
Drosophila as a Model of Unconventional Translation in Spinocerebellar Ataxia Type 3

 , and
, and 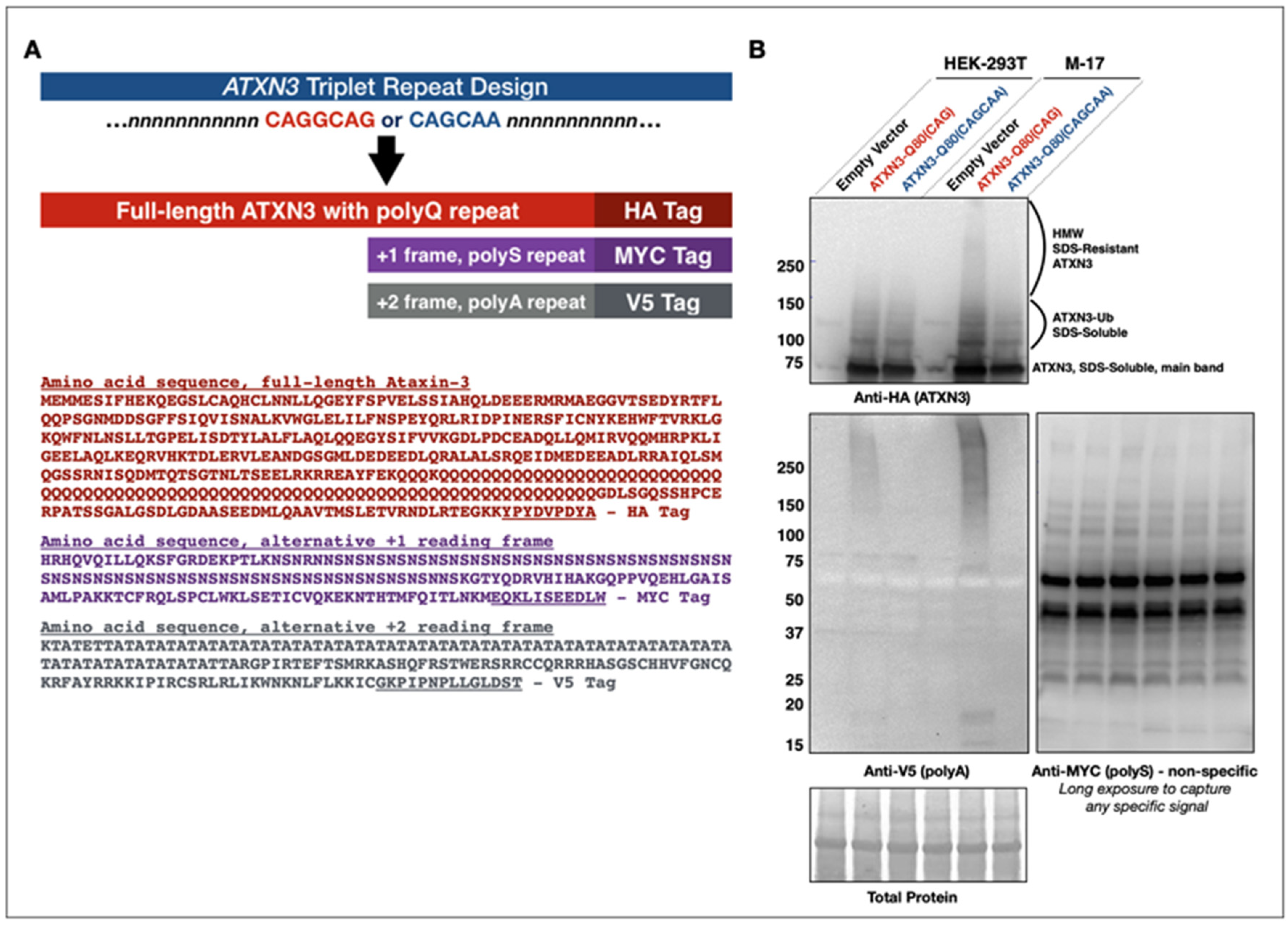{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construct Design
2.2. Antibodies
2.3. Mammalian Cells and Assays
2.4. Drosophila Stocks and Husbandry
2.5. Quantitative Real-Time Polymerase Chain Reaction
2.6. Drosophila Examinations
2.7. Western Blotting and Quantification
2.8. Filter-Trap Assays
2.9. Co-Immunopurification Assays
2.10. Statistics
3. Results
3.1. Development of New Constructs to Examine RAN in SCA3/MJD
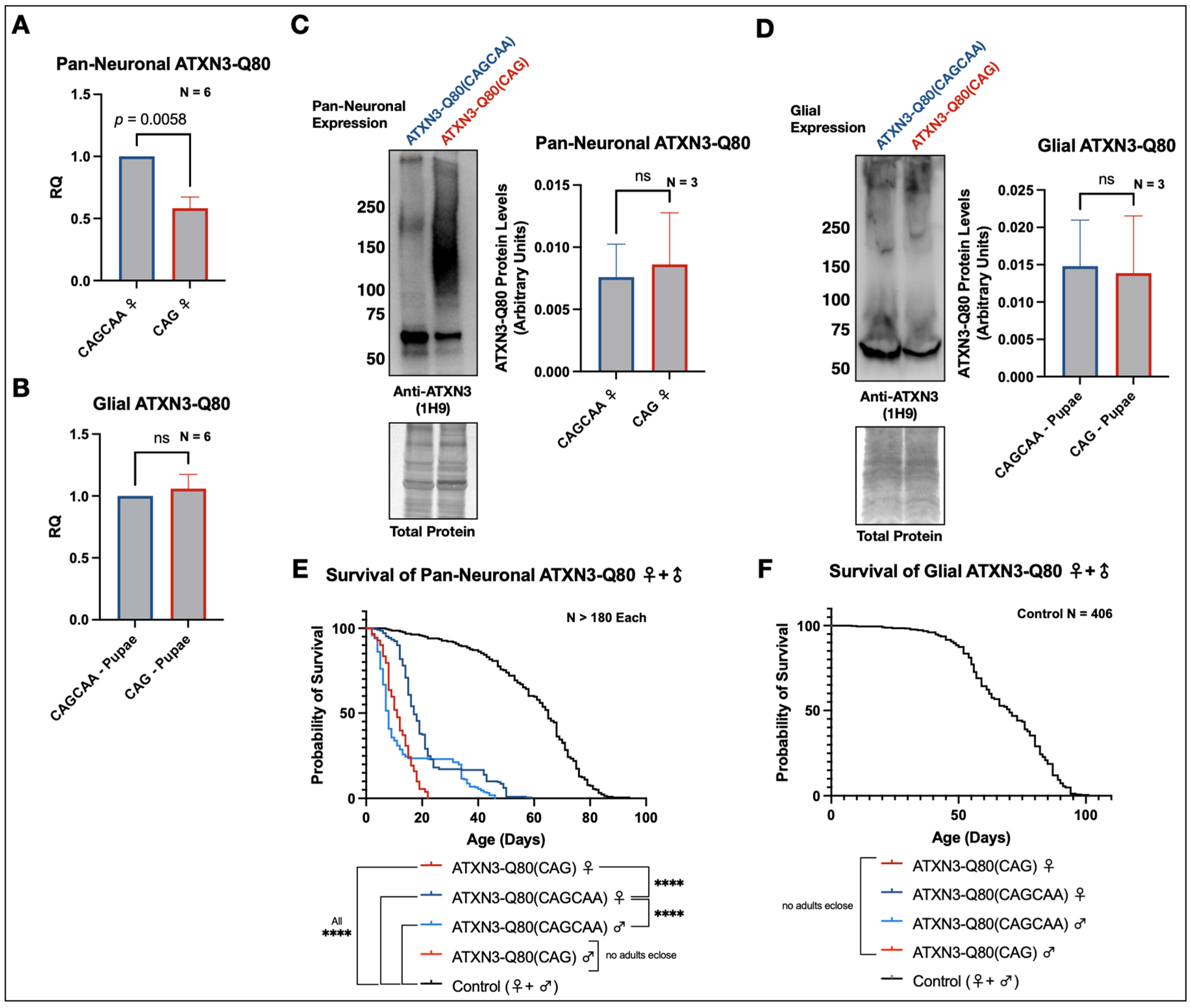
3.2. Homomeric and Alternating Pathogenic Ataxin-3 PolyQ Repeats Are Differentially Toxic in Drosophila melanogaster
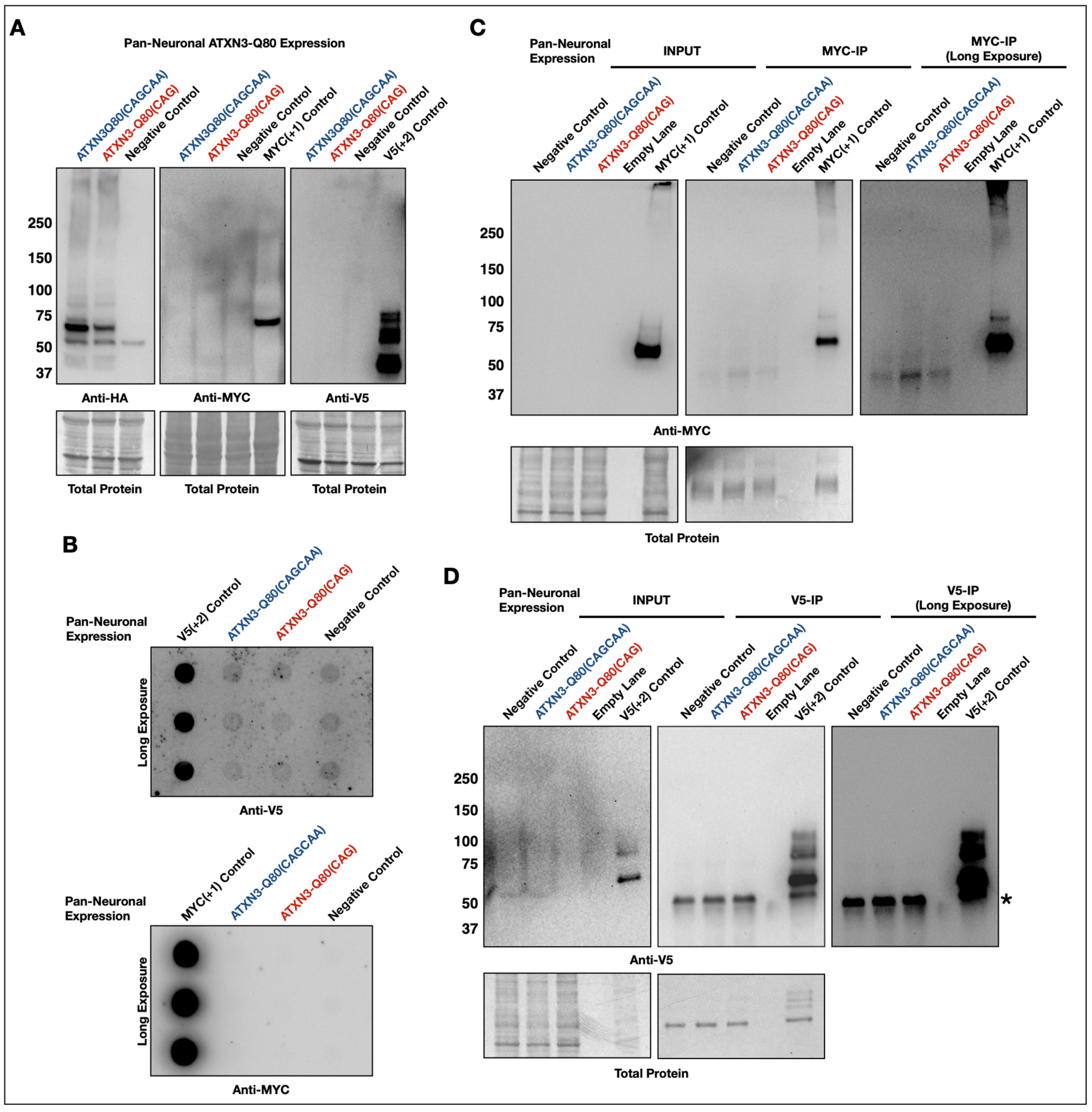
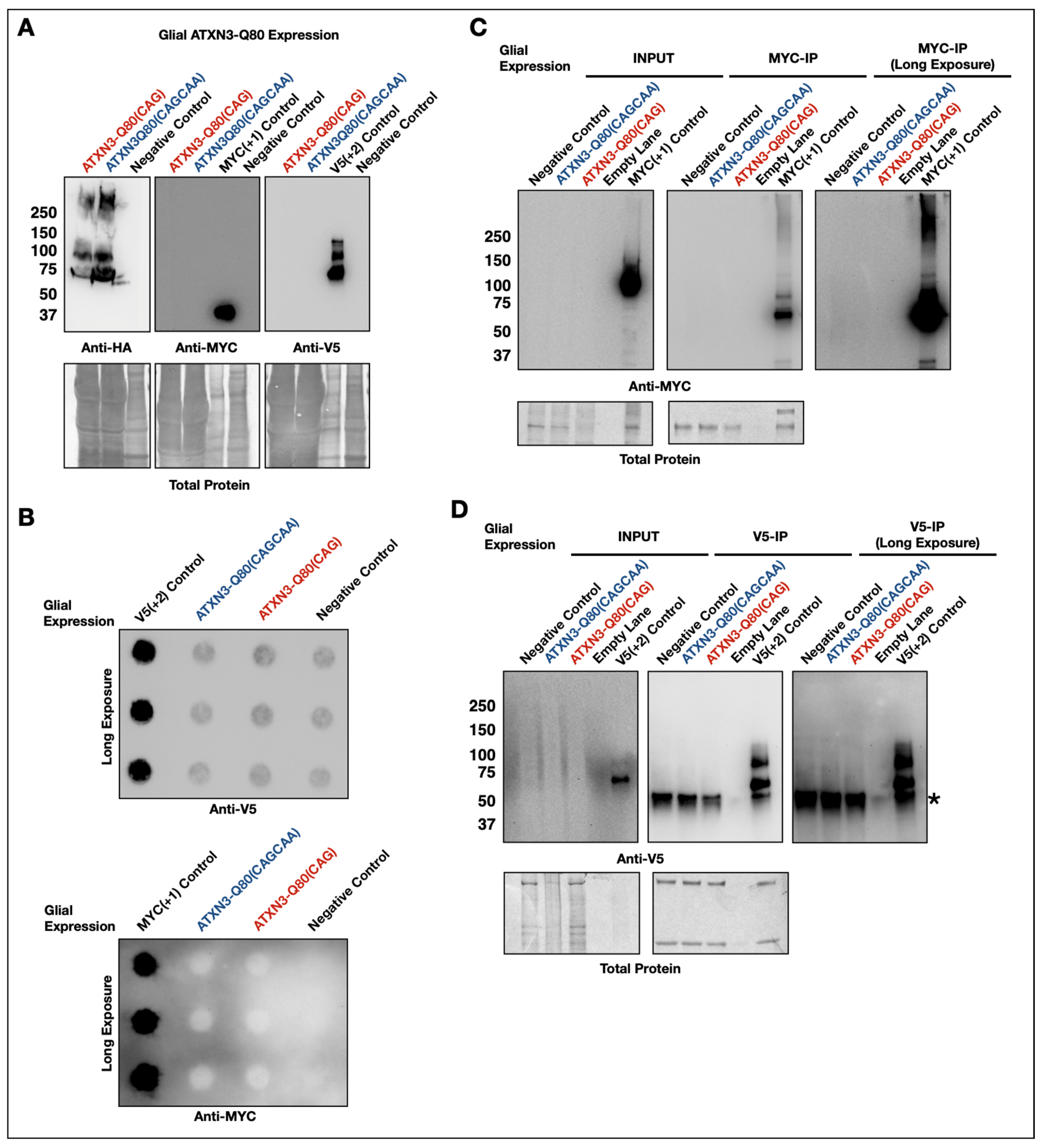
3.3. Lack of Evidence of RAN Peptides in Drosophila from Pathogenic ATXN3
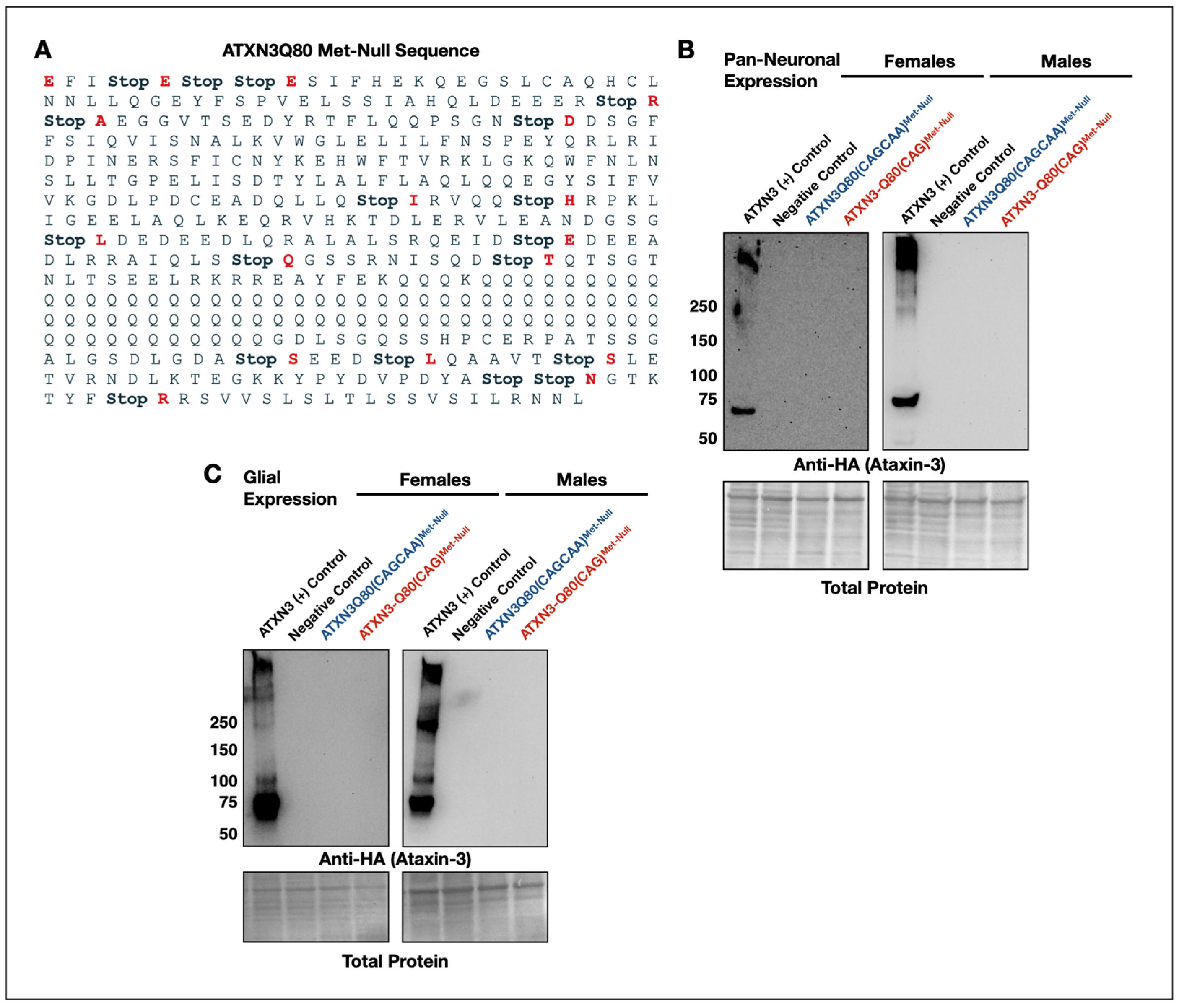
3.4. New Transgenes to Investigate mRNA-Based Toxicity in Drosophila Models of SCA3/MJD
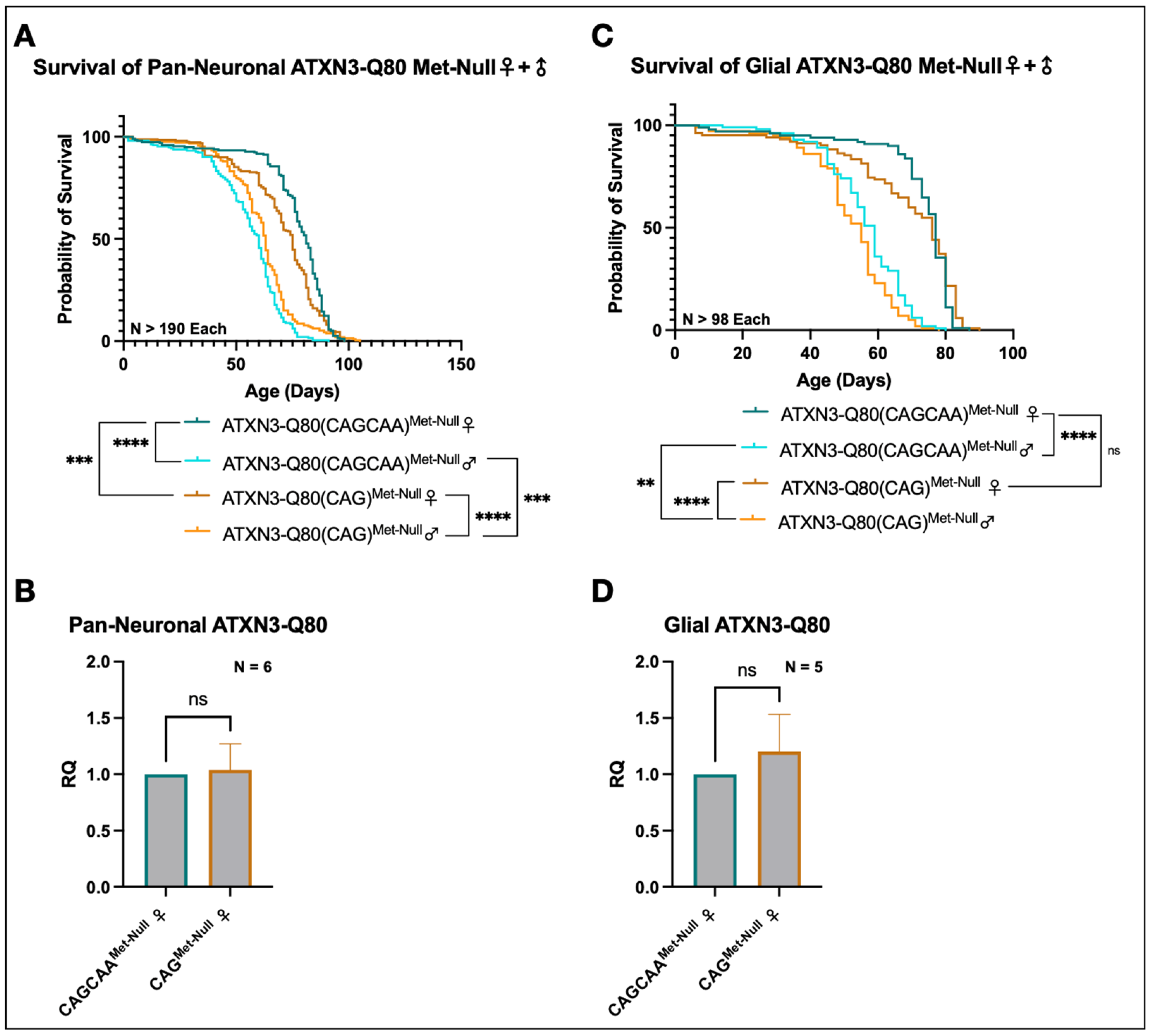
3.5. Minimal Survival Difference in Flies Expressing Alternating or Homomeric, Met-Null CAG/A Repeats
3.6. Alternative Mechanisms of Homomeric CAG Toxicity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Usdin, K. The biological effects of simple tandem repeats: Lessons from the repeat expansion diseases. Genome Res. 2008, 18, 1011–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Li, L.B.; Bonini, N.M. Roles of trinucleotide-repeat RNA in neurological disease and degeneration. Trends Neurosci. 2010, 33, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, K.M.; Linsalata, A.E.; Todd, P.K. RAN translation—What makes it run? Brain Res. 2016, 1647, 30–42. [Google Scholar] [CrossRef] [Green Version]
- Bañez-Coronel, M.; Ayhan, F.; Tarabochia, A.D.; Zu, T.; Perez, B.A.; Tusi, S.K.; Pletnikova, O.; Borchelt, D.R.; Ross, C.A.; Margolis, R.L.; et al. RAN Translation in Huntington Disease. Neuron 2015, 88, 667–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, T.; Pattamatta, A.; Ranum, L.P.W. Repeat-Associated Non-ATG Translation in Neurological Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033019. [Google Scholar] [CrossRef]
- Zu, T.; Liu, Y.; Bañez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, T.; Sato, N.; Ueyama, M.; Fujikake, N.; Sellier, C.; Kanegami, A.; Tokuda, E.; Zamiri, B.; Gall-Duncan, T.; Mirceta, M.; et al. Regulatory Role of RNA Chaperone TDP-43 for RNA Misfolding and Repeat-Associated Translation in SCA31. Neuron 2017, 94, 108–124.e7. [Google Scholar] [CrossRef] [Green Version]
