
Altered Spinal Homeostasis and Maladaptive Plasticity in GFAP Null Mice Following Peripheral Nerve Injury

 , , ,
, , ,  , and
, and 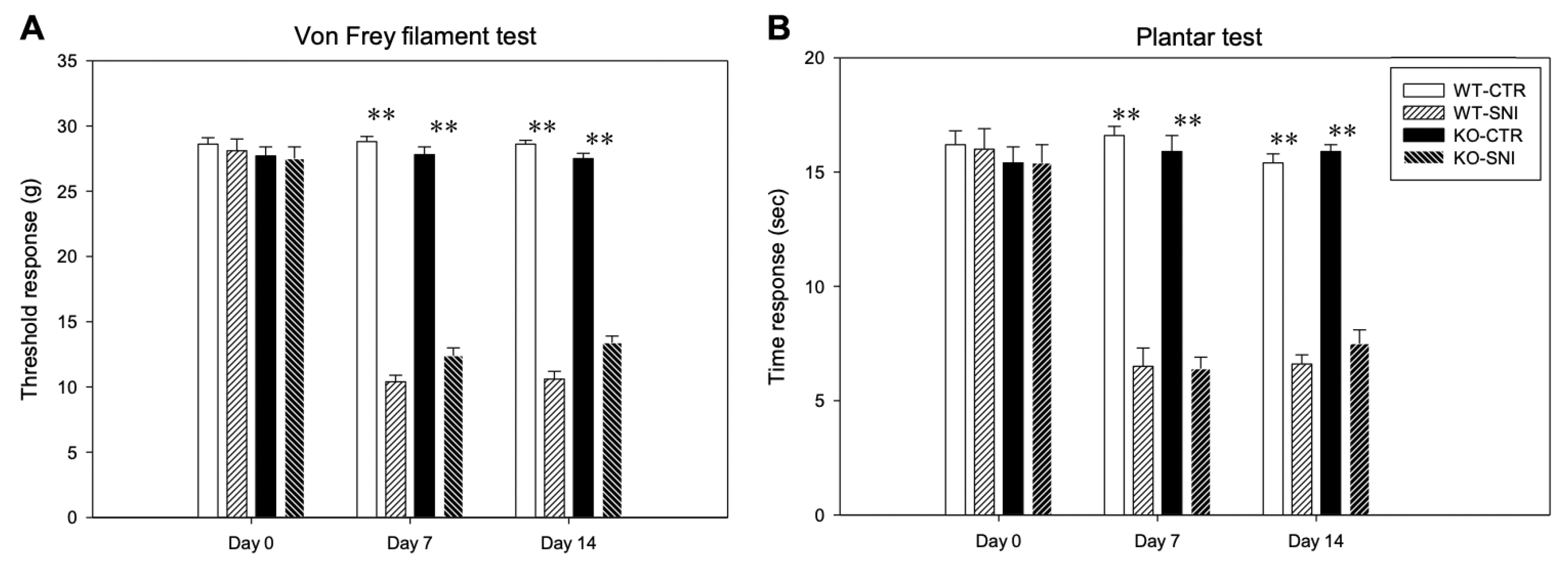{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and SNI Model
2.2. Behavioral Testing
2.3. Tissue Preparation
2.4. Immunohistochemistry
2.5. Western Blotting
2.6. Standard Light and Confocal Microscopy
2.7. Measurements and Statistical Analysis
3. Results
3.1. Behavioral Analysis
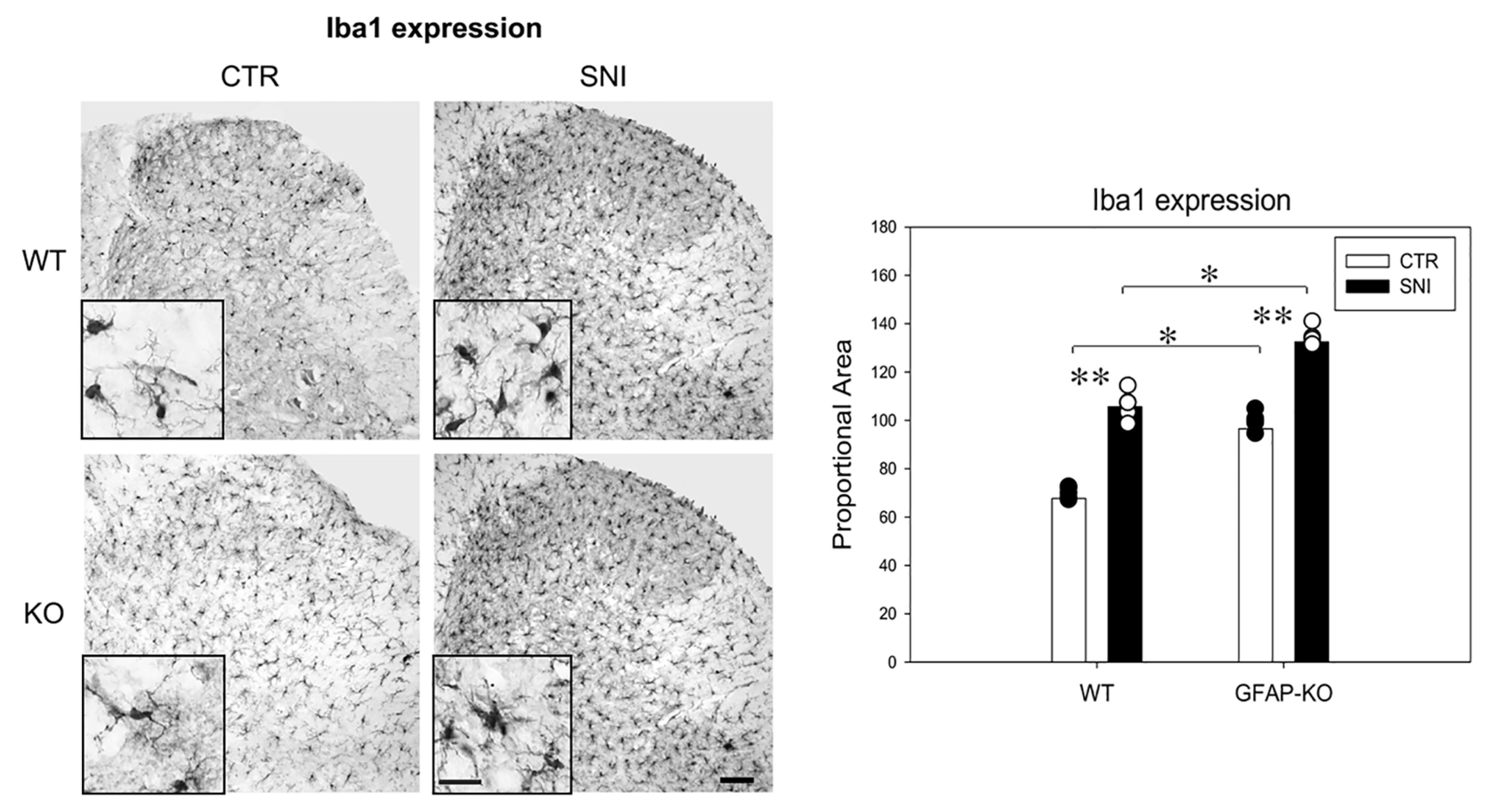
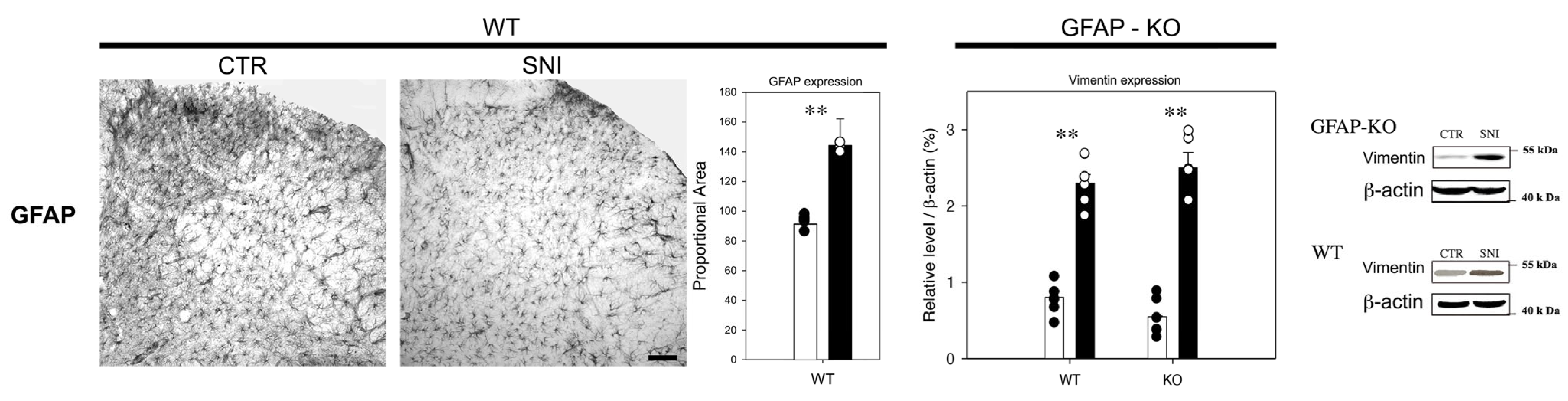
3.2. Reactive Gliosis Induction in the Lumbar Spinal Cord Following SNI
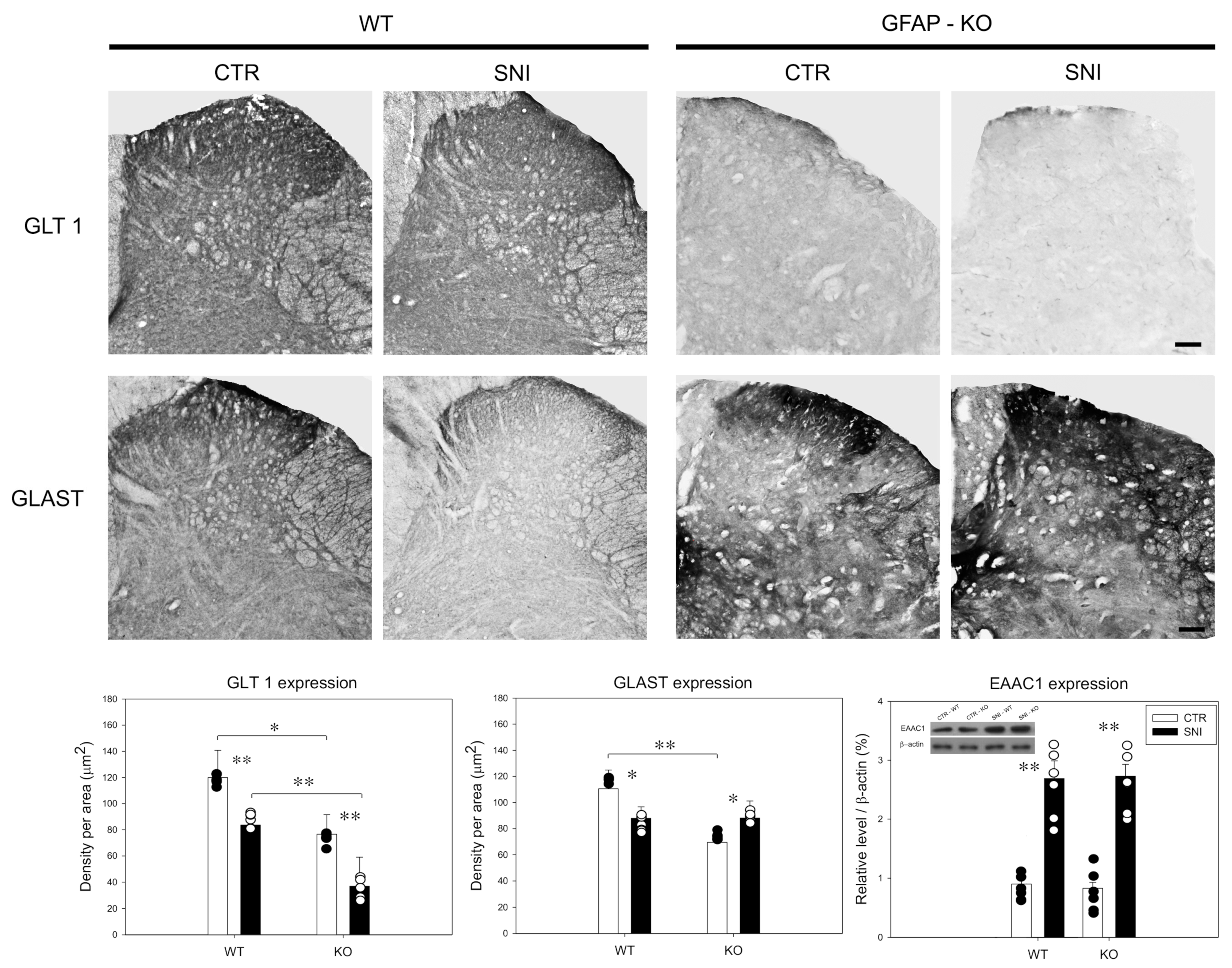
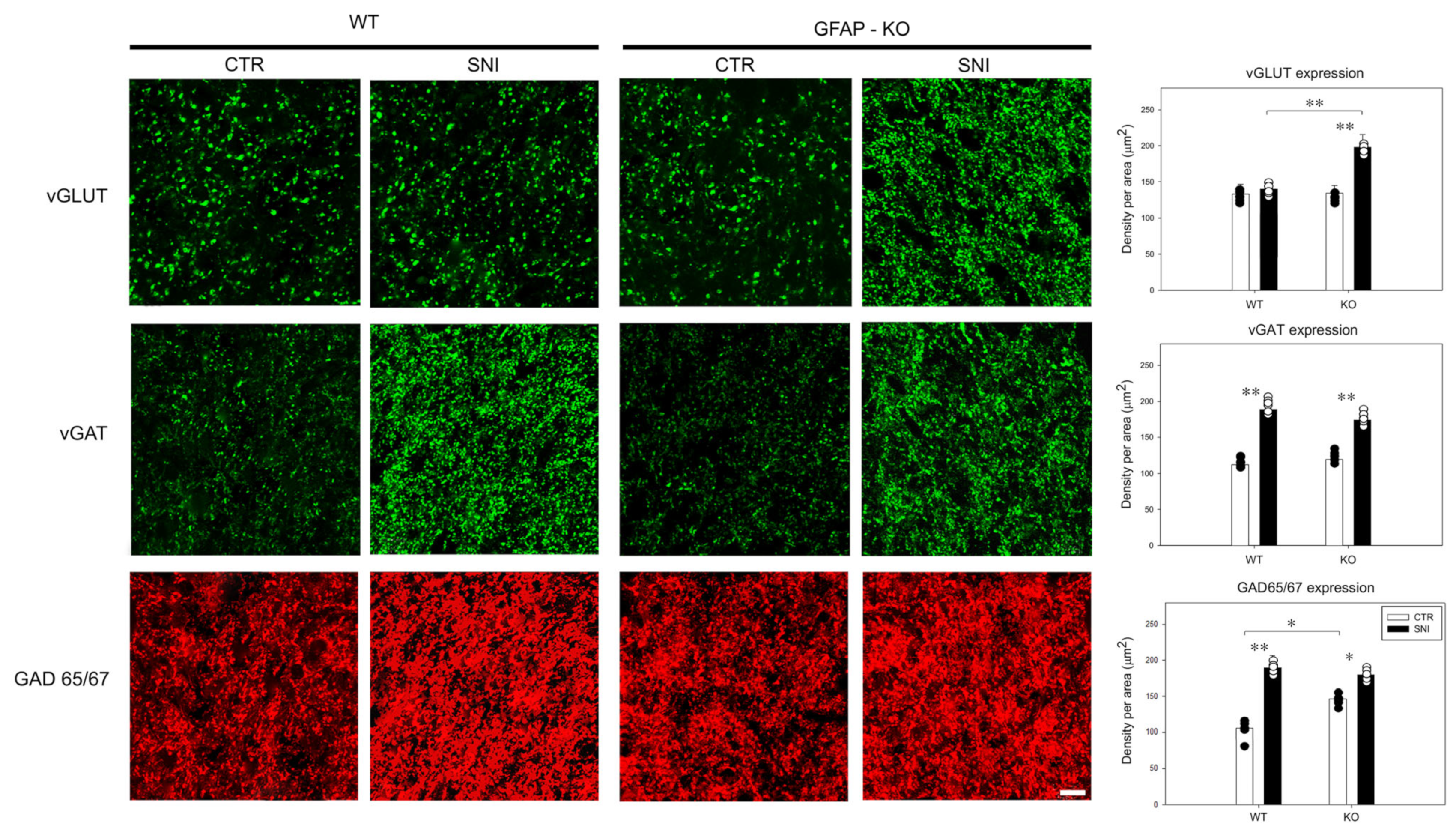
3.3. Remodeling of Glial/Neuronal Glutamate/GABA Transporters in GFAP-KO Animals Following SNI
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Papa, M.; De Luca, C.; Petta, F.; Alberghina, L.; Cirillo, G. Astrocyte-neuron interplay in maladaptive plasticity. Neurosci. Biobehav. Rev. 2014, 42, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M.; Ransom, B.; Goldman, S.A. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003, 26, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Volterra, A.; Meldolesi, J. Astrocytes, from brain glue to communication elements: The revolution continues. Nat. Rev. Neurosci. 2005, 6, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Haydon, P.G. Glia: Listening and talking to the synapse. Nat. Rev. Neurosci. 2001, 2, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Fellin, T.; Carmignoto, G. Neurone-to-astrocyte signalling in the brain represents a distinct multifunctional unit. J. Physiol. 2004, 559, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirillo, G.; De Luca, D.; Papa, M. Calcium Imaging of Living Astrocytes in the Mouse Spinal Cord following Sensory Stimulation. Neural Plast. 