
Ubiquitin Proteasome System and Microtubules Are Master Regulators of Central and Peripheral Nervous System Axon Degeneration



{kind=link}
{kind=link}
Abstract
:1. Introduction
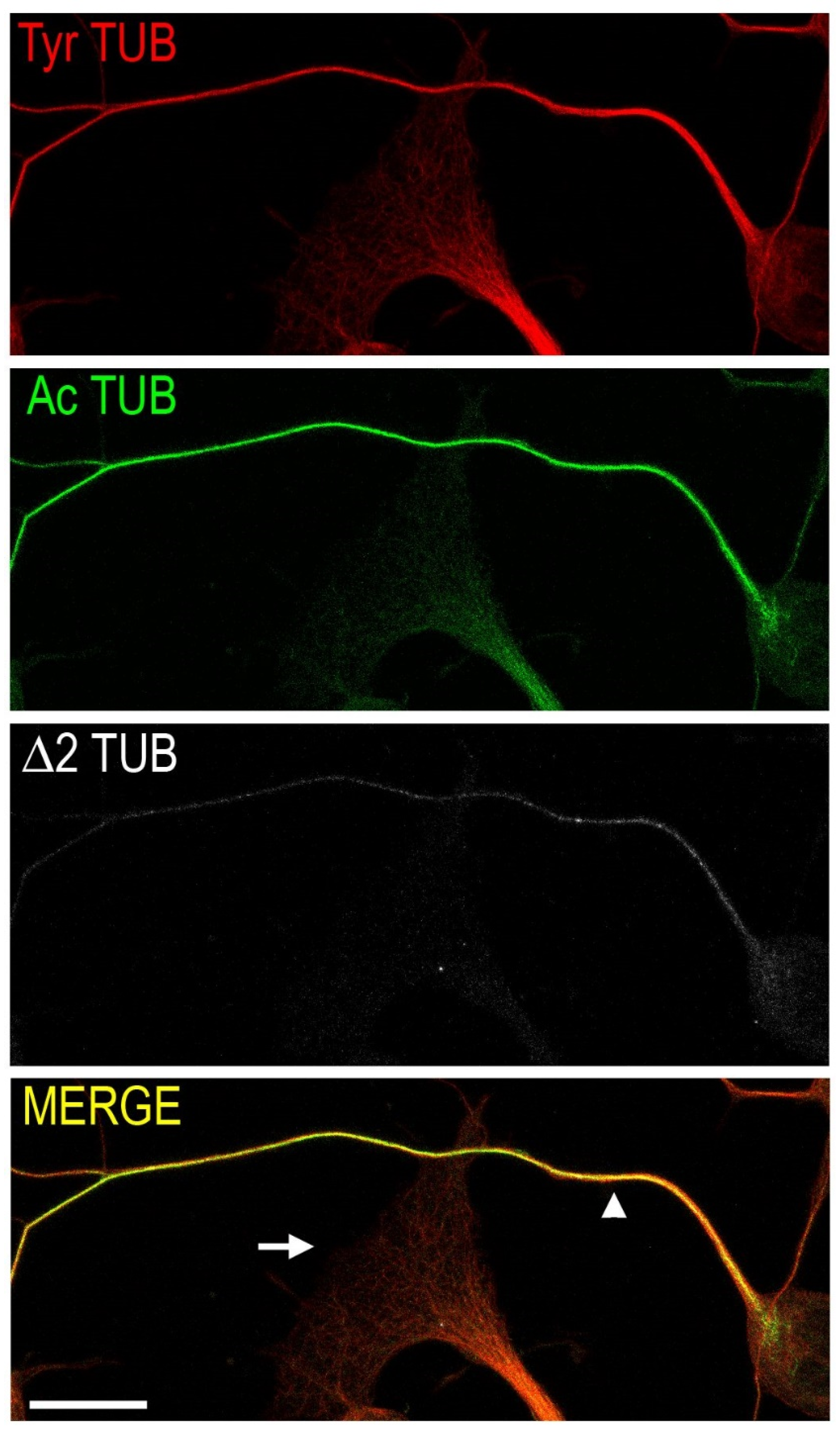
1.1. Microtubules and Axonal Transport
1.2. Ubiquitin-Proteasome System and Protein Quality Control
1.3. Axon Degeneration
2. Parkinson’s Disease
3. Peripheral Neuropathies and Chemotherapy-Induced Peripheral Neuropathy
4. Regenerative Potentialities
5. Closing Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whitmore, A.V.; Lindsten, T.; Raff, M.C.; Thompson, C.B. The proapoptotic proteins Bax and Bak are not involved in Wallerian degeneration. Cell Death Differ. 2003, 10, 260–261. [Google Scholar] [CrossRef] [PubMed]
- Krantic, S.; Mechawar, N.; Reix, S.; Quirion, R. Molecular basis of programmed cell death involved in neurodegeneration. Trends Neurosci. 2005, 28, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.S.; Fang, G.; Culver, D.G.; Davis, A.A.; Rich, M.M.; Glass, J.D. The WldS protein protects against axonal degeneration: A model of gene therapy for peripheral neuropathy. Ann. Neurol. 2001, 50, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Conforti, L.; Gilley, J.; Coleman, M.P. Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 2014, 15, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Arthur-Farraj, P.; Coleman, M.P. Lessons from injury: How nerve injury studies reveal basic biological mechanisms and therapeutic opportunities for peripheral nerve diseases. Neurotherapeutics 2021, 18, 2200–2221. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M. Axon degeneration mechanisms: Commonality amid diversity. Nat. Rev. Neurosci. 2005, 6, 889–898. [Google Scholar] [CrossRef]
- Coleman, M.P. The challenges of axon survival: Introduction to the special issue on axonal degeneration. Exp. Neurol. 2013, 246, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Nolano, M.; Provitera, V.; Manganelli, F.; Iodice, R.; Stancanelli, A.; Caporaso, G.; Saltalamacchia, A.; Califano, F.; Lanzillo, B.; Picillo, M.; et al. Loss of cutaneous large and small fibers in naive and l-dopa-treated PD patients. Neurology 2017, 89, 776–784. [Google Scholar] [CrossRef]
- Yau, K.W.; Schätzle, P.; Tortosa, E.; Pagès, S.; Holtmaat, A.; Kapitein, L.C.; Hoogenraad, C.C. Dendrites In vitro and in vivo contain microtubules of opposite polarity and axon formation correlates with uniform plus-end-out microtubule orientation. J. Neurosci. 2016, 36, 1071–1085. [Google Scholar] [CrossRef] [Green Version]
- Kapitein, L.C.; Schlager, M.A.; Kuijpers, M.; Wulf, P.S.; Van Spronsen, M.; MacKintosh, F.C.; Hoogenraad, C.C. Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr. Biol. 2010, 20, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Janke, C.; Magiera, M.M. The tubulin code and its role in controlling microtubule properties and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.; Kapitein, L.C.; Gouveia, S.M.; Dortland, B.R.; Wulf, P.S.; Grigoriev, I.; Camera, P.; Spangler, S.A.; Di Stefano, P.; Demmers, J.; et al. Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron 2009, 61, 85–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bär, J.; Popp, Y.; Bucher, M.; Mikhaylova, M. Direct and indirect effects of tubulin post-translational modifications on microtubule stability: Insights and regulations. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119241. [Google Scholar] [CrossRef] [PubMed]
