanson, M.S.; Molkentin, J.D. MBNL1-mediated regulation of differentiation RNAs promotes myofibroblast transformation and the fibrotic response. Nat. Commun. 2015, 6, 10084. [Google Scholar] [CrossRef] [Green Version]
- Valiente-Alandi, I.; Potter, S.J.; Salvador, A.M.; Schafer, A.E.; Schips, T.; Carrillo-Salinas, F.; Gibson, A.M.; Nieman, M.L.; Perkins, C.; Sargent, M.A.; et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation 2018, 138, 1236–1252. [Google Scholar] [CrossRef]
- Wu, X.; Reboll, M.R.; Korf-Klingebiel, M.; Wollert, K.C. Angiogenesis after acute myocardial infarction. Cardiovasc. Res. 2021, 117, 1257–1273. [Google Scholar] [CrossRef]
- Mouton, A.J.; Ma, Y.; Rivera Gonzalez, O.J.; Daseke, M.J., 2nd; Flynn, E.R.; Freeman, T.C.; Garrett, M.R.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res. Cardiol. 2019, 114, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraswati, S.; Marrow, S.M.W.; Watch, L.A.; Young, P.P. Identification of a pro-angiogenic functional role for FSP1-positive fibroblast subtype in wound healing. Nat. Commun. 2019, 10, 3027. [Google Scholar] [CrossRef] [PubMed]
- Ubil, E.; Duan, J.; Pillai, I.C.; Rosa-Garrido, M.; Wu, Y.; Bargiacchi, F.; Lu, Y.; Stanbouly, S.; Huang, J.; Rojas, M.; et al. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature 2014, 514, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Meng, S.; Lv, J.; Chanda, P.K.; Owusu, I.; Chen, K.; Cooke, J.P. Reservoir of Fibroblasts Promotes Recovery From Limb Ischemia. Circulation 2020, 142, 1647–1662. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jambusaria, A.; Hong, Z.; Marsboom, G.; Toth, P.T.; Herbert, B.S.; Malik, A.B.; Rehman, J. SOX17 Regulates Conversion of Human Fibroblasts Into Endothelial Cells and Erythroblasts by Dedifferentiation Into CD34(+) Progenitor Cells. Circulation 2017, 135, 2505–2523. [Google Scholar] [CrossRef]
- He, L.; Huang, X.; Kanisicak, O.; Li, Y.; Wang, Y.; Li, Y.; Pu, W.; Liu, Q.; Zhang, H.; Tian, X.; et al. Preexisting endothelial cells mediate cardiac neovascularization after injury. J. Clin. Investig. 2017, 127, 2968–2981. [Google Scholar] [CrossRef]
- Hayakawa, K.; Takemura, G.; Kanoh, M.; Li, Y.; Koda, M.; Kawase, Y.; Maruyama, R.; Okada, H.; Minatoguchi, S.; Fujiwara, T.; et al. Inhibition of granulation tissue cell apoptosis during the subacute stage of myocardial infarction improves cardiac remodeling and dysfunction at the chronic stage. Circulation 2003, 108, 104–109. [Google Scholar] [CrossRef] [Green Version]
- Hanyu, A.; Ishidou, Y.; Ebisawa, T.; Shimanuki, T.; Imamura, T.; Miyazono, K. The N domain of Smad7 is essential for specific inhibition of transforming growth factor-beta signaling. J. Cell Biol. 2001, 155, 1017–1027. [Google Scholar] [CrossRef]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef] [Green Version]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef] [Green Version]
- Cunnington, R.H.; Wang, B.; Ghavami, S.; Bathe, K.L.; Rattan, S.G.; Dixon, I.M. Antifibrotic properties of c-Ski and its regulation of cardiac myofibroblast phenotype and contractility. Am. J. Physiol. Cell Physiol. 2011, 300, C176–C186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landry, N.M.; Rattan, S.G.; Filomeno, K.L.; Meier, T.W.; Meier, S.C.; Foran, S.J.; Meier, C.F.; Koleini, N.; Fandrich, R.R.; Kardami, E.; et al. SKI activates the Hippo pathway via LIMD1 to inhibit cardiac fibroblast activation. Basic Res. Cardiol. 2021, 116, 25. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; McCourt, J.; Ma, F.; Ren, S.; Li, S.; Kim, T.H.; Kurmangaliyev, Y.Z.; Nasiri, R.; Ahadian, S.; Nguyen, T.; et al. Type V Collagen in Scar Tissue Regulates the Size of Scar after Heart Injury. Cell 2020, 182, 545–562.e523. [Google Scholar] [CrossRef] [PubMed]