2012, 2012, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, G.; Bianco, M.R.; Colangelo, A.M.; Cavaliere, C.; Daniele, D.L.; Zaccaro, L.; Alberghina, L.; Papa, M. Reactive astrocytosis-induced perturbation of synaptic homeostasis is restored by nerve growth factor. Neurobiol. Dis. 2011, 41, 630–639. [Google Scholar] [CrossRef]
- De Luca, C.; Savarese, L.; Colangelo, A.M.; Bianco, M.R.; Cirillo, G.; Alberghina, L.; Papa, M. Astrocytes and Microglia-Mediated Immune Response in Maladaptive Plasticity is Differently Modulated by NGF in the Ventral Horn of the Spinal Cord Following Peripheral Nerve Injury. Cell. Mol. Neurobiol. 2016, 36, 37–46. [Google Scholar] [CrossRef]
- Cirillo, G.; Colangelo, A.M.; Berbenni, M.; Ippolito, V.M.; De Luca, C.; Verdesca, F.; Savarese, L.; Alberghina, L.; Maggio, N.; Papa, M. Purinergic Modulation of Spinal Neuroglial Maladaptive Plasticity Following Peripheral Nerve Injury. Mol. Neurobiol. 2015, 52, 1440–1457. [Google Scholar] [CrossRef]
- Cirillo, G.; Colangelo, A.M.; De Luca, C.; Savarese, L.; Barillari, M.R.; Alberghina, L.; Papa, M. Modulation of matrix metalloproteinases activity in the ventral horn of the spinal cord re-stores neuroglial synaptic homeostasis and neurotrophic support following peripheral nerve injury. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New Roles for the Synaptic Stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcello, L.; Cavaliere, C.; Colangelo, A.M.; Bianco, M.R.; Cirillo, G.; Alberghina, L.; Papa, M. Remodelling of supraspinal neuroglial network in neuropathic pain is featured by a reactive gliosis of the nociceptive amygdala. Eur. J. Pain 