- Sharp, D.J.; Ross, J.L. Microtubule-severing enzymes at the cutting edge. J. Cell Sci. 2012, 125, 2561–2569. [Google Scholar] [CrossRef] [Green Version]
- Sirajuddin, M.; Rice, L.M.; Vale, R.D. Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat. Cell Biol. 2014, 16, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Portran, D.; Schaedel, L.; Xu, Z.; Théry, M.; Nachury, M.V. Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat. Cell Biol. 2017, 19, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Nirschl, J.J.; Ghiretti, A.E.; Holzbaur, E.L.F. The impact of cytoskeletal organization on the local regulation of neuronal transport. Nat. Rev. Neurosci. 2017, 18, 585–597. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Rossor, A.M.; Fellows, A.D.; Tosolini, A.P.; Schiavo, G. Axonal transport and neurological disease. Nat. Rev. Neurol. 2019, 15, 691–703. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Zilberman, Y.; Ballestrem, C.; Carramusa, L.; Mazitschek, R.; Khochbin, S.; Bershadsky, A. Regulation of microtubule dynamics by inhibition of the tubulin deacetylase HDAC6. J. Cell Sci. 2009, 122, 3531–3541. [Google Scholar] [CrossRef] [Green Version]
- Simões-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin-proteasome system (UPS) as a target for anticancer treatment. Arch. Pharm Res. 2020, 43, 1144–1161. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.F.; Gan, Z.Y.; Komander, D.; Dewson, G. Ubiquitin signalling in neurodegeneration: Mechanisms and therapeutic opportunities. Cell Death Differ. 2021, 28, 570–590. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Ambasta, R.K.; Kumar, P. Ubiquitin biology in neurodegenerative disorders: From impairment to therapeutic strategies. Ageing Res. Rev. 2020, 61, 101078. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Cruz, I.; Reynaud, E. Proteasome subunits involved in neurodegenerative diseases. Arch. Med. Res. 2021, 52, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Babetto, E.; Beirowski, B.; Russler, E.V.; Milbrandt, J.; DiAntonio, A. The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep. 2013, 3, 1422–1429. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Hao, Y.; Sun, K.; Li, J.; Li, X.; Mishra, B.; Soppina, P.; Wu, C.; Hume, R.I.; Collins, C.A. The highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein. PLoS Biol. 2012, 10, e1001440. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Ferguson, T.A.; Schoch, K.M.; Li, J.; Qian, Y.; Shofer, F.S.; Saatman, K.E.; Neumar, R.W. Calpains mediate axonal cytoskeleton disintegration during Wallerian degeneration. Neurobiol. Dis. 2013, 56, 34–46. [Google Scholar] [CrossRef] [Green Version]