- Ash, P.E.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.L.; Dejesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W.; Rademakers, R.; et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.J.; Jansen-West, K.; Ash, P.E.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Arzberger, T.; Grässer, F.A.; Gijselinck, I.; May, S.; Rentzsch, K.; Weng, S.M.; Schludi, M.H.; van der Zee, J.; Cruts, M.; et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013, 126, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Weng, S.M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Todd, P.K.; Oh, S.Y.; Krans, A.; He, F.; Sellier, C.; Frazer, M.; Renoux, A.J.; Chen, K.C.; Scaglione, K.M.; Basrur, V.; et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 2013, 78, 440–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todi, S.V.; Williams, A.J.; Paulson, H.L. Polyglutamine Disorders Including Huntington’s Disease. In Molecular Neurology; Waxman, S., Ed.; Academic Press: Cambridge, MA, USA, 2007; pp. 257–276. [Google Scholar]
- Costa, M.o.C.; Paulson, H.L. Toward understanding Machado-Joseph disease. Prog. Neurobiol. 2012, 97, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutinho, P.; Andrade, C. Autosomal dominant system degeneration in Portuguese families of the Azores Islands. A new genetic disorder involving cerebellar, pyramidal, extrapyramidal and spinal cord motor functions. Neurology 1978, 28, 703–709. [Google Scholar] [CrossRef]
- Li, X.; Liu, H.; Fischhaber, P.L.; Tang, T.S. Toward therapeutic targets for SCA3: Insight into the role of Machado-Joseph disease protein ataxin-3 in misfolded proteins clearance. Prog. Neurobiol. 2015, 132, 34–58. [Google Scholar] [CrossRef]
- Matos, C.A.; de Almeida, L.P.; Nóbrega, C. Machado-Joseph disease/spinocerebellar ataxia type 3: Lessons from disease pathogenesis and clues into therapy. J. Neurochem. 2019, 148, 8–28. [Google Scholar] [CrossRef] [Green Version]
- Paulson, H. Machado-Joseph disease/spinocerebellar ataxia type 3. Handb. Clin. Neurol. 2012, 103, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Ranum, L.P.; Lundgren, J.K.; Schut, L.J.; Ahrens, M.J.; Perlman, S.; Aita, J.; Bird, T.D.; Gomez, C.; Orr, H.T. Spinocerebellar ataxia type 1 and Machado-Joseph disease: Incidence of CAG expansions among adult-onset ataxia patients from 311 families with dominant, recessive, or sporadic ataxia. Am. J. Hum. Genet. 1995, 57, 603–608. [Google Scholar]
