
Identification of Transcription Factors Responsible for a Transforming Growth Factor-β-Driven Hypertrophy-like Phenotype in Human Osteoarthritic Chondrocytes

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Material
2.2. Primary Chondrocyte Cell Culture and Stimulation
2.3. Construction Reporter Plasmids, Virus Production and Transduction
2.4. Stimulation and Reporter Gene Assays
2.5. RNA Isolation and Quantitative Real-Time PCR
2.6. Statistical Analysis
3. Results
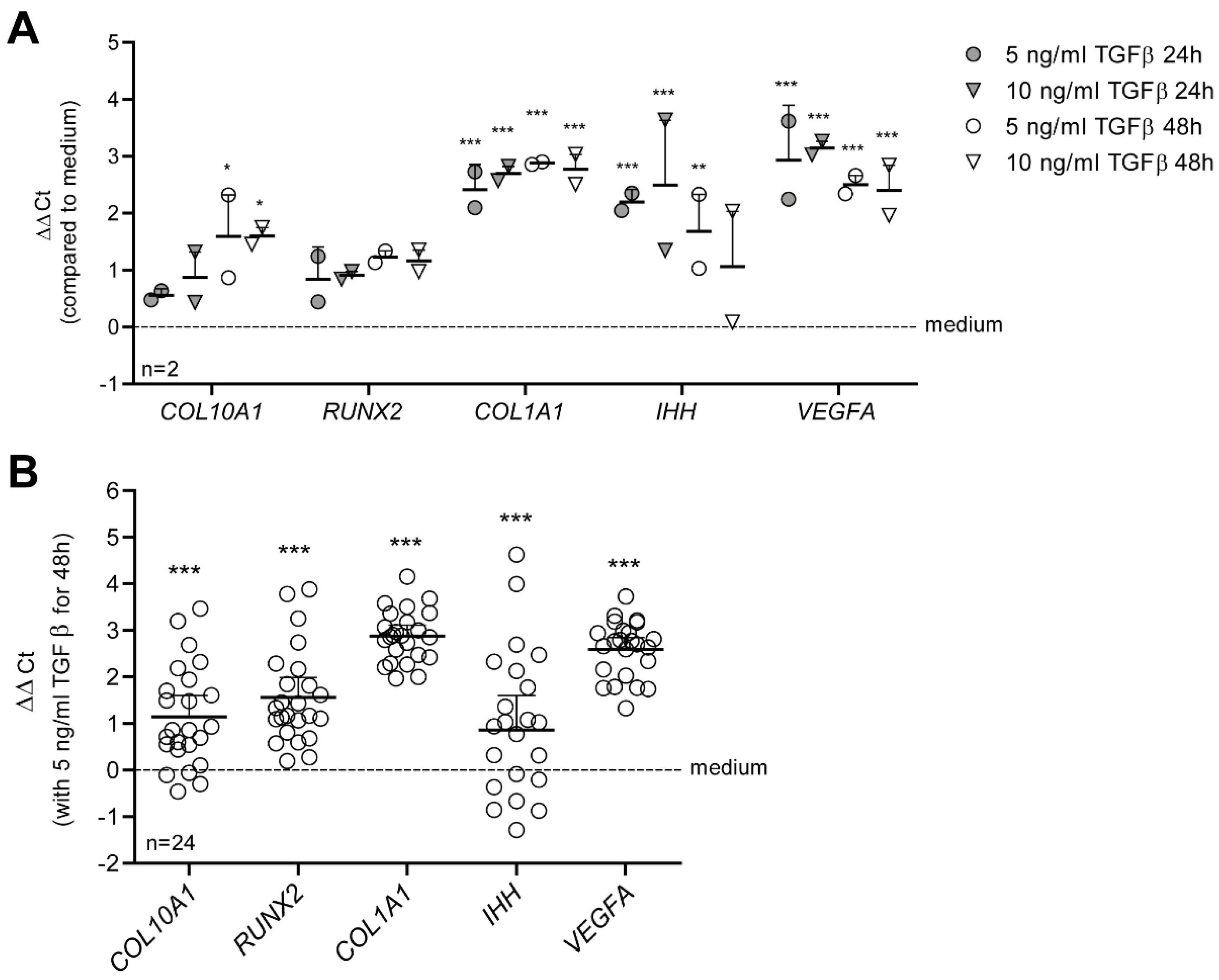
3.1. TGF-β Drives a Hypertrophy-like Phenotype in Primary Human OA Chondrocytes
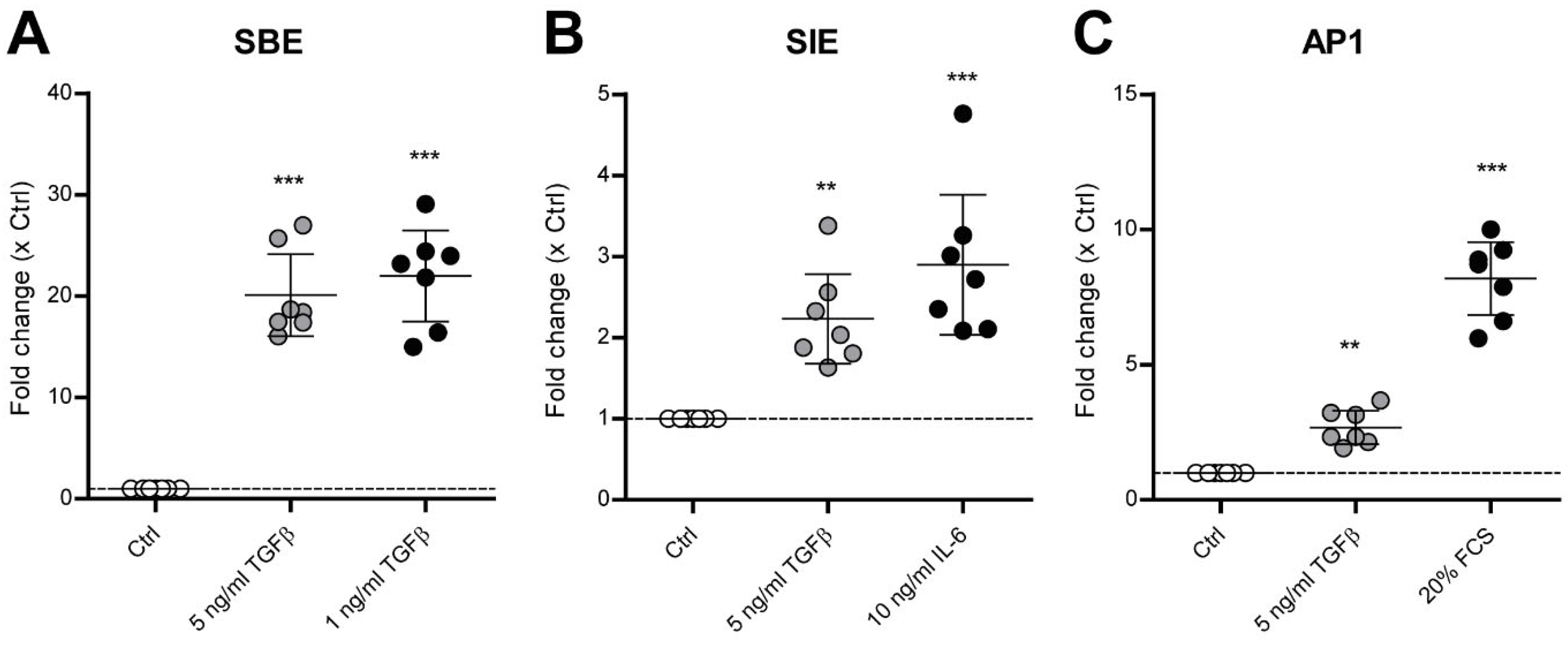
3.2. TGF-β Activates SMAD3:4, STAT3 and AP1 Signaling in Human OA Chondrocytes
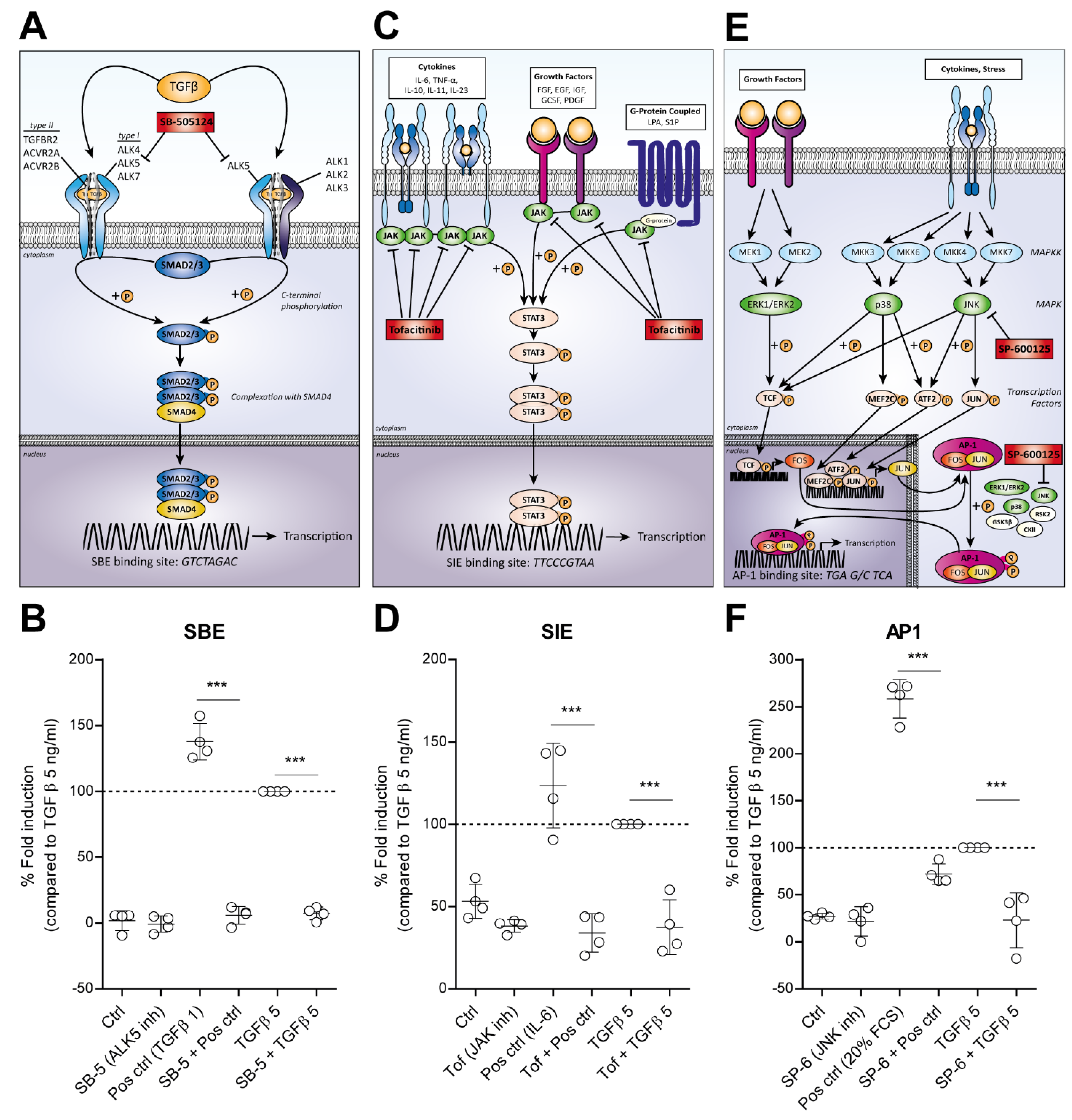
3.3. TGF-β Activated SMAD3:4, STAT3 and AP1 Signaling Is Blocked by Specific Inhibitors
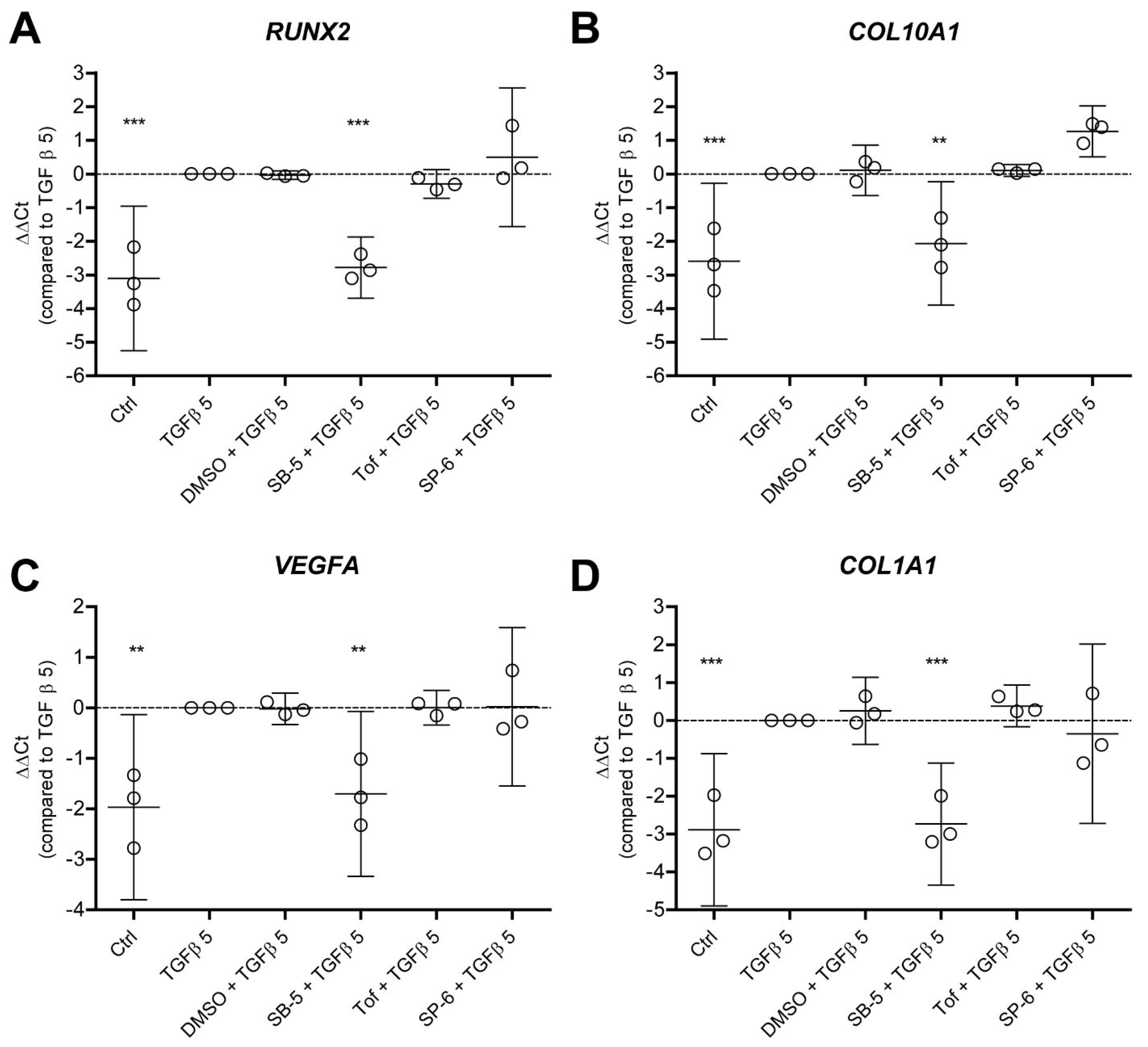
3.4. TGF-β-Driven Hypertrophy-like Phenotype in OA Chondrocytes Is Dependent on ALK5 and Is Not Reliant on JAK and JNK
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mariani, E.; Pulsatelli, L.; Facchini, A. Signaling Pathways in Cartilage Repair. Int. J. Mol. Sci. 2014, 15, 8667–8698. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M.; Van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripmeester, E.G.J.; Timur, U.T.; Caron, M.M.J.; Welting, T.J.M. Recent Insights into the Contribution of the Changing Hypertrophic Chondrocyte Phenotype in the Development and Progression of Osteoarthritis. Front. Bioeng. Biotechnol. 2018, 6, 18. [Google Scholar] [CrossRef]
- Singh, P.; Marcu, K.B.; Goldring, M.B.; Otero, M. Phenotypic instability of chondrocytes in osteoarthritis: On a path to hypertrophy. Ann. New York Acad. Sci. 2018, 1442, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.K.; Park, H.-R.; Cho, H.-J.; Jang, J.-A.; Lee, E.-J.; Han, M.-S.; Kim, G.-W.; Han, S. Degrading products of chondroitin sulfate can induce hypertrophy-like changes and MMP-13/ADAMTS5 production in chondrocytes. Sci. Rep. 2019, 9, 15846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thielen, N.G.M.; van der Kraan, P.M.; van Caam, A.P.M. TGFbeta/BMP Signaling Pathway in Cartilage Homeostasis. Cells 2019, 8, 969. [Google Scholar] [CrossRef] [Green Version]
- Van der Kraan, P.M. Differential Role of Transforming Growth Factor-beta in an Osteoarthritic or a Healthy Joint. J. Bone Metab. 2018, 25, 65–72. [Google Scholar] [CrossRef]
- Zhong, L.; Huang, X.; Karperien, M.; Post, J.N. The Regulatory Role of Signaling Crosstalk in Hypertrophy of MSCs and Human Articular Chondrocytes. Int. J. Mol. Sci. 2015, 16, 19225–19247. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.G.; Thuillier, D.; Chin, E.N.; Alliston, T. Chondrocyte-intrinsic Smad3 represses Runx2-inducible matrix metalloproteinase 13 expression to maintain articular cartilage and prevent osteoarthritis. Arthritis Care Res. 2012, 64, 3278–3289. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.O.; Sampson, E.R.; Maynard, R.D.; O’Keefe, R.J.; Chen, D.; Drissi, H.; Rosier, R.N.; Hilton, M.J.; Zuscik, M.J. Ski inhibits TGF-beta/phospho-Smad3 signaling and accelerates hypertrophic differentiation in chondrocytes. J. Cell Biochem. 2012, 113, 2156–2166. [Google Scholar] [CrossRef] [Green Version]
- Horiki, M.; Imamura, T.; Okamoto, M.; Hayashi, M.; Murai, J.; Myoui, A.; Ochi, T.; Miyazono, K.; Yoshikawa, H.; Tsumaki, N. Smad6/Smurf1 overexpression in cartilage delays chondrocyte hypertrophy and causes dwarfism with osteopenia. J. Cell Biol. 2004, 165, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Javed, A.; Bae, J.-S.; Afzal, F.; Gutierrez, S.; Pratap, J.; Zaidi, S.K.; Lou, Y.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Structural Coupling of Smad and Runx2 for Execution of the BMP2 Osteogenic Signal. J. Biol. Chem. 2008, 283, 8412–8422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leboy, P.; Grasso-Knight, G.; D’Angelo, M.; Volk, S.W.; Lian, J.V.; Drissi, H.; Stein, G.S.; Adams, S.L. Smad-Runx interactions during chondrocyte maturation. J. Bone. Joint. Surg. Am. 2001, 83-A Pt 1 (Suppl. 1), S15–S22. [Google Scholar] [CrossRef] [Green Version]
- Li, T.F.; O’Keefe, R.J.; Chen, D. TGF-beta signaling in chondrocytes. Front. Biosci. 2005, 10, 681–688. [Google Scholar] [CrossRef]