2013, 17, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Cragnolini, A.; Lampitella, G.; Virtuoso, A.; Viscovo, I.; Panetsos, F.; Papa, M.; Cirillo, G. Regional brain susceptibility to neurodegeneration: What is the role of glial cells? Neural Regen. Res. 2020, 15, 838. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, G.; Cirillo, M.; Panetsos, F.; Virtuoso, A.; Papa, M. Selective vulnerability of basal ganglia: Insights into the mechanisms of bilateral striatal necrosis. J. Neuropathol. Exp. Neurol. 2019, 78, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Li, B.; Scuderi, C.; Parpura, V. Principles of Astrogliopathology. In Astrocytes in Psychiatric Disorders; Springer: Berlin/Heidelberg, Germany, 2021; pp. 55–73. [Google Scholar]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Potokar, M.; Morita, M.; Wiche, G.; Jorgačevski, J. The Diversity of Intermediate Filaments in Astrocytes. Cells 2020, 9, 1604. [Google Scholar] [CrossRef]
- Brenner, M. Role of GFAP in CNS injuries. Neurosci. Lett. 2014, 565, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Virtuoso, A.; Giovannoni, R.; De Luca, C.; Gargano, F.; Cerasuolo, M.; Maggio, N.; Lavitrano, M.; Papa, M. The Glioblastoma Microenvironment: Morphology, Metabolism, and Molecular Signature of Glial Dynamics to Discover Metabolic Rewiring Sequence. Int. J. Mol. Sci. 2021, 22, 3301. [Google Scholar] [CrossRef]
- Middeldorp, J.; Hol, E.M. GFAP in health and disease. Prog. Neurobiol. 2011, 93, 421–443. [Google Scholar] [CrossRef]
- Hol, E.M.; Pekny, M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr. Opin. Cell Biol. 2015, 32, 121–130. [Google Scholar] [CrossRef]