- Osterloh, J.M.; Yang, J.; Rooney, T.M.; Fox, A.N.; Adalbert, R.; Powell, E.H.; Sheehan, A.E.; Avery, M.A.; Hackett, R.; Logan, M.A.; et al. DSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012, 337, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Czopka, T.; López-Schier, H. Systemic loss of Sarm1 protects Schwann cells from chemotoxicity by delaying axon degeneration. Commun. Biol. 2020, 3, 49. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, A.; Kumari, N.; Mukherjee, P. The curious case of SARM1: Dr. Jekyll and Mr. Hyde in cell death and immunity? FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Peters, O.M.; Lewis, E.A.; Osterloh, J.M.; Weiss, A.; Salameh, J.S.; Metterville, J.; Brown, R.H.; Freeman, M.R. Loss of Sarm1 does not suppress motor neuron degeneration in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2018, 27, 3761–3771. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; Press, C.; Daniels, R.W.; Sasaki, Y.; Milbrandt, J.; DiAntonio, A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat. Neurosci. 2009, 12, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Oh, Y.; Cho, E.; DiAntonio, A.; Cavalli, V.; Shin, J.E.; Choi, H.W.; Cho, Y. FK506-binding protein-like and FK506-binding protein 8 regulate dual leucine zipper kinase degradation and neuronal responses to axon injury. J. Biol. Chem. 2022, 298, 101647. [Google Scholar] [CrossRef]
- Chen, Y.H.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 is required in human derived sensory neurons for injury-induced and neurotoxic axon degeneration. Exp. Neurol. 2021, 339, 113636. [Google Scholar] [CrossRef]
- Zhai, Q.; Wang, J.; Kim, A.; Liu, Q.; Watts, R.; Hoopfer, E.; Mitchison, T.; Luo, L.; He, Z. Involvement of the ubiquitin-proteasome system in the early stages of wallerian degeneration. Neuron 2003, 39, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Ertürk, A.; Hellal, F.; Enes, J.; Bradke, F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J. Neurosci. 2007, 27, 9169–9180. [Google Scholar] [CrossRef] [Green Version]
- Hellal, F.; Hurtado, A.; Ruschel, J.; Flynn, K.C.; Laskowski, C.J.; Umlauf, M.; Kapitein, L.C.; Strikis, D.; Lemmon, V.; Bixby, J.; et al. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 2011, 331, 928–931. [Google Scholar] [CrossRef] [Green Version]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.; Halliday, G. Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Kuzuhara, S.; Mori, H.; Izumiyama, N.; Yoshimura, M.; Ihara, Y. Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol. 1988, 75, 345–353. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunn, B.H.; Cragg, S.J.; Bolam, J.P.; Spillantini, M.G.; Wade-Martins, R. Impaired intracellular trafficking defines early Parkinson’s disease. Trends Neurosci. 2015, 38, 178–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartelli, D.; Cappelletti, G. Microtubule destabilization paves the way to Parkinson’s disease. Mol. Neurobiol. 2017, 54, 6762–6774. [Google Scholar] [CrossRef]
- Peters, O.M.; Weiss, A.; Metterville, J.; Song, L.; Logan, R.; Smith, G.A.; Schwarzschild, M.A.; Mueller, C.; Brown, R.H.; Freeman, M. Genetic diversity of axon degenerative mechanisms in models of Parkinson’s disease. Neurobiol. Dis. 2021, 155, 105368. [Google Scholar] [CrossRef]