- Qiao, B.; Padilla, S.R.; Benya, P.D. Transforming growth factor (TGF)-beta-activated kinase 1 mimics and mediates TGF-beta-induced stimulation of type II collagen synthesis in chondrocytes independent of Col2a1 transcription and Smad3 signaling. J. Biol. Chem. 2005, 280, 17562–17571. [Google Scholar] [CrossRef] [Green Version]
- Coricor, G.; Serra, R. TGF-beta regulates phosphorylation and stabilization of Sox9 protein in chondrocytes through p38 and Smad dependent mechanisms. Sci. Rep. 2016, 6, 38616. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.F.; Soung, D.Y.; Chang, Y.; Enomoto-Iwamoto, M.; Paris, M.; O’Keefe, R.J.; Schwarz, E.M.; Drissi, H. Transforming growth factor-beta and Wnt signals regulate chondrocyte differentiation through Twist1 in a stage-specific manner. Mol. Endocrinol. 2007, 21, 2805–2820. [Google Scholar] [CrossRef] [Green Version]
- Ionescu, A.M.; Schwarz, E.M.; Zuscik, M.J.; Drissi, H.; Puzas, J.E.; Rosier, R.N.; O’Keefe, R.J. ATF-2 cooperates with Smad3 to mediate TGF-beta effects on chondrocyte maturation. Exp. Cell. Res. 2003, 288, 198–207. [Google Scholar] [CrossRef]
- Van den Bosch, M.H.; Blom, A.B.; van Lent, P.L.; van Beuningen, H.M.; Davidson, E.N.B.; van der Kraan, P.M.; van den Berg, W.B. Canonical Wnt signaling skews TGF-beta signaling in chondrocytes towards signaling via ALK1 and Smad 1/5/8. Cell. Signal. 2014, 26, 951–958. [Google Scholar] [CrossRef]
- Van Beuningen, H.M.; Van Der Kraan, P.M.; Arntz, O.J.; Berg, W.B.V.D. Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. Lab. Investig. 1994, 71, 279–290. [Google Scholar]
- Bakker, A.; van de Loo, F.; van Beuningen, H.; Sime, P.; van Lent, P.; van der Kraan, P.; Richards, C.; Berg, W.V.D. Overexpression of active TGF-beta-1 in the murine knee joint: Evidence for synovial-layer-dependent chondro-osteophyte formation. Osteoarthr. Cartil. 2001, 9, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharstuhl, A.; Glansbeek, H.L.; van Beuningen, H.M.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Inhibition of endogenous TGF-beta during experimental osteoarthritis prevents osteophyte formation and impairs cartilage repair. J. Immunol. 2002, 169, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Q.; Zheng, Y.; Zhang, G.; Hu, Y.; Fan, X.; Hou, Y.; Wen, L.; Li, L.; Xu, Y.; Wang, Y.; et al. Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann. Rheum. Dis. 2019, 78, 100–110. [Google Scholar] [CrossRef]
- Shen, J.; Li, S.; Chen, D. TGF-beta signaling and the development of osteoarthritis. Bone Res. 2014, 2, 14002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.; Wan, F.; Zhou, Z.; Tao, R.; Lu, Y.; Zhou, M.; Liu, F.; Liu, Y. Identification of key regulators responsible for dysregulated networks in osteoarthritis by large-scale expression analysis. J. Orthop. Surg. Res. 2021, 16, 259. [Google Scholar] [CrossRef]
- Boer, C.G.; Hatzikotoulas, K.; Southam, L.; Stefánsdóttir, L.; Zhang, Y.; Coutinho de Almeida, R.; Wu, T.T.; Zheng, J.; Hartley, A.; Teder-Laving, M.; et al. Deciphering osteoarthritis genetics across 826,690 individuals from 9 populations. Cell 2021, 184, 4784–4818.e17. [Google Scholar] [CrossRef] [PubMed]
- Fava, R.; Olsen, N.; Keski-Oja, J.; Moses, H.; Pincus, T. Active and latent forms of transforming growth factor beta activity in synovial effusions. J. Exp. Med. 1989, 169, 291–296. [Google Scholar] [CrossRef]
- Koli, K.; Saharinen, J.; Hyytiäinen, M.; Penttinen, C.; Keski-Oja, J. Latency, activation, and binding proteins of TGF-beta. Microsc. Res. Tech. 2001, 52, 354–362. [Google Scholar] [CrossRef]
- Jiang, Q.; Qiu, Y.-T.; Chen, M.-J.; Zhang, Z.-Y.; Yang, C. Synovial TGF-beta1 and MMP-3 levels and their correlation with the progression of temporomandibular joint osteoarthritis combined with disc displacement: A preliminary study. Biomed. Rep. 2013, 1, 218–222. [Google Scholar] [CrossRef] [Green Version]
- Sandy, J.; Chan, D.; Trevino, R.; Wimmer, M.; Plaas, A. Human genome-wide expression analysis reorients the study of inflammatory mediators and biomechanics in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1939–1945. [Google Scholar] [CrossRef] [Green Version]