- Krstevski, C.; Cohen, C.D.; Dona, M.S.I.; Pinto, A.R. New perspectives of the cardiac cellular landscape: Mapping cellular mediators of cardiac fibrosis using single-cell transcriptomics. Biochem. Soc. Trans. 2020, 48, 2483–2493. [Google Scholar] [CrossRef]
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.; Nordon, R.E.; Harvey, R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. eLife 2019, 8, e43882. [Google Scholar] [CrossRef] [PubMed]
- Pyagay, P.; Heroult, M.; Wang, Q.; Lehnert, W.; Belden, J.; Liaw, L.; Friesel, R.E.; Lindner, V. Collagen triple helix repeat containing 1, a novel secreted protein in injured and diseased arteries, inhibits collagen expression and promotes cell migration. Circ. Res. 2005, 96, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Villalba, A.; Romero, J.P.; Hernandez, S.C.; Vilas-Zornoza, A.; Fortelny, N.; Castro-Labrador, L.; San Martin-Uriz, P.; Lorenzo-Vivas, E.; Garcia-Olloqui, P.; Palacio, M.; et al. Single-Cell RNA Sequencing Analysis Reveals a Crucial Role for CTHRC1 (Collagen Triple Helix Repeat Containing 1) Cardiac Fibroblasts After Myocardial Infarction. Circulation 2020, 142, 1831–1847. [Google Scholar] [CrossRef]
- McLellan, M.A.; Skelly, D.A.; Dona, M.S.I.; Squiers, G.T.; Farrugia, G.E.; Gaynor, T.L.; Cohen, C.D.; Pandey, R.; Diep, H.; Vinh, A.; et al. High-Resolution Transcriptomic Profiling of the Heart During Chronic Stress Reveals Cellular Drivers of Cardiac Fibrosis and Hypertrophy. Circulation 2020, 142, 1448–1463. [Google Scholar] [CrossRef]
- Langer, L.B.N.; Hess, A.; Korkmaz, Z.; Tillmanns, J.; Reffert, L.M.; Bankstahl, J.P.; Bengel, F.M.; Thackeray, J.T.; Ross, T.L. Molecular imaging of fibroblast activation protein after myocardial infarction using the novel radiotracer [(68)Ga]MHLL1. Theranostics 2021, 11, 7755–7766. [Google Scholar] [CrossRef]
- Shah, H.; Hacker, A.; Langburt, D.; Dewar, M.; McFadden, M.J.; Zhang, H.; Kuzmanov, U.; Zhou, Y.Q.; Hussain, B.; Ehsan, F.; et al. Myocardial Infarction Induces Cardiac Fibroblast Transformation within Injured and Noninjured Regions of the Mouse Heart. J. Proteome Res. 2021, 20, 2867–2881. [Google Scholar] [CrossRef]
- Nagaraju, C.K.; Dries, E.; Popovic, N.; Singh, A.A.; Haemers, P.; Roderick, H.L.; Claus, P.; Sipido, K.R.; Driesen, R.B. Global fibroblast activation throughout the left ventricle but localized fibrosis after myocardial infarction. Sci. Rep. 2017, 7, 10801. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.Q.; Zhang, J.; Lamparter, S. Cardiac remodeling by fibrous tissue after infarction in rats. J. Lab. Clin. Med. 2000, 135, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Kweon, H.J.; Chatila, K.; Li, N.; Taffet, G.; Frangogiannis, N.G. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J. Am. Coll. Cardiol. 2008, 51, 1384–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.D.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598. [Google Scholar] [CrossRef]
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef] [Green Version]
- Keepers, B.; Liu, J.; Qian, L. What’s in a cardiomyocyte—And how do we make one through reprogramming? Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118464. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, J.; Qian, L. Direct cardiac reprogramming comes of age: Recent advance and remaining challenges. Semin. Cell Dev. Biol. 2022, 122, 37–43. [Google Scholar] [CrossRef]



| Cytokine/Species | Effect | Mechanism | Ref. |
|---|---|---|---|
| IL-6/rat cardiac fibroblasts | IL-6 enhances collagen synthesis and myofibroblast formation. | IL-6 binds to gp130, leading to the phosphorylation of Janus kinase, activating cellular events. | [86] |