- Okonkwo, D.O.; Puffer, R.C.; Puccio, A.M.; Yuh, E.L.; Yue, J.K.; Diaz-Arrastia, R.; Korley, F.K.; Wang, K.K.W.; Sun, X.; Taylor, S.R.; et al. Point-of-Care Platform Blood Biomarker Testing of Glial Fibrillary Acidic Protein versus S100 Calcium-Binding Protein B for Prediction of Traumatic Brain Injuries: A Transforming Research and Clinical Knowledge in Traumatic Brain Injury Study. J. Neurotrauma 2020, 37, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Bianco, M.R.; Cirillo, G.; Petrosino, V.; Marcello, L.; Soleti, A.; Merizzi, G.; Cavaliere, C.; Papa, M. Neuropathic pain and reactive gliosis are reversed by dialdehydic compound in neuropathic pain rat models. Neurosci. Lett. 2012, 530. [Google Scholar] [CrossRef] [PubMed]
- Bianco, M.R.; Berbenni, M.; Amara, F.; Viggiani, S.; Fragni, M.; Galimberti, V.; Colombo, D.; Cirillo, G.; Papa, M.; Alberghina, L.; et al. Cross-talk between cell cycle induction and mitochondrial dysfunction during oxidative stress and nerve growth factor withdrawal in differentiated PC12 cells. J. Neurosci. Res. 2011, 89. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, G.; Papa, M. Beyond peripheral nerve injury: Spinal gliopathy and maladaptive synaptic plasticity. Neural Regen. Res. 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Messing, A. Refining the concept of GFAP toxicity in Alexander disease. J. Neurodev. Disord. 2019, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Helman, G.; Takanohashi, A.; Hagemann, T.L.; Perng, M.D.; Walkiewicz, M.; Woidill, S.; Sase, S.; Cross, Z.; Du, Y.; Zhao, L.; et al. Type II Alexander disease caused by splicing errors and aberrant overexpression of an uncharacterized GFAP isoform. Hum. Mutat. 2020, 41, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Ribotta, M.G.; Menet, V.; Privat, A. Glial scar and axonal regeneration in the CNS: Lessons from GFAP and vimentin transgenic mice. In Mechanisms of Secondary Brain Damage from Trauma and Ischemia; Springer: Vienna, Austria, 2004; pp. 87–92. [Google Scholar]