- Rosenberg, R.N. Machado-Joseph disease: An autosomal dominant motor system degeneration. Mov. Disord. 1992, 7, 193–203. [Google Scholar] [CrossRef]
- Schöls, L.; Meyer, C.; Schmid, G.; Wilhelms, I.; Przuntek, H. Therapeutic strategies in Friedreich’s ataxia. J. Neural. Transm. Suppl. 2004, 68, 135–145. [Google Scholar]
- Kawaguchi, Y.; Okamoto, T.; Taniwaki, M.; Aizawa, M.; Inoue, M.; Katayama, S.; Kawakami, H.; Nakamura, S.; Nishimura, M.; Akiguchi, I. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994, 8, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Takiyama, Y.; Nishizawa, M.; Tanaka, H.; Kawashima, S.; Sakamoto, H.; Karube, Y.; Shimazaki, H.; Soutome, M.; Endo, K.; Ohta, S. The gene for Machado-Joseph disease maps to human chromosome 14q. Nat. Genet. 1993, 4, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Li, L.B.; Yu, Z.; Teng, X.; Bonini, N.M. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 2008, 453, 1107–1111. [Google Scholar] [CrossRef] [Green Version]
- Figura, G.; Koscianska, E.; Krzyzosiak, W.J. In Vitro Expansion of CAG, CAA, and Mixed CAG/CAA Repeats. Int. J. Mol. Sci. 2015, 16, 18741–18751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shieh, S.Y.; Bonini, N.M. Genes and pathways affected by CAG-repeat RNA-based toxicity in Drosophila. Hum. Mol. Genet. 2011, 20, 4810–4821. [Google Scholar] [CrossRef] [Green Version]
- Martí, E. RNA toxicity induced by expanded CAG repeats in Huntington’s disease. Brain Pathol. 2016, 26, 779–786. [Google Scholar] [CrossRef]
- Zhang, N.; Ashizawa, T. RNA toxicity and foci formation in microsatellite expansion diseases. Curr. Opin. Genet. Dev. 2017, 44, 17–29. [Google Scholar] [CrossRef]
- Gaspar, C.; Jannatipour, M.; Dion, P.; Laganière, J.; Sequeiros, J.; Brais, B.; Rouleau, G.A. CAG tract of MJD-1 may be prone to frameshifts causing polyalanine accumulation. Hum. Mol. Genet. 2000, 9, 1957–1966. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.E.; Rubinsztein, D.C. Polyalanine and polyserine frameshift products in Huntington’s disease. J. Med. Genet. 2006, 43, 893–896. [Google Scholar] [CrossRef] [Green Version]
- Stochmanski, S.J.; Therrien, M.; Laganière, J.; Rochefort, D.; Laurent, S.; Karemera, L.; Gaudet, R.; Vyboh, K.; Van Meyel, D.J.; Di Cristo, G.; et al. Expanded ATXN3 frameshifting events are toxic in Drosophila and mammalian neuron models. Hum. Mol. Genet. 2012, 21, 2211–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulouse, A.; Au-Yeung, F.; Gaspar, C.; Roussel, J.; Dion, P.; Rouleau, G.A. Ribosomal frameshifting on MJD-1 transcripts with long CAG tracts. Hum. Mol. Genet. 