| IL-6/mouse cardiac fibroblasts | IL-6 promotes myofibroblast differentiation and the release of proinflammatory cytokines in culture. In vivo, treatment with an anti-IL-6 blocking antibody reduced myofibroblast infiltration in the infarct (and also attenuated neutrophil infiltration) but also increased the size of the infarct. | IL-6-induced phosphorylation of STAT3 upregulates the expression of hyaluronan synthesis that supports a myofibroblast phenotype in cultured fibroblasts. | [87] |
| IL-6/mouse cardiac fibroblasts | IL-6 promotes fibroblast activation and collagen synthesis. | In a co-culture model, macrophages stimulate cardiac fibroblasts to produce IL-6, which promotes TGF-β production and the downstream activation of Smad3 in fibroblasts. | [80] |
| IL-6/neonatal rat ventricular fibroblasts | IL-6 stimulates fibroblast proliferation and myofibroblast differentiation under hypoxia. Inhibition of IL-6 signaling with an IL-6 receptor inhibitor attenuates hypoxia-induced fibroblast proliferation and differentiation and collagen I expression. | In cultured fibroblasts exposed to hypoxia, the effects of IL-6 are attributed to the activation of TGFβ1, MMP2, and MMP9. | [88] |
| IL-6/human cardiac fibroblasts | IL-6 in endothelial cell-derived conditioned media increases collagen type I and fibronectin gene expression in cardiac fibroblasts. The addition of soluble gp130 to endothelial cell-derived conditioned media prevents IL-6-dependent collagen type I and fibronectin gene expression. | IL-6 in conditioned media from endothelial cells binds to soluble IL-6R to induce trans-IL-6 signaling in cardiac fibroblasts. | [89] |
| IL-6/rat cardiac fibroblasts | In contrast to the promigratory effects of IL-1β and TNF-α, IL-6 has no effect on fibroblast migration. | IL-6 does not stimulate the activation of mitogen-activated protein kinases that are involved in the regulation of cell migration. | [40] |
| IL-6/adult mouse ventricular fibroblasts. | IL-6 increases fibroblast adhesion and proliferation. | Cardiomyocyte-derived IL-6 acts in a paracrine manner to promote fibroblast proliferation in a cardiomyocyte/fibroblast co-culture model. | [90] |
| IL-6/neonatal mouse cardiac fibroblasts | IL-6 loss decreases fibroblast-myocyte adhesion in vitro and markedly upregulates fibroblast proliferation. | In a fibroblast/cardiomyocyte coculture system, IL-6/soluble IL-6R trans-signaling activates STAT3 in fibroblasts to modulate fibroblast proliferation and adhesion to cardiomyocytes. | [91] |
| IL1-β/mouse cardiac fibroblasts | IL1-β: (a) attenuates TGF-β-induced α-SMA expression and incorporation into stress fibers, (b) abrogates fibroblast-mediated collagen pad contraction and expression of periostin, and (c) promotes a matrix-degrading phenotype via stimulating MMP3 and MMP8 expression. | IL1-β acts via IL-1R1 to upregulate BAMBI, which acts to negatively regulate TGFβ signaling. IL1-β also downregulates endoglin signaling receptors. | [35] |
| IL1-β/adult rat cardiac fibroblasts | IL1-β augments the expression and activity of MMP-2, 3, and 9, alongside an increase in TIMP-1 expression, and enhances fibroblast migration. | IL1-β activates p38, ERK, and JNK MAP kinase pathways to stimulate MMP expression and migration. | [92] |
| IL-1β/neonatal and adult rat cardiac fibroblasts | IL1-β selectively downregulates the expression and synthesis of fibrillar collagens. Increases total MMP activity, with an increase in the expression of MMP-2, 9, and 13. | No mechanism is studied. | [93] |
| IL-1β/mouse cardiac fibroblasts | IL-1β stimulates proinflammatory gene expression. It promotes ECM remodeling. | IL-1β acts via IL-R1 to promote ECM remodeling via enhancing the fibroblast expression of MMPs (MMP-3, 8, and 9) and downregulating the expression of TIMP-2 and TIMP-4. | [94] |
| IL-1β/neonatal rat cardiac fibroblasts | IL-1β induces AT1 receptor upregulation in fibroblasts, contributing to ECM remodeling. | IL-1β acts via an NFκB-dependent mechanism to upregulate AT1R expression. | [95] |