- Zielinski, S.; Bartels, K.; Cebulski, K.; Kühne, C.; Kekow, J. Evidence of proteolytic activation of transforming growth factor beta in synovial fluid. Adv. Exp. Med. Biol. 2000, 477, 477–481. [Google Scholar] [PubMed]
- Gao, X.; Sun, Y.; Li, X. Identification of key gene modules and transcription factors for human osteoarthritis by weighted gene co-expression network analysis. Exp. Ther. Med. 2019, 18, 2479–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisch, K.; Gamini, R.; Alvarez-Garcia, O.; Akagi, R.; Saito, M.; Muramatsu, Y.; Sasho, T.; Koziol, J.; Su, A.I.; Lotz, M. Identification of transcription factors responsible for dysregulated networks in human osteoarthritis cartilage by global gene expression analysis. Osteoarthr. Cartil. 2018, 26, 1531–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neefjes, M.; van Caam, A.P.M.; van der Kraan, P.M. Transcription Factors in Cartilage Homeostasis and Osteoarthritis. Biology 2020, 9, 290. [Google Scholar] [CrossRef] [PubMed]
- Neefjes, M.; Housmans, B.A.C.; van den Akker, G.G.H.; van Rhijn, L.W.; Welting, T.J.M.; van der Kraan, P.M. Reporter gene comparison demonstrates interference of complex body fluids with secreted luciferase activity. Sci. Rep. 2021, 11, 1359. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.-X.; Zou, F.-M.; Li, Y.; Liu, A.-M.; Tu, M. JNK pathway in osteoarthritis: Pathological and therapeutic aspects. J. Recept. Signal Transduct. 2017, 37, 431–436. [Google Scholar] [CrossRef]
- Kameda, T.; Watanabe, H.; Iba, H. C-Jun and JunD suppress maturation of chondrocytes. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1997, 8, 495–503. [Google Scholar]
- Ionescu, A.M.; Schwarz, E.M.; Vinson, C.; Puzas, J.E.; Rosier, R.; Reynolds, P.R.; O’Keefe, R.J. PTHrP Modulates Chondrocyte Differentiation through AP-1 and CREB Signaling. J. Biol. Chem. 2001, 276, 11639–11647. [Google Scholar] [CrossRef] [Green Version]
- Moritani, N.H.; Kubota, S.; Eguchi, T.; Fukunaga, T.; Yamashiro, T.; Takano-Yamamoto, T.; Tahara, H.; Ohyama, K.; Sugahara, T.; Takigawa, M. Interaction of AP-1 and the ctgf gene: A possible driver of chondrocyte hypertrophy in growth cartilage. J. Bone Miner. Metab. 2003, 21, 205–210. [Google Scholar]
- Haseeb, A.; Kc, R.; Angelozzi, M.; de Charleroy, C.; Rux, D.; Tower, R.J.; Yao, L.; da Silva, R.P.; Pacifici, M.; Qin, L.; et al. SOX9 keeps growth plates and articular cartilage healthy by inhibiting chondrocyte dedifferentiation/osteoblastic redifferentiation. Proc. Natl. Acad. Sci. USA 2021, 118, e2019152118. [Google Scholar] [CrossRef]
- Liang, B.; Mamidi, M.K.; Samsa, W.E.; Chen, Y.; Lee, B.; Zheng, Q.; Zhou, G. Targeted and sustained Sox9 expression in mouse hypertrophic chondrocytes causes severe and spontaneous osteoarthritis by perturbing cartilage homeostasis. Am. J. Transl. Res. 2020, 12, 1056–1069. [Google Scholar] [PubMed]
- Ikegami, D.; Akiyama, H.; Suzuki, A.; Nakamura, T.; Nakano, T.; Yoshikawa, H.; Tsumaki, N. Sox9 sustains chondrocyte survival and hypertrophy in part through Pik3ca-Akt pathways. Development 2011, 138, 1507–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.C.; Jo, J.; Park, J.; Kang, H.K.; Park, Y. NF-kappaB Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells 2019, 8, 734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Tanaka, S. Molecular mechanisms underlying osteoarthritis development: Notch and NF-kappaB. Arthritis Res. Ther. 2017, 19, 94. [Google Scholar] [CrossRef]
- Olivotto, E.; Otero, M.; Marcu, K.B.; Goldring, M.B. Pathophysiology of osteoarthritis: Canonical NF-kappaB/IKKbeta-dependent and kinase-independent effects of IKKalpha in cartilage degradation and chondrocyte differentiation. RMD Open 2015, 1 (Suppl. 1), e000061. [Google Scholar] [CrossRef] [Green Version]
- Sassi, N.; Gadgadi, N.; Laadhar, L.; Allouche, M.; Mourali, S.; Zandieh-Doulabi, B.; Hamdoun, M.; Nulend, J.K.; Makni, S.; Sellami, S. Notch signaling is involved in human articular chondrocytes de-differentiation during osteoarthritis. J. Recept. Signal Transduct. 2013, 34, 48–57. [Google Scholar] [CrossRef]
- Sassi, N.; Laadhar, L.; Driss, M.; Kallel-Sellami, M.; Sellami, S.; Makni, S. The role of the Notch pathway in healthy and osteoarthritic articular cartilage: From experimental models to ex vivo studies. Arthritis Res. Ther. 2011, 13, 208. [Google Scholar] [CrossRef] [Green Version]