- Ren, Y.; Liu, W.; Jiang, H.; Jiang, Q.; Feng, J. Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J. Biol. Chem. 2005, 280, 34105–34112. [Google Scholar] [CrossRef] [Green Version]
- Cartelli, D.; Ronchi, C.; Maggioni, M.G.; Rodighiero, S.; Giavini, E.; Cappelletti, G. Microtubule dysfunction precedes transport impairment and mitochondria damage in MPP+ -induced neurodegeneration. J. Neurochem. 2010, 115, 247–258. [Google Scholar] [CrossRef]
- Casagrande, F.V.M.; Amadeo, A.; Cartelli, D.; Calogero, A.M.; Modena, D.; Costa, I.; Cantele, F.; Onelli, E.; Moscatelli, A.; Ascagni, M.; et al. The imbalance between dynamic and stable microtubules underlies neurodegeneration induced by 2,5-hexanedione. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165581. [Google Scholar] [CrossRef]
- Cartelli, D.; Casagrande, F.; Busceti, C.L.; Bucci, D.; Molinaro, G.; Traficante, A.; Passarella, D.; Giavini, E.; Pezzoli, G.; Battaglia, G.; et al. Microtubule alterations occur early in experimental parkinsonism and the microtubule stabilizer epothilone D is neuroprotective. Sci. Rep. 2013, 3, 1837. [Google Scholar] [CrossRef] [Green Version]
- Hasbani, D.M.; O’Malley, K.L. Wld(S) mice are protected against the Parkinsonian mimetic MPTP. Exp. Neurol. 2006, 202, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Sajadi, A.; Schneider, B.L.; Aebischer, P. Wlds-mediated protection of dopaminergic fibers in an animal model of Parkinson disease. Curr. Biol. 2004, 14, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Godena, V.K.; Brookes-Hocking, N.; Moller, A.; Shaw, G.; Oswald, M.; Sancho, R.M.; Miller, C.C.; Whitworth, A.J.; De Vos, K.J. Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun. 2014, 5, 5245. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Jiang, H.; Hu, Z.; Fan, K.; Wang, J.; Janoschka, S.; Wang, X.; Ge, S.; Feng, J. Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. Stem Cells 2015, 33, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Cartelli, D.; Goldwurm, S.; Casagrande, F.; Pezzoli, G.; Cappelletti, G. Microtubule destabilization is shared by genetic and idiopathic Parkinson’s disease patient fibroblasts. PLoS ONE 2012, 7, e37467. [Google Scholar] [CrossRef]
- Cartelli, D.; Amadeo, A.; Calogero, A.M.; Casagrande, F.V.M.; De Gregorio, C.; Gioria, M.; Kuzumaki, N.; Costa, I.; Sassone, J.; Ciammola, A.; et al. Parkin absence accelerates microtubule aging in dopaminergic neurons. Neurobiol. Aging 2018, 61, 66–74. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef]
- Shimura, H.; Schlossmacher, M.G.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 2001, 293, 263–269. [Google Scholar] [CrossRef]
- Lu, X.H.; Fleming, S.M.; Meurers, B.; Ackerson, L.C.; Mortazavi, F.; Lo, V.; Hernandez, D.; Sulzer, D.; Jackson, G.R.; Maidment, N.T.; et al. Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K-resistant alpha-synuclein. J. Neurosci. 2009, 29, 1962–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; De Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Zhao, J.; Feng, J. Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J. Neurosci. 2003, 23, 3316–3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, R.; Van Helleputte, L.; Benoy, V.; Van Den Bosch, L. Defective axonal transport: A common pathological mechanism in inherited and acquired peripheral neuropathies. Neurobiol. Dis. 2017, 105, 300–320. [Google Scholar] [CrossRef] [PubMed]