- Pekny, M.; Johansson, C.B.; Eliasson, C.; Stakeberg, J.; Wallén, Å.; Perlmann, T.; Lendahl, U.; Betsholtz, C.; Berthold, C.-H.; Frisén, J. Abnormal Reaction to Central Nervous System Injury in Mice Lacking Glial Fibrillary Acidic Protein and Vimentin. J. Cell Biol. 1999, 145, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Kamphuis, W.; Kooijman, L.; Orre, M.; Stassen, O.; Pekny, M.; Hol, E.M. GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild-type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer’s disease. Glia 2015, 63, 1036–1056. [Google Scholar] [CrossRef]
- Otani, N.; Nawashiro, H.; Nomura, N.; Fukui, S.; Tsuzuki, N.; Ishihara, S.; Shima, K. A role of glial fibrillary acidic protein in hippocampal degeneration after cerebral trauma or kainate-induced seizure. In Brain Edema XII; Springer: Vienna, Austria, 2003; pp. 267–269. [Google Scholar]
- McCall, M.A.; Gregg, R.G.; Behringer, R.R.; Brenner, M.; Delaney, C.L.; Galbreath, E.J.; Zhang, C.L.; Pearce, R.A.; Chiu, S.Y.; Messing, A. Targeted deletion in astrocyte intermediate filament (Gfap) alters neuronal physiology. Proc. Natl. Acad. Sci. USA 1996, 93, 6361–6366. [Google Scholar] [CrossRef] [Green Version]
- Decosterd, I.; Woolf, C.J. Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 2000, 87, 149–158. [Google Scholar] [CrossRef]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Yamanaka, H.; Kobayashi, K.; Okubo, M.; Noguchi, K. Annexin A2 in primary afferents contributes to neuropathic pain associated with tissue type plasminogen activator. Neuroscience 2016, 314, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.; Dubner, R.; Brown, F.; Flores, C.; Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988, 32, 77–88. [Google Scholar] [CrossRef]