2005, 14, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Girstmair, H.; Saffert, P.; Rode, S.; Czech, A.; Holland, G.; Bannert, N.; Ignatova, Z. Depletion of cognate charged transfer RNA causes translational frameshifting within the expanded CAG stretch in huntingtin. Cell Rep. 2013, 3, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berke, S.J.; Chai, Y.; Marrs, G.L.; Wen, H.; Paulson, H.L. Defining the role of ubiquitin-interacting motifs in the polyglutamine disease protein, ataxin-3. J. Biol. Chem. 2005, 280, 32026–32034. [Google Scholar] [CrossRef] [Green Version]
- Nicastro, G.; Masino, L.; Esposito, V.; Menon, R.P.; De Simone, A.; Fraternali, F.; Pastore, A. Josephin domain of ataxin-3 contains two distinct ubiquitin-binding sites. Biopolymers 2009, 91, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, G.; Todi, S.V.; Karaca, E.; Bonvin, A.M.; Paulson, H.L.; Pastore, A. Understanding the role of the Josephin domain in the PolyUb binding and cleavage properties of ataxin-3. PLoS ONE 2010, 5, e12430. [Google Scholar] [CrossRef]
- Blount, J.R.; Burr, A.A.; Denuc, A.; Marfany, G.; Todi, S.V. Ubiquitin-specific protease 25 functions in Endoplasmic Reticulum-associated degradation. PLoS ONE 2012, 7, e36542. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.L.; Blount, J.R.; Libohova, K.; Ranxhi, B.; Paulson, H.L.; Tsou, W.L.; Todi, S.V. Differential toxicity of ataxin-3 isoforms in Drosophila models of Spinocerebellar Ataxia Type 3. Neurobiol. Dis. 2019, 132, 104535. [Google Scholar] [CrossRef]
- Johnson, S.L.; Ranxhi, B.; Libohova, K.; Tsou, W.L.; Todi, S.V. Ubiquitin-interacting motifs of ataxin-3 regulate its polyglutamine toxicity through Hsc70-4-dependent aggregation. eLife 2020, 9, e60742. [Google Scholar] [CrossRef]
- Johnson, S.L.; Libohova, K.; Blount, J.R.; Sujkowski, A.L.; Prifti, M.V.; Tsou, W.L.; Todi, S.V. Targeting the VCP-binding motif of ataxin-3 improves phenotypes in Drosophila models of Spinocerebellar Ataxia Type 3. Neurobiol. Dis. 2021, 160, 105516. [Google Scholar] [CrossRef]
- Ristic, G.; Sutton, J.R.; Libohova, K.; Todi, S.V. Toxicity and aggregation of the polyglutamine disease protein, ataxin-3 is regulated by its binding to VCP/p97 in Drosophila melanogaster. Neurobiol. Dis. 2018, 116, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J.R.; Blount, J.R.; Libohova, K.; Tsou, W.L.; Joshi, G.S.; Paulson, H.L.; Costa, M.D.C.; Scaglione, K.M.; Todi, S.V. Interaction of the polyglutamine protein ataxin-3 with Rad23 regulates toxicity in Drosophila models of Spinocerebellar Ataxia Type 3. Hum. Mol. Genet. 