- Cavaletti, G. Chemotherapy-induced peripheral neurotoxicity (CIPN): What we need and what we know. J. Peripher. Nerv. Syst. 2014, 19, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Tofthagen, C. Patient perceptions associated with chemotherapy-induced peripheral neuropathy. Clin. J. Oncol. Nurs. 2010, 14, E22–E28. [Google Scholar] [CrossRef] [Green Version]
- Argyriou, A.A.; Bruna, J.; Marmiroli, P.; Cavaletti, G. Chemotherapy-induced peripheral neurotoxicity (CIPN): An update. Crit. Rev. Oncol. Hematol. 2012, 82, 51–77. [Google Scholar] [CrossRef]
- Geisler, S. Vincristine- and bortezomib-induced neuropathies—From bedside to bench and back. Exp. Neurol. 2021, 336, 113519. [Google Scholar] [CrossRef]
- Cashman, C.R.; Höke, A. Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci. Lett. 2015, 596, 33–50. [Google Scholar] [CrossRef] [Green Version]
- Hammond, J.W.; Huang, C.F.; Kaech, S.; Jacobson, C.; Banker, G.; Verhey, K.J. Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol. Biol. Cell 2010, 21, 572–583. [Google Scholar] [CrossRef] [Green Version]
- Gornstein, E.; Schwarz, T.L. The paradox of paclitaxel neurotoxicity: Mechanisms and unanswered questions. Neuropharmacology 2014, 76, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.S.; Davis, A.A.; Culver, D.G.; Glass, J.D. WldS mice are resistant to paclitaxel (taxol) neuropathy. Ann. Neurol. 2002, 52, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Doan, R.A.; Cheng, G.C.; Cetinkaya-Fisgin, A.; Huang, S.X.; Höke, A.; Milbrandt, J.; DiAntonio, A. Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. JCI Insight 2019, 4, e129920. [Google Scholar] [CrossRef] [PubMed]
- Ketschek, A.; Holland, S.M.; Gallo, G. SARM1 suppresses axon branching through attenuation of axonal cytoskeletal dynamics. Front. Mol. Neurosci. 2022, 15, 726962. [Google Scholar] [CrossRef] [PubMed]
- Malacrida, A.; Meregalli, C.; Rodriguez-Menendez, V.; Nicolini, G. Chemotherapy-induced peripheral neuropathy and changes in cytoskeleton. Int. J. Mol. Sci. 2019, 20, 2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staff, N.P.; Podratz, J.L.; Grassner, L.; Bader, M.; Paz, J.; Knight, A.M.; Loprinzi, C.L.; Trushina, E.; Windebank, A.J. Bortezomib alters microtubule polymerization and axonal transport in rat dorsal root ganglion neurons. Neurotoxicology 2013, 39, 124–131. [Google Scholar] [CrossRef] [Green Version]
- LaPointe, N.E.; Morfini, G.; Brady, S.T.; Feinstein, S.C.; Wilson, L.; Jordan, M.A. Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: Implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology 2013, 37, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Topp, K.S.; Tanner, K.D.; Levine, J.D. Damage to the cytoskeleton of large diameter sensory neurons and myelinated axons in vincristine-induced painful peripheral neuropathy in the rat. J. Comp. Neurol. 2000, 424, 563–576. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, M.; Su, R.; Zhang, D.; Yang, G. The efficacy and mechanism of proteasome inhibitors in solid tumor treatment. Recent Pat. Anticancer Drug Discov. 2021. [Google Scholar] [CrossRef]