- Xiao, D.; Bi, R.; Liu, X.; Mei, J.; Jiang, N.; Zhu, S. Notch Signaling Regulates MMP-13 Expression via Runx2 in Chondrocytes. Sci. Rep. 2019, 9, 15596. [Google Scholar] [CrossRef]
- Zieba, J.T.; Chen, Y.-T.; Lee, B.H.; Bae, Y. Notch Signaling in Skeletal Development, Homeostasis and Pathogenesis. Biomolecules 2020, 10, 332. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, E.; Seibel, M.J.; Zhou, H. Arthritis and the role of endogenous glucocorticoids. Bone Res. 2020, 8, 33. [Google Scholar] [CrossRef]
- Nalesso, G.; Sherwood, J.; Bertrand, J.; Pap, T.; Ramachandran, M.; De Bari, C.; Pitzalis, C.; Dell’Accio, F. WNT-3A modulates articular chondrocyte phenotype by activating both canonical and noncanonical pathways. J. Cell Biol. 2011, 193, 551–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savvidou, O.; Milonaki, M.; Goumenos, S.; Flevas, D.; Papagelopoulos, P.; Moutsatsou, P. Glucocorticoid signaling and osteoarthritis. Mol. Cell. Endocrinol. 2019, 480, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Koenen, M.; Schauer, S.; Wittig-Blaich, S.; Ahmad, M.; Baschant, U.; Tuckermann, J.P. Molecular Actions of Glucocorticoids in Cartilage and Bone During Health, Disease, and Steroid Therapy. Physiol. Rev. 2016, 96, 409–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, J.; Zhang, P.; Ji, Z.; Henneicke, H.; Li, J.; Kim, S.; Swarbrick, M.M.; Wu, Y.; Little, C.B.; Seibel, M.J.; et al. Disruption of glucocorticoid signalling in osteoblasts attenuates age-related surgically induced osteoarthritis. Osteoarthr. Cartil. 2019, 27, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Latourte, A.; Cherifi, C.; Maillet, J.; Ea, H.-K.; Bouaziz, W.; Funck-Brentano, T.; Cohen-Solal, M.; Hay, E.; Richette, P. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann. Rheum. Dis. 2017, 76, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Held, A.; Glas, A.; Dietrich, L.; Bollmann, M.; Brandstädter, K.; Grossmann, T.N.; Lohmann, C.H.; Pap, T.; Bertrand, J. Targeting beta-catenin dependent Wnt signaling via peptidomimetic inhibitors in murine chondrocytes and OA cartilage. Osteoarthr. Cartil. 2018, 26, 818–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.-G.; Yu, S.-S.; Lee, S.-W.; Chun, J.-S. Wnt-3a regulates chondrocyte differentiation via c-Jun/AP-1 pathway. FEBS Lett. 2005, 579, 4837–4842. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Koga, T.; Isobe, M.; Kern, B.E.; Yokochi, T.; Chin, Y.E.; Karsenty, G.; Taniguchi, T.; Takayanagi, H. Stat1 functions as a cytoplasmic attenuator of Runx2 in the transcriptional program of osteoblast differentiation. Genes Dev. 2003, 17, 1979–1991. [Google Scholar] [CrossRef] [Green Version]
- Damerau, A.; Gaber, T.; Ohrndorf, S.; Hoff, P. JAK/STAT Activation: A General Mechanism for Bone Development, Homeostasis, and Regeneration. Int. J. Mol. Sci. 2020, 21, 9004. [Google Scholar] [CrossRef]
- Kondo, M.; Yamaoka, K.; Sakata, K.; Sonomoto, K.; Lin, L.; Nakano, K.; Tanaka, Y. Contribution of the Interleukin-6/STAT-3 Signaling Pathway to Chondrogenic Differentiation of Human Mesenchymal Stem Cells. Arthritis Rheumatol. 2015, 67, 1250–1260. [Google Scholar] [CrossRef]
- Litherland, G.J.; Elias, M.S.; Hui, W.; Macdonald, C.D.; Catterall, J.B.; Barter, M.J.; Farren, M.J.; Jefferson, M.; Rowan, A.D. Protein kinase C isoforms zeta and iota mediate collagenase expression and cartilage destruction via STAT3- and ERK-dependent c-fos induction. J. Biol. Chem. 2010, 285, 22414–22425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.Q.; Lin, Y.; Li, L.; Lu, J.; Geng, D.; Zhang, J.; Jashashvili, T.; Buser, Z.; Magallanes, J.; Tassey, J.; et al. gp130/STAT3 signaling is required for homeostatic proliferation and anabolism in postnatal growth plate and articular chondrocytes. Commun. Biol. 2022, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Housmans, B.; Neefjes, M.; Surtel, D.; Vitík, M.; Cremers, A.; van Rhijn, L.; van der Kraan, P.; Akker, G.v.D.; Welting, T. Synovial fluid from end-stage osteoarthritis induces proliferation and fibrosis of articular chondrocytes via MAPK and RhoGTPase signaling. Osteoarthr. Cartil. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Prasadam, I.; van Gennip, S.; Friis, T.; Shi, W.; Crawford, R.; Xiao, Y. ERK-1/2 and p38 in the regulation of hypertrophic changes of normal articular cartilage chondrocytes induced by osteoarthritic subchondral osteoblasts. Arthritis Rheum. 2010, 62, 1349–1360. [Google Scholar] [CrossRef] [Green Version]