- Maggio, N.; Sellitti, S.; Capano, C.P.; Papa, M. Tissue-transglutaminase in rat and human brain: Light and electron immunocytochemical analysis and in situ hybridization study. Brain Res. Bull. 2001, 56, 173–182. [Google Scholar] [CrossRef]
- Papa, M.; Boscia, F.; Canitano, A.; Castaldo, P.; Sellitti, S.; Annunziato, L.; Taglialatela, M. Expression pattern of the ether-a-gogo-related (ERG) K+ channel-encoding genes ERG1, ERG2, and ERG3 in the adult rat central nervous system. J. Comp. Neurol. 2003, 466, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, G.; Colangelo, A.M.; Bianco, M.R.; Cavaliere, C.; Zaccaro, L.; Sarmientos, P.; Alberghina, L.; Papa, M. BB14, a Nerve Growth Factor (NGF)-like peptide shown to be effective in reducing reactive astrogliosis and restoring synaptic homeostasis in a rat model of peripheral nerve injury. Biotechnol. Adv. 2012, 30, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Nawashiro, H.; Messing, A.; Azzam, N.; Brenner, M. Mice lacking GFAP are hypersensitive to traumatic cerebrospinal injury. Neuroreport 1998, 9, 1691–1696. [Google Scholar] [CrossRef]
- Nawashiro, H.; Brenner, M.; Fukui, S.; Shima, K.; Hallenbeck, J.M. High Susceptibility to Cerebral Ischemia in GFAP-Null Mice. J. Cereb. Blood Flow Metab. 2000, 20, 1040–1044. [Google Scholar] [CrossRef] [Green Version]