- Poruchynsky, M.S.; Sackett, D.L.; Robey, R.W.; Ward, Y.; Annunziata, C.; Fojo, T. Proteasome inhibitors increase tubulin polymerization and stabilization in tissue culture cells: A possible mechanism contributing to peripheral neuropathy and cellular toxicity following proteasome inhibition. Cell Cycle 2008, 7, 940–949. [Google Scholar] [CrossRef] [Green Version]
- Hrstka, S.C.L.; Ankam, S.; Agac, B.; Klein, J.P.; Moore, R.A.; Narapureddy, B.; Schneider, I.; Hrstka, R.F.; Dasari, S.; Staff, N.P. Proteomic analysis of human iPSC-derived sensory neurons implicates cell stress and microtubule dynamics dysfunction in bortezomib-induced peripheral neurotoxicity. Exp. Neurol. 2021, 335, 113520. [Google Scholar] [CrossRef] [PubMed]
- Meregalli, C.; Chiorazzi, A.; Carozzi, V.A.; Canta, A.; Sala, B.; Colombo, M.; Oggioni, N.; Ceresa, C.; Foudah, D.; La Russa, F.; et al. Evaluation of tubulin polymerization and chronic inhibition of proteasome as citotoxicity mechanisms in bortezomib-induced peripheral neuropathy. Cell Cycle 2014, 13, 612–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pero, M.E.; Meregalli, C.; Qu, X.; Shin, G.J.; Kumar, A.; Shorey, M.; Rolls, M.M.; Tanji, K.; Brannagan, T.H.; Alberti, P.; et al. Pathogenic role of delta 2 tubulin in bortezomib-induced peripheral neuropathy. Proc. Natl. Acad. Sci. USA 2021, 118, e2012685118. [Google Scholar] [CrossRef] [PubMed]
- Jannuzzi, A.T.; Arslan, S.; Yilmaz, A.M.; Sari, G.; Beklen, H.; Méndez, L.; Fedorova, M.; Arga, K.Y.; Karademir Yilmaz, B.; Alpertunga, B. Higher proteotoxic stress rather than mitochondrial damage is involved in higher neurotoxicity of bortezomib compared to carfilzomib. Redox Biol. 2020, 32, 101502. [Google Scholar] [CrossRef]
- Beider, K.; Rosenberg, E.; Dimenshtein-Voevoda, V.; Sirovsky, Y.; Vladimirsky, J.; Magen, H.; Ostrovsky, O.; Shimoni, A.; Bromberg, Z.; Weiss, L.; et al. Blocking of Transient Receptor Potential Vanilloid 1 (TRPV1) promotes terminal mitophagy in multiple myeloma, disturbing calcium homeostasis and targeting ubiquitin pathway and bortezomib-induced unfolded protein response. J. Hematol. Oncol. 2020, 13, 158. [Google Scholar] [CrossRef]
- Kim, Y.Y.; Yoon, J.H.; Um, J.H.; Jeong, D.J.; Shin, D.J.; Hong, Y.B.; Kim, J.K.; Kim, D.H.; Kim, C.; Chung, C.G.; et al. PINK1 alleviates thermal hypersensitivity in a paclitaxel-induced Drosophila model of peripheral neuropathy. PLoS ONE 2020, 15, e0239126. [Google Scholar] [CrossRef]
- Russell, J.W.; Windebank, A.J.; McNiven, M.A.; Brat, D.J.; Brimijoin, W.S. Effect of cisplatin and ACTH4-9 on neural transport in cisplatin induced neurotoxicity. Brain Res. 1995, 676, 258–267. [Google Scholar] [CrossRef]