- Öztürk, E.; Despot-Slade, E.; Pichler, M.; Zenobi-Wong, M. RhoA activation and nuclearization marks loss of chondrocyte phenotype in crosstalk with Wnt pathway. Exp. Cell Res. 2017, 360, 113–124. [Google Scholar] [CrossRef]
- Kubo, Y.; Beckmann, R.; Fragoulis, A.; Conrads, C.; Pavanram, P.; Nebelung, S.; Wolf, M.; Wruck, C.J.; Jahr, H.; Pufe, T. Nrf2/ARE Signaling Directly Regulates SOX9 to Potentially Alter Age-Dependent Cartilage Degeneration. Antioxidants 2022, 11, 263. [Google Scholar] [CrossRef]
- Morita, K.; Miyamoto, T.; Fujita, N.; Kubota, Y.; Ito, K.; Takubo, K.; Miyamoto, K.; Ninomiya, K.; Suzuki, T.; Iwasaki, R.; et al. Reactive oxygen species induce chondrocyte hypertrophy in endochondral ossification. J. Exp. Med. 2007, 204, 1613–1623. [Google Scholar] [CrossRef]
- Bolduc, J.A.; Collins, J.A.; Loeser, R.F. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic. Biol. Med. 2019, 132, 73–82. [Google Scholar] [CrossRef]
- Erickson, G.R.; Alexopoulos, L.G.; Guilak, F. Hyper-osmotic stress induces volume change and calcium transients in chondrocytes by transmembrane, phospholipid, and G-protein pathways. J. Biomech. 2001, 34, 1527–1535. [Google Scholar] [CrossRef]
- Van der Windt, A.E.; Haak, E.; Das, R.H.J.; Kops, N.; Welting, T.J.M.; Caron, M.M.J.; van Til, N.P.; Verhaar, J.A.N.; Weinans, H.; Jahr, H. Physiological tonicity improves human chondrogenic marker expression through nuclear factor of activated T-cells 5 in vitro. Arthritis Res. Ther. 2010, 12, R100. [Google Scholar] [CrossRef] [Green Version]
- Johnson, Z.I.; Shapiro, I.M.; Risbud, M.V. Extracellular osmolarity regulates matrix homeostasis in the intervertebral disc and articular cartilage: Evolving role of TonEBP. Matrix Biol. 2014, 40, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Francois, M.; Richette, P.; Tsagris, L.; Raymondjean, M.; Fulchignoni-Lataud, M.-C.; Forest, C.; Savouret, J.-F.; Corvol, M.-T. Peroxisome proliferator-activated receptor-gamma down-regulates chondrocyte matrix metalloproteinase-1 via a novel composite element. J. Biol. Chem. 2004, 279, 28411–28418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poleni, P.E.; Bianchi, A.; Etienne, S.; Koufany, M.; Sebillaud, S.; Netter, P.; Terlain, B.; Jouzeau, J.Y. Agonists of peroxisome proliferators-activated receptors (PPAR) alpha, beta/delta or gamma reduce transforming growth factor (TGF)-beta-induced proteoglycans’ production in chondrocytes. Osteoarthr. Cartil. 2007, 15, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Poleni, P.E.; Etienne, S.; Velot, E.; Netter, P.; Bianch, A. Activation of PPARs alpha, beta/delta, and gamma Impairs TGF-beta1-Induced Collagens’ Production and Modulates the TIMP-1/MMPs Balance in Three-Dimensional Cultured Chondrocytes. PPAR Res. 2010, 2010, 635912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Huang, X.; Karperien, M.; Post, J.N. Correlation between Gene Expression and Osteoarthritis Progression in Human. Int. J. Mol. Sci. 2016, 17, 1126. [Google Scholar] [CrossRef] [Green Version]
- Truong, L.-H.; Kuliwaba, J.S.; Tsangari, H.; Fazzalari, N.L. Differential gene expression of bone anabolic factors and trabecular bone architectural changes in the proximal femoral shaft of primary hip osteoarthritis patients. Arthritis Res. Ther. 2006, 8, R188. [Google Scholar] [CrossRef] [Green Version]
- DaCosta Byfield, S.; Major, C.; Laping, N.J.; Roberts, A.B. SB-505124 is a selective inhibitor of transforming growth factor-beta type I receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2004, 65, 744–752. [Google Scholar] [CrossRef]
- Lau, Y.-T.K.; Ramaiyer, M.; Johnson, D.E.; Grandis, J.R. Targeting STAT3 in Cancer with Nucleotide Therapeutics. Cancers 2019, 11, 1681. [Google Scholar] [CrossRef] [Green Version]
- Adis Editorial. Tofacitinib. Drugs R. D. 2010, 10, 271–284. [Google Scholar]