2017, 26, 1419–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todi, S.V.; Winborn, B.J.; Scaglione, K.M.; Blount, J.R.; Travis, S.M.; Paulson, H.L. Ubiquitination directly enhances activity of the deubiquitinating enzyme ataxin-3. EMBO J. 2009, 28, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Tsou, W.L.; Ouyang, M.; Hosking, R.R.; Sutton, J.R.; Blount, J.R.; Burr, A.A.; Todi, S.V. The deubiquitinase ataxin-3 requires Rad23 and DnaJ-1 for its neuroprotective role in Drosophila melanogaster. Neurobiol. Dis. 2015, 82, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Winborn, B.J.; Travis, S.M.; Todi, S.V.; Scaglione, K.M.; Xu, P.; Williams, A.J.; Cohen, R.E.; Peng, J.; Paulson, H.L. The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J. Biol. Chem. 2008, 283, 26436–26443. [Google Scholar] [CrossRef] [Green Version]
- Tsou, W.L.; Qiblawi, S.H.; Hosking, R.R.; Gomez, C.M.; Todi, S.V. Polyglutamine length-dependent toxicity from α1ACT in Drosophila models of spinocerebellar ataxia type 6. Biol. Open 2016, 5, 1770–1775. [Google Scholar] [CrossRef]
- Fish, M.P.; Groth, A.C.; Calos, M.P.; Nusse, R. Creating transgenic Drosophila by microinjecting the site-specific phiC31 integrase mRNA and a transgene-containing donor plasmid. Nat. Protoc. 2007, 2, 2325–2331. [Google Scholar] [CrossRef]
- Groth, A.C.; Fish, M.; Nusse, R.; Calos, M.P. Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics 2004, 166, 1775–1782. [Google Scholar] [CrossRef] [Green Version]
- Blount, J.R.; Libohova, K.; Silva, G.M.; Todi, S.V. Isoleucine 44 Hydrophobic Patch Controls Toxicity of Unanchored, Linear Ubiquitin Chains through NF-κB Signaling. Cells 2020, 9, 1519. [Google Scholar] [CrossRef]
- Blount, J.R.; Libohova, K.; Marsh, G.B.; Sutton, J.R.; Todi, S.V. Expression and Regulation of Deubiquitinase-Resistant, Unanchored Ubiquitin Chains in Drosophila. Sci. Rep. 2018, 8, 8513. [Google Scholar] [CrossRef]
- Paulson, H.L.; Das, S.S.; Crino, P.B.; Perez, M.K.; Patel, S.C.; Gotsdiner, D.; Fischbeck, K.H.; Pittman, R.N. Machado-Joseph disease gene product is a cytoplasmic protein widely expressed in brain. Ann. Neurol. 1997, 41, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Stork, T.; Bernardos, R.; Freeman, M.R. Analysis of glial cell development and function in Drosophila. Cold Spring Harb. Protoc. 2012, 2012, pdb-top067587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepp, K.J.; Auld, V.J. RhoA and Rac1 GTPases mediate the dynamic rearrangement of actin in peripheral glia. Development 2003, 130, 1825–1835. [Google Scholar] [CrossRef] [Green Version]