- Podratz, J.L.; Lee, H.; Knorr, P.; Koehler, S.; Forsythe, S.; Lambrecht, K.; Arias, S.; Schmidt, K.; Steinhoff, G.; Yudintsev, G.; et al. Cisplatin induces mitochondrial deficits in Drosophila larval segmental nerve. Neurobiol. Dis. 2017, 97, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Krukowski, K.; Ma, J.; Golonzhka, O.; Laumet, G.O.; Gutti, T.; Van Duzer, J.H.; Mazitschek, R.; Jarpe, M.B.; Heijnen, C.J.; Kavelaars, A. HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy. Pain 2017, 158, 1126–1137. [Google Scholar] [CrossRef]
- Gilley, J.; Mayer, P.R.; Yu, G.; Coleman, M.P. Low levels of NMNAT2 compromise axon development and survival. Hum. Mol. Genet. 2019, 28, 448–458. [Google Scholar] [CrossRef]
- Christie, K.J.; Martinez, J.A.; Zochodne, D.W. Disruption of E3 ligase NEDD4 in peripheral neurons interrupts axon outgrowth: Linkage to PTEN. Mol. Cell Neurosci. 2012, 50, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Meyer Zu Reckendorf, S.; Moser, D.; Blechschmidt, A.; Joga, V.N.; Sinske, D.; Hegler, J.; Deininger, S.; Catanese, A.; Vettorazzi, S.; Antoniadis, G.; et al. Motoneuron-specific PTEN deletion in mice induces neuronal hypertrophy and also regeneration after facial nerve injury. J. Neurosci. 2022, 42, 2474–2491. [Google Scholar] [CrossRef]
- Deftu, A.F.; Chu Sin Chung, P.; Laedermann, C.J.; Gillet, L.; Pertin, M.; Kirschmann, G.; Décosterd, I. The anti-diabetic drug metformin regulates voltage-gated sodium channel Na. Eneuro 2022, 9, ENEURO.0409-21.2022. [Google Scholar] [CrossRef] [PubMed]
- Turkiew, E.; Falconer, D.; Reed, N.; Höke, A. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J. Peripher. Nerv. Syst. 2017, 22, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosanac, T.; Hughes, R.O.; Engber, T.; Devraj, R.; Brearley, A.; Danker, K.; Young, K.; Kopatz, J.; Hermann, M.; Berthemy, A.; et al. Pharmacological SARM1 inhibition protects axon structure and function in paclitaxel-induced peripheral neuropathy. Brain 2021, 144, 3226–3238. [Google Scholar] [CrossRef]
- Baas, P.W.; Ahmad, F.J. Beyond taxol: Microtubule-based treatment of disease and injury of the nervous system. Brain 2013, 136, 2937–2951. [Google Scholar] [CrossRef] [Green Version]
- Brunden, K.R.; Zhang, B.; Carroll, J.; Yao, Y.; Potuzak, J.S.; Hogan, A.M.; Iba, M.; James, M.J.; Xie, S.X.; Ballatore, C.; et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J. Neurosci. 2010, 30, 13861–13866. [Google Scholar] [CrossRef] [Green Version]
- Boxer, A.L.; Lang, A.E.; Grossman, M.; Knopman, D.S.; Miller, B.L.; Schneider, L.S.; Doody, R.S.; Lees, A.; Golbe, L.I.; Williams, D.R.; et al. Davunetide in patients with progressive supranuclear palsy: A randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol. 2014, 13, 676–685. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Cho, Y.; Watt, D.; Cavalli, V. Tubulin-tyrosine ligase (TTL)-mediated increase in tyrosinated α-tubulin in injured axons is required for retrograde injury signaling and axon regeneration. J. Biol. Chem. 2015, 290, 14765–14775. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Jin, Y.; Shapiro, T.M.; Hinduja, A.; Baas, P.W.; Tom, V.J. Chronic neuronal activation increases dynamic microtubules to enhance functional axon regeneration after dorsal root crush injury. Nat. Commun. 2020, 11, 6131. [Google Scholar] [CrossRef]