- Gazon, H.; Barbeau, B.; Mesnard, J.-M.; Peloponese, J.-M., Jr. Hijacking of the AP-1 Signaling Pathway during Development of ATL. Front. Microbiol. 2017, 8, 2686. [Google Scholar] [CrossRef] [Green Version]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Albro, M.B.; Nims, R.J.; Cigan, A.D.; Yeroushalmi, K.J.; Shim, J.J.; Hung, C.T.; Ateshian, G.A. Dynamic mechanical compression of devitalized articular cartilage does not activate latent TGF-beta. J. Biomech. 2013, 46, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Ther. Adv. Musculoskelet. Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Remst, D.F.G.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.-J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, E.N.B.; Scharstuhl, A.; Vitters, E.L.; Van Der Kraan, P.M.; Berg, W.B.V.D. Reduced transforming growth factor-beta signaling in cartilage of old mice: Role in impaired repair capacity. Arthritis Res. Ther. 2005, 7, R1338–R1347. [Google Scholar] [CrossRef] [Green Version]
- Cherifi, C.; Monteagudo, S.; Lories, R.J. Promising targets for therapy of osteoarthritis: A review on the Wnt and TGF-beta signalling pathways. Ther. Adv. Musculoskelet. Dis. 2021, 13, 1759720X211006959. [Google Scholar] [CrossRef]
- Van der Kraan, P.M. The changing role of TGFbeta in healthy, ageing and osteoarthritic joints. Nat. Rev. Rheumatol. 2017, 13, 155–163. [Google Scholar] [CrossRef]
- Allas, L.; Rochoux, Q.; Leclercq, S.; Boumediene, K.; Baugé, C. Development of a simple osteoarthritis model useful to predict in vitro the anti-hypertrophic action of drugs. Lab. Investig. 2020, 100, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Babur, B.K.; Futrega, K.; Lott, W.; Klein, T.; Cooper-White, J.; Doran, M.R. High-throughput bone and cartilage micropellet manufacture, followed by assembly of micropellets into biphasic osteochondral tissue. Cell Tissue Res. 2015, 361, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Antoniou, J.; Wang, H.T.; Alaseem, A.M.; Haglund, L.; Roughley, P.J.; Mwale, F. The effect of Link N on differentiation of human bone marrow-derived mesenchymal stem cells. Arthritis Res. Ther. 2012, 14, R267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, M.B.; Tuan, R.S. Functional characterization of hypertrophy in chondrogenesis of human mesenchymal stem cells. Arthritis Care Res. 2008, 58, 1377–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirsch, T.; von der Mark, K. Remodelling of collagen types I, II and X and calcification of human fetal cartilage. Bone Miner. 1992, 18, 107–117. [Google Scholar] [CrossRef]
- Nurminskaya, M.; Linsenmayer, T.F. Identification and characterization of up-regulated genes during chondrocyte hypertrophy. Dev. Dyn. 1996, 206, 260–271. [Google Scholar] [CrossRef]
- Li, J.; Dong, S. The Signaling Pathways Involved in Chondrocyte Differentiation and Hypertrophic Differentiation. Stem Cells Int. 2016, 2016, 2470351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, H.; Chen, H.; Javed, A. Runx2 is required for hypertrophic chondrocyte mediated degradation of cartilage matrix during endochondral ossification. Matrix Biol. Plus 2021, 12, 10008. [Google Scholar] [CrossRef] [PubMed]
- Solomon, L.A.; Bérubé, N.G.; Beier, F. Transcriptional regulators of chondrocyte hypertrophy. Birth Defects Res. Part C Embryo Today Rev. 2008, 84, 123–130. [Google Scholar] [CrossRef]
- Amano, K.; Densmore, M.; Nishimura, R.; Lanske, B. Indian hedgehog signaling regulates transcription and expression of collagen type X via Runx2/Smads interactions. J. Biol. Chem. 2014, 289, 24898–24910. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wei, X.; Wei, L. Indian Hedgehog, a critical modulator in osteoarthritis, could be a potential therapeutic target for attenuating cartilage degeneration disease. Connect. Tissue Res. 2014, 55, 257–261. [Google Scholar] [CrossRef]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of Rate of Cartilage Differentiation by Indian Hedgehog and PTH-Related Protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef]
- Gerber, H.-P.; Vu, T.H.; Ryan, A.M.; Kowalski, J.; Werb, Z.; Ferrara, N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999, 5, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Nagao, M.; Hamilton, J.L.; Kc, R.; Berendsen, A.D.; Duan, X.; Cheong, C.W.; Li, X.; Im, H.-J.; Olsen, B.R. Vascular Endothelial Growth Factor in Cartilage Development and Osteoarthritis. Sci. Rep. 2017, 7, 13027. [Google Scholar] [CrossRef] [PubMed]