- De Luca, C.; Colangelo, A.M.; Virtuoso, A.; Alberghina, L.; Papa, M. Neurons, glia, extracellular matrix and neurovascular unit: A systems biology approach to the complexity of synaptic plasticity in health and disease. Int. J. Mol. Sci. 2020, 21, 1539. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Cavaliere, C.; Cirillo, G.; Rosaria Bianco, M.; Rossi, F.; De Novellis, V.; Maione, S.; Papa, M. Gliosis alters expression and uptake of spinal glial amino acid transporters in a mouse neuropathic pain model. Neuron Glia Biol. 2007, 3. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.C.; Skipp, P.; O’Connor, V.M.; Perry, V.H. Increased expression of glial fibrillary acidic protein fragments and μ-calpain activation within the hippocampus of prion-infected mice. Biochem. Soc. Trans. 2006, 34, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.B.; Du, S.; Rhim, H.; Lee, E.B.; Markelonis, G.J.; Oh, T.H. Rapid increase in immunoreactivity to GFAP in astrocytes in vitro induced by acidic pH is mediated by calcium influx and calpain I. Brain Res. 2000, 864, 220–229. [Google Scholar] [CrossRef]
- Baliova, M.; Jursky, F. Calpain Sensitive Regions in the N-terminal Cytoplasmic Domains of Glycine Transporters GlyT1A and GlyT1B. Neurochem. Res. 2005, 30, 1093–1100. [Google Scholar] [CrossRef]
- Giovannoni, R.; Maggio, N.; Rosaria Bianco, M.; Cavaliere, C.; Cirillo, G.; Lavitrano, M.; Papa, M. Reactive astrocytosis and glial glutamate transporter clustering are early changes in a spinocerebellar ataxia type 1 transgenic mouse model. Neuron Glia Biol. 2007, 3. [Google Scholar] [CrossRef] [Green Version]
- Woller, S.A.; Choi, S.-H.; An, E.J.; Low, H.; Schneider, D.A.; Ramachandran, R.; Kim, J.; Bae, Y.S.; Sviridov, D.; Corr, M.; et al. Inhibition of Neuroinflammation by AIBP: Spinal Effects upon Facilitated Pain States. Cell Rep. 2018, 23, 2667–2677. [Google Scholar] [CrossRef]