| IL-1α/mouse neonatal ventricular fibroblasts | IL-1R antagonism and the administration of an anti-IL-1α blocking antibody show that the conditioned medium of necrotic cardiomyocytes activates proinflammatory signaling in fibroblasts through IL-1α. | IL-1α acts via an MyD88-dependent and NLRP3-independent pathway to promote pro-inflammatory gene expression in cardiac fibroblasts. | [43] |
| IL-1α/human cardiac fibroblasts | IL-1α markedly increases the expression of MMP-1, 3, 9, and 10, with a minimal effect on the mRNA expression of structural ECM proteins, and reduces the expression of ADAMTS1. | IL-1α acts via distinct P38 MAPK subtypes α/β/γ/δ to regulate the expression of MMPs and metalloproteinases in fibroblasts. | [96] |
| IL-1α/human cardiac fibroblasts | IL-1α stimulates proinflammatory gene expression in fibroblasts via upregulation of IL-1β, TNF-α, and IL-6. | ERK, JNK, and p38 MAPKs, along with nuclear factor (NF)-kB signaling, distinctly regulates IL1-β, TNF-α, and IL-6 expression. | [39] |
| IL-1α/human cardiac fibroblasts | Cardiac fibroblasts express neutrophil-binding adhesion molecules and neutrophil chemoattractants in response to IL-1α, promoting neutrophil recruitment into the infarcted myocardium. | IL-1α acts via a p38- and NF-κB-dependent mechanism to promote the expression of ICAM-1, E-selectin, and CXC chemokines in fibroblasts. | [97] |
| IL-1α/human cardiac fibroblasts | IL-1α has opposing effects on the expression of connective tissue growth factor (CTGF) and tenascin-C (TNC) expression. | Stimulates NFκB, PI3K/AKT, and p38 MAPK pathways to upregulate the expression of TNC while downregulating CTGF expression. | [83] |
| TNF-α/neonatal and adult rat cardiac fibroblasts | TNF-α promotes matrix degradation via mediating a decrease in collagen synthesis with an increase in MMP-2, MMP-9, and MMP-13. It has no effect on cell proliferation and total protein synthesis. | No mechanism studied. | [93] |
| TNF-α/neonatal rat cardiac fibroblasts | TNF-α increases AT1 receptor density in cardiac fibroblasts. | TNF-α acts via NFκB to increase AT1 receptor expression. | [95] |
| TNF-α/primary human fibroblasts from patient biopsies with dilated cardiomyopathy | (a) TNF-α increases cytokine expression at the transcriptome level; however, this increase was not reflected in the cytokine secretome. (b) TNF-α treatment has no effect on collagen/MMP/TIMP gene expression. | TNF-α effects are mediated via the transcriptional activation of NFκB. | [98] |
| TNF-α/rat cardiac fibroblasts | TNF-α stimulates a concentration-dependent increase in fibroblast migration. | TNF-α-dependent migration is regulated by the activation of MAPKs–ERK1/2, JNK, and p38. | [40] |
| TNF-α/human cardiac fibroblasts | TNF-α promotes fibroblast MMP-9 expression that is abrogated following treatment with an anti-TNF-α blocking antibody. | TNF-α acts via NFκB to promote the expression of MMP-9. | [99] |
| TNF-α/human cardiac fibroblasts | TNF-α promotes the proliferation of cardiac fibroblasts. | TNF-α-dependent activation of ERK1/2 and NFκB drives fibroblast cell proliferation. | [100] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venugopal, H.; Hanna, A.; Humeres, C.; Frangogiannis, N.G. Properties and Functions of Fibroblasts and Myofibroblasts in Myocardial Infarction. Cells 2022, 11, 1386. https://doi.org/10.3390/cells11091386
Venugopal H, Hanna A, Humeres C, Frangogiannis NG. Properties and Functions of Fibroblasts and Myofibroblasts in Myocardial Infarction. Cells. 2022; 11(9):1386. https://doi.org/10.3390/cells11091386
Chicago/Turabian StyleVenugopal, Harikrishnan, Anis Hanna, Claudio Humeres, and Nikolaos G. Frangogiannis. 2022. "Properties and Functions of Fibroblasts and Myofibroblasts in Myocardial Infarction" Cells 11, no. 9: 1386. https://doi.org/10.3390/cells11091386