- Takano, S.; Uchida, K.; Shoji, S.; Itakura, M.; Iwase, D.; Aikawa, J.; Mukai, M.; Sekiguchi, H.; Inoue, G.; Takaso, M. Vascular Endothelial Growth Factor Is Regulated by the Canonical and Noncanonical Transforming Growth Factor-beta Pathway in Synovial Fibroblasts Derived from Osteoarthritis Patients. Biomed. Res. Int. 2019, 2019, 6959056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlier, E.; Deroyer, C.; Ciregia, F.; Malaise, O.; Neuville, S.; Plener, Z.; Malaise, M.; de Seny, D. Chondrocyte dedifferentiation and osteoarthritis (OA). Biochem. Pharmacol. 2019, 165, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Rim, Y.A.; Ju, J.H. The Role of Fibrosis in Osteoarthritis Progression. Life 2020, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Deroyer, C.; Charlier, E.; Neuville, S.; Malaise, O.; Gillet, P.; Kurth, W.; Chariot, A.; Malaise, M.; De Seny, D. CEMIP (KIAA1199) induces a fibrosis-like process in osteoarthritic chondrocytes. Cell Death Dis. 2019, 10, 103. [Google Scholar] [CrossRef] [Green Version]
- Miosge, N.; Hartmann, M.; Maelicke, C.; Herken, R. Expression of collagen type I and type II in consecutive stages of human osteoarthritis. Histochem. Cell Biol. 2004, 122, 229–236. [Google Scholar]
- Liu, C.F.; Samsa, W.E.; Zhou, G.; Lefebvre, V. Transcriptional control of chondrocyte specification and differentiation. Semin. Cell. Dev. Biol. 2017, 62, 34–49. [Google Scholar] [CrossRef] [Green Version]
- Wiegertjes, R.; van Caam, A.; van Beuningen, H.; Koenders, M.; van Lent, P.; van der Kraan, P.; van de Loo, F.; Davidson, E.B. TGF-beta dampens IL-6 signaling in articular chondrocytes by decreasing IL-6 receptor expression. Osteoarthr. Cartil. 2019, 27, 1197–1207. [Google Scholar] [CrossRef]
- Wiegertjes, R.; Loo, F.A.J.V.D.; Davidson, E.N.B. A roadmap to target interleukin-6 in osteoarthritis. Rheumatology 2020, 59, 2681–2694. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Kontzias, A.; Yamaoka, K.; Tanaka, Y.; Laurence, A. Janus kinase inhibitors in autoimmune diseases. Ann. Rheum. Dis. 2013, 72, ii111–ii115. [Google Scholar] [CrossRef] [PubMed]
- Kjelgaard-Petersen, C.F.; Sharma, N.; Kayed, A.; Karsdal, M.A.; Mobasheri, A.; Hägglund, P.; Bay-Jensen, A.-C.; Thudium, C.S. Tofacitinib and TPCA-1 exert chondroprotective effects on extracellular matrix turnover in bovine articular cartilage ex vivo. Biochem. Pharmacol. 2019, 165, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Blanco, M.F.; Garcia, H.D.; Legeai-Mallet, L.; van Osch, G. Tyrosine kinases regulate chondrocyte hypertrophy: Promising drug targets for Osteoarthritis. Osteoarthr. Cartil. 2021, 29, 1389–1398. [Google Scholar] [CrossRef]
- He, X.; Ohba, S.; Hojo, H.; McMahon, A.P. AP-1 family members act with Sox9 to promote chondrocyte hypertrophy. Development 2016, 143, 3012–3023. [Google Scholar] [CrossRef] [Green Version]
- Hess, J.; Porte, D.; Munz, C.; Ange, P. AP-1 and Cbfa/runt physically interact and regulate parathyroid hormone-dependent MMP13 expression in osteoblasts through a new osteoblast-specific element 2/AP-1 composite element. J. Biol. Chem. 2001, 276, 20029–20038. [Google Scholar] [CrossRef] [Green Version]
- Ye, N.; Ding, Y.; Wild, C.; Shen, Q.; Zhou, J. Small Molecule Inhibitors Targeting Activator Protein 1 (AP-1). J. Med. Chem. 2014, 57, 6930–6948. [Google Scholar] [CrossRef]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; ten Dijke, P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell. 2003, 12, 817–828. [Google Scholar] [CrossRef]
- Qu, Z.; Zhang, F.; Chen, W.; Lin, T.; Sun, Y. High-dose TGF-beta1 degrades human nucleus pulposus cells via ALK1-Smad1/5/8 activation. Exp. Ther. Med. 2020, 20, 3661–3668. [Google Scholar] [PubMed]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell. Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Van Caam, A.; Madej, W.; de Vinuesa, A.G.; Goumans, M.