- Miller, Y.I.; Navia-Pelaez, J.M.; Corr, M.; Yaksh, T.L. Lipid rafts in glial cells: Role in neuroinflammation and pain processing. J. Lipid Res. 2020, 61, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Virtuoso, A.; Colangelo, A.M.; Maggio, N.; Fennig, U.; Weinberg, N.; Papa, M.; De Luca, C. The Spatiotemporal Coupling: Regional Energy Failure and Aberrant Proteins in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 11304. [Google Scholar] [CrossRef]
- Viedma-Poyatos, Á.; de Pablo, Y.; Pekny, M.; Pérez-Sala, D. The cysteine residue of glial fibrillary acidic protein is a critical target for lipoxidation and required for efficient network organization. Free Radic. Biol. Med. 2018, 120, 380–394. [Google Scholar] [CrossRef]
- Yuan, W.; Lu, L.; Rao, M.; Huang, Y.; Liu, C.; Liu, S.; Zhao, Y.; Liu, H.; Zhu, J.; Chao, T.; et al. GFAP hyperpalmitoylation exacerbates astrogliosis and neurodegenerative pathology in PPT1-deficient mice. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Luca, C.; Virtuoso, A.; Korai, S.A.; Cirillo, R.; Gargano, F.; Papa, M.; Cirillo, G. Altered Spinal Homeostasis and Maladaptive Plasticity in GFAP Null Mice Following Peripheral Nerve Injury. Cells 2022, 11, 1224. https://doi.org/10.3390/cells11071224
De Luca C, Virtuoso A, Korai SA, Cirillo R, Gargano F, Papa M, Cirillo G. Altered Spinal Homeostasis and Maladaptive Plasticity in GFAP Null Mice Following Peripheral Nerve Injury. Cells. 2022; 11(7):1224. https://doi.org/10.3390/cells11071224
Chicago/Turabian StyleDe Luca, Ciro, Assunta Virtuoso, Sohaib Ali Korai, Raffaella Cirillo, Francesca Gargano, Michele Papa, and Giovanni Cirillo. 2022. "Altered Spinal Homeostasis and Maladaptive Plasticity in GFAP Null Mice Following Peripheral Nerve Injury" Cells 11, no. 7: 1224. https://doi.org/10.3390/cells11071224