- Sepp, K.J.; Schulte, J.; Auld, V.J. Peripheral glia direct axon guidance across the CNS/PNS transition zone. Dev. Biol. 2001, 238, 47–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, J.D.; Boury, A.L.; Gerald, N.J.; Kiehart, D.P. Native nonmuscle myosin II stability and light chain binding in Drosophila melanogaster. Cell Motil. Cytoskeleton. 2006, 63, 604–622. [Google Scholar] [CrossRef]
- Franke, J.D.; Dong, F.; Rickoll, W.L.; Kelley, M.J.; Kiehart, D.P. Rod mutations associated with MYH9-related disorders disrupt nonmuscle myosin-IIA assembly. Blood 2005, 105, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Franke, J.D.; Montague, R.A.; Kiehart, D.P. Nonmuscle myosin II generates forces that transmit tension and drive contraction in multiple tissues during dorsal closure. Curr. Biol. 2005, 15, 2208–2221. [Google Scholar] [CrossRef] [Green Version]
- Franke, J.D.; Montague, R.A.; Kiehart, D.P. Nonmuscle myosin II is required for cell proliferation, cell sheet adhesion and wing hair morphology during wing morphogenesis. Dev. Biol. 2010, 345, 117–132. [Google Scholar] [CrossRef] [Green Version]
- Franke, J.D.; Montague, R.A.; Rickoll, W.L.; Kiehart, D.P. An MYH9 human disease model in flies: Site-directed mutagenesis of the Drosophila non-muscle myosin II results in hypomorphic alleles with dominant character. Hum. Mol. Genet. 2007, 16, 3160–3173. [Google Scholar] [CrossRef] [Green Version]
- Kiehart, D.P.; Franke, J.D. Actin dynamics: The arp2/3 complex branches out. Curr. Biol. 2002, 12, R557–R559. [Google Scholar] [CrossRef] [Green Version]
- Kiehart, D.P.; Franke, J.D.; Chee, M.K.; Montague, R.A.; Chen, T.L.; Roote, J.; Ashburner, M. Drosophila crinkled, mutations of which disrupt morphogenesis and cause lethality, encodes fly myosin VIIA. Genetics 2004, 168, 1337–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todi, S.V.; Franke, J.D.; Kiehart, D.P.; Eberl, D.F. Myosin VIIA defects, which underlie the Usher 1B syndrome in humans, lead to deafness in Drosophila. Curr. Biol. 2005, 15, 862–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, A.R.; Rosen, D.R.; Robinow, S.N.; White, K. Molecular analysis of the locus elav in Drosophila melanogaster: A gene whose embryonic expression is neural specific. EMBO J. 1987, 6, 425–431. [Google Scholar] [CrossRef]