- Gobrecht, P.; Andreadaki, A.; Diekmann, H.; Heskamp, A.; Leibinger, M.; Fischer, D. Promotion of functional nerve regeneration by inhibition of microtubule detyrosination. J. Neurosci. 2016, 36, 3890–3902. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Koike, T. Mammalian Sir2-related protein (SIRT) 2-mediated modulation of resistance to axonal degeneration in slow Wallerian degeneration mice: A crucial role of tubulin deacetylation. Neuroscience 2007, 147, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Palma, A.M.; Gomes, R.; Santos, D.; Silva, D.F.; Cardoso, S.M. Acetylation as a major determinant to microtubule-dependent autophagy: Relevance to Alzheimer’s and Parkinson disease pathology. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2008–2023. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Cavalli, V. HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J. 2012, 31, 3063–3078. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Zhou, Y.; Ye, L.; Lu, Q.; Zhang, K.; Zhang, J.; Xie, L.; Wu, Y.; Xu, K.; Zhang, H.; et al. Histone deacetylase 6 inhibition restores autophagic flux to promote functional recovery after spinal cord injury. Exp. Neurol. 2020, 324, 113138. [Google Scholar] [CrossRef]
- El-Saiy, K.A.; Sayed, R.H.; El-Sahar, A.E.; Kandil, E.A. Modulation of histone deacetylase, the ubiquitin proteasome system, and autophagy underlies the neuroprotective effects of venlafaxine in a rotenone-induced Parkinson’s disease model in rats. Chem. Biol. Interact. 2022, 354, 109841. [Google Scholar] [CrossRef]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelières, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, C.; Wu, J.; Tao, J.J.; Sui, X.L.; Yao, Z.G.; Xu, Y.F.; Huang, L.; Zhu, H.; Sheng, S.L.; et al. Tubastatin A/ACY-1215 improves cognition in Alzheimer’s disease transgenic mice. J. Alzheimers Dis. 2014, 41, 1193–1205. [Google Scholar] [CrossRef] [Green Version]
- Rossaert, E.; Van Den Bosch, L. HDAC6 inhibitors: Translating genetic and molecular insights into a therapy for axonal CMT. Brain Res. 2020, 1733, 146692. [Google Scholar] [CrossRef]
- Dai, Z.; Chu, H.; Ma, J.; Yan, Y.; Zhang, X.; Liang, Y. The regulatory mechanisms and therapeutic potential of microRNAs: From chronic pain to morphine tolerance. Front. Mol. Neurosci. 2018, 11, 80. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cartelli, D.; Cavaletti, G.; Lauria, G.; Meregalli, C. Ubiquitin Proteasome System and Microtubules Are Master Regulators of Central and Peripheral Nervous System Axon Degeneration. Cells 2022, 11, 1358. https://doi.org/10.3390/cells11081358
Cartelli D, Cavaletti G, Lauria G, Meregalli C. Ubiquitin Proteasome System and Microtubules Are Master Regulators of Central and Peripheral Nervous System Axon Degeneration. Cells. 2022; 11(8):1358. https://doi.org/10.3390/cells11081358
Chicago/Turabian StyleCartelli, Daniele, Guido Cavaletti, Giuseppe Lauria, and Cristina Meregalli. 2022. "Ubiquitin Proteasome System and Microtubules Are Master Regulators of Central and Peripheral Nervous System Axon Degeneration" Cells 11, no. 8: 1358. https://doi.org/10.3390/cells11081358