-J.; ten Dijke, P.; Blaney Davidson, E.; van der Kraan, P. TGFbeta1-induced SMAD2/3 and SMAD1/5 phosphorylation are both ALK5-kinase-dependent in primary chondrocytes and mediated by TAK1 kinase activity. Arthritis. Res. Ther. 2017, 19, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korchynskyi, O.; ten Dijke, P. Identification and Functional Characterization of Distinct Critically Important Bone Morphogenetic Protein-specific Response Elements in the Id1 Promoter. J. Biol. Chem. 2002, 277, 4883–4891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilberberg, L.; ten Dijke, P.; Sakai, L.Y.; Rifkin, D.B. A rapid and sensitive bioassay to measure bone morphogenetic protein activity. BMC Cell Biol. 2007, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Garcia de Vinuesa, A.; Sanchez-Duffhues, G.; Blaney-Davidson, E.; van Caam, A.; Lodder, K.; Ramos, Y.; Kloppenburg, M.; Meulenbelt, I.; van der Kraan, P.; Goumans, M.-J.; et al. Cripto favors chondrocyte hypertrophy via TGF-beta SMAD1/5 signaling during development of osteoarthritis. J. Pathol. 2021, 255, 330–342. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | TF(s) | TF Element (Abbreviation Construct) | Positive Control | References |
|---|---|---|---|---|
| MAPK/JNK | c-FOS:c-JUN | Activator Protein 1 response element (AP1) | FCS (20%) | [36,37,38,39] |
| SOX9 | SOX9 | SRY-box transcription factor 9 response element (SOX9) | Forskolin (10 µM) | [40,41,42] |
| NFκB | NFκB:p65 | Nuclear factor κ B response element (NFκB) | IL-1β (1 ng/mL) | [43,44,45] |
| Notch | CSL | CBF1/RBPJκ/Suppressor of Hairless/Lag-1 response element (CSL) | FCS (20%) | [46,47,48,49] |
| cAMP/PKA | CREB | Cyclic AMP response element (CRE) | Forskolin (10 µM) | [41,50,51] |
| Glucocorticoid signaling | Glucocorticoid receptor | Glucocorticoid receptor response element (GRE) | Dexamethasone (10 µM) | [50,52,53,54] |
| TGF-β | SMAD3:SMAD4 | SMAD binding element (SBE) | TGF-β1 (1 ng/mL) | [8,9,55] |
| Wnt | TCF/LEF | T-cell factor/lymphoid enhancer factor family response element (TCF/LEF) | Wnt3a (200 ng/mL) | [51,56,57] |
| INF-α | STAT1:STAT2 | Interferon-stimulated response element (ISRE) | IFN-α (100 ng/mL) | [58,59] |
| IL-6 | STAT3:STAT3 | Sis-inducible element (SIE) | IL-6 (10 ng/mL) | [55,60,61,62] |
| MAPK/ERK | ELK-1:SRF | Serum response element (SRE) | FCS (20%) | [63,64] |
| RhoA | SRF | Serum response factor (SRF) | FCS (20%) | [63,65] |
| Oxidative stress | NRF2 | Antioxidant response element (ARE) | H2O2 (50 µM) | [66,67,68] |
| Calcium/calcineurin: hyperosmotic signaling | NFAT5 | Nuclear factor of activated T-cells 5 response element (NFAT5) | +100 milliosmole | [69,70,71] |
| PPARγ | PPARγ | Peroxisome Proliferator activated receptor-γ response element (PPRE) | Rosiglitazone (20 µM) | [72,73,74] |
| Gene | Forward Sequence | Reverse Sequence |
|---|---|---|
| GAPDH | ATCTTCTTTTGCGTCGCCAG | TTCCCCATGGTGTCTGAGC |
| RPL22 | TCGCTCACCTCCCTTTCTAA | TCACGGTGATCTTGCTCTTG |
| RPS27A | TGGCTGTCCTGAAATATTATAAGGT | CCCCAGCACCACATTCATCA |
| RUNX2 | GCAAGGTTCAACGATCTGAGA | TTCCCGAGGTCCATCTACTG |
| COL10A1 | TTTTACGCTGAACGATACCAAATG | CTGTGTCTTGGTGTTGGGTAGTG |
| COL1A1 | AGATCGAGAACATCCGGAG | AGTACTCTCCACTCTTCCAG |
| VEGFA | CAGGGAAGAGGAGGAGATGAGA | GCTGGGTTTGTCGGTGTTC |
| IHH | CAATTACAATCCAGACATCATCTTCA | CGAGATAGCCAGCGAGTTCAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thielen, N.G.M.; Neefjes, M.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Koenders, M.I.; van Lent, P.L.E.M.; van de Loo, F.A.J.; Blaney Davidson, E.N.; van Caam, A.P.M.; et al. Identification of Transcription Factors Responsible for a Transforming Growth Factor-β-Driven Hypertrophy-like Phenotype in Human Osteoarthritic Chondrocytes. Cells 2022, 11, 1232. https://doi.org/10.3390/cells11071232
Thielen NGM, Neefjes M, Vitters EL, van Beuningen HM, Blom AB, Koenders MI, van Lent PLEM, van de Loo FAJ, Blaney Davidson EN, van Caam APM, et al. Identification of Transcription Factors Responsible for a Transforming Growth Factor-β-Driven Hypertrophy-like Phenotype in Human Osteoarthritic Chondrocytes. Cells. 2022; 11(7):1232. https://doi.org/10.3390/cells11071232
Chicago/Turabian StyleThielen, Nathalie G. M., Margot Neefjes, Elly L. Vitters, Henk M. van Beuningen, Arjen B. Blom, Marije I. Koenders, Peter L. E. M. van Lent, Fons A. J. van de Loo, Esmeralda N. Blaney Davidson, Arjan P. M. van Caam, and et al. 2022. "Identification of Transcription Factors Responsible for a Transforming Growth Factor-β-Driven Hypertrophy-like Phenotype in Human Osteoarthritic Chondrocytes" Cells 11, no. 7: 1232. https://doi.org/10.3390/cells11071232