- Robinow, S.; Campos, A.R.; Yao, K.M.; White, K. The elav gene product of Drosophila, required in neurons, has three RNP consensus motifs. Science 1988, 242, 1570–1572. [Google Scholar] [CrossRef]
- Robinow, S.; White, K. The locus elav of Drosophila melanogaster is expressed in neurons at all developmental stages. Dev. Biol. 1988, 126, 294–303. [Google Scholar] [CrossRef]
- Robinow, S.; White, K. Characterization and spatial distribution of the ELAV protein during Drosophila melanogaster development. J. Neurobiol. 1991, 22, 443–461. [Google Scholar] [CrossRef]
- Sobczak, K.; de Mezer, M.; Michlewski, G.; Krol, J.; Krzyzosiak, W.J. RNA structure of trinucleotide repeats associated with human neurological diseases. Nucleic Acids Res. 2003, 31, 5469–5482. [Google Scholar] [CrossRef]
- Sobczak, K.; Krzyzosiak, W.J. CAG repeats containing CAA interruptions form branched hairpin structures in spinocerebellar ataxia type 2 transcripts. J. Biol. Chem. 2005, 280, 3898–3910. [Google Scholar] [CrossRef] [Green Version]
- Blount, J.R.; Tsou, W.L.; Ristic, G.; Burr, A.A.; Ouyang, M.; Galante, H.; Scaglione, K.M.; Todi, S.V. Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23. Nat. Commun. 2014, 5, 4638. [Google Scholar] [CrossRef] [Green Version]
- Tsou, W.L.; Burr, A.A.; Ouyang, M.; Blount, J.R.; Scaglione, K.M.; Todi, S.V. Ubiquitination regulates the neuroprotective function of the deubiquitinase ataxin-3 in vivo. J. Biol. Chem. 2013, 288, 34460–34469. [Google Scholar] [CrossRef] [Green Version]
- Burr, A.A.; Tsou, W.L.; Ristic, G.; Todi, S.V. Using membrane-targeted green fluorescent protein to monitor neurotoxic protein-dependent degeneration of Drosophila eyes. J. Neurosci. Res. 2014, 92, 1100–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsou, W.L.; Hosking, R.R.; Burr, A.A.; Sutton, J.R.; Ouyang, M.; Du, X.; Gomez, C.M.; Todi, S.V. DnaJ-1 and karyopherin α3 suppress degeneration in a new Drosophila model of Spinocerebellar Ataxia Type 6. Hum. Mol. Genet. 2015, 24, 4385–4396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, K.H.; Zalon, A.J.; Zhang, H.; DiFranco, D.M.; Stec, N.R.; Haque, Z.; Blumenstein, K.G.; Pierce, A.M.; Guan, Y.; Paulson, H.L.; et al. Impaired oligodendrocyte maturation is an early feature in SCA3 disease pathogenesis. J. Neurosci. 2022, 42, 1604–1617. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Kuret, J. Detection and quantification of tau aggregation using a membrane filter assay. Anal. Biochem. 2008, 373, 330–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Gonzales, V.; Borchelt, D.R. Rapid detection of protein aggregates in the brains of Alzheimer patients and transgenic mouse models of amyloidosis. Alzheimer Dis. Assoc. Disord. 2002, 16, 191–195. [Google Scholar] [CrossRef]
- Weishäupl, D.; Schneider, J.; Peixoto Pinheiro, B.; Ruess, C.; Dold, S.M.; von Zweydorf, F.; Gloeckner, C.J.; Schmidt, J.; Riess, O.; Schmidt, T. Physiological and pathophysiological characteristics of ataxin-3 isoforms. J. Biol. Chem. 2019, 294, 644–661. [Google Scholar] [CrossRef] [Green Version]
- Buhr, F.; Ciryam, P.S.; Vendruscolo, M. A mistranslation-prone transcriptome underlying polyglutamine expansion diseases. Nat. Rev. Mol. Cell Biol. 2021, 22, 583–584. [Google Scholar] [CrossRef]
- Piper, M.D.; Blanc, E.; Leitão-Gonçalves, R.; Yang, M.; He, X.; Linford, N.J.; Hoddinott, M.P.; Hopfen, C.; Soultoukis, G.A.; Niemeyer, C.; et al. A holidic medium for Drosophila melanogaster. Nat. Methods 2014, 11, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Piper, M.D.W.; Soultoukis, G.A.; Blanc, E.; Mesaros, A.; Herbert, S.L.; Juricic, P.; He, X.; Atanassov, I.; Salmonowicz, H.; Yang, M.; et al. Matching Dietary Amino Acid Balance to the In Silico-Translated Exome Optimizes Growth and Reproduction without Cost to Lifespan. Cell Metab. 2017, 25, 610–621. [Google Scholar] [CrossRef] [Green Version]
- Eshraghi, M.; Karunadharma, P.P.; Blin, J.; Shahani, N.; Ricci, E.P.; Michel, A.; Urban, N.T.; Galli, N.; Sharma, M.; Ramírez-Jarquín, U.N.; et al. Mutant Huntingtin stalls ribosomes and represses protein synthesis in a cellular model of Huntington disease. Nat. Commun. 2021, 12, 1461. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, S.L.; Prifti, M.V.; Sujkowski, A.; Libohova, K.; Blount, J.R.; Hong, L.; Tsou, W.-L.; Todi, S.V. Drosophila as a Model of Unconventional Translation in Spinocerebellar Ataxia Type 3. Cells 2022, 11, 1223. https://doi.org/10.3390/cells11071223
Johnson SL, Prifti MV, Sujkowski A, Libohova K, Blount JR, Hong L, Tsou W-L, Todi SV. Drosophila as a Model of Unconventional Translation in Spinocerebellar Ataxia Type 3. Cells. 2022; 11(7):1223. https://doi.org/10.3390/cells11071223
Chicago/Turabian StyleJohnson, Sean L., Matthew V. Prifti, Alyson Sujkowski, Kozeta Libohova, Jessica R. Blount, Luke Hong, Wei-Ling Tsou, and Sokol V. Todi. 2022. "Drosophila as a Model of Unconventional Translation in Spinocerebellar Ataxia Type 3" Cells 11, no. 7: 1223. https://doi.org/10.3390/cells11071223