
Activin A Causes Muscle Atrophy through MEF2C-Dependent Impaired Myogenesis

 , , and
, , and
Abstract
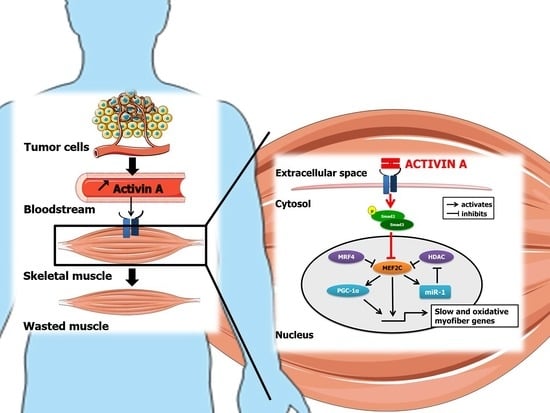
:
1. Introduction
2. Experimental Procedure
2.1. Cell Culture, Treatment, and Transfection
2.2. Cell Viability
2.3. Myotube Morphological Analysis
2.4. Direct miRNA or mRNA Quantification by RT-qPCR
2.5. Western Blotting
2.6. Mouse Models of Cancer Cachexia
2.6.1. C26 and Baf3 Models
2.6.2. KPC Model
2.7. Statistical Analysis
3. Results
3.1. Activin A Causes Atrophy of Human Skeletal Muscle Cells
3.2. Activin A-Induced Myotube Atrophy Is Characterized by a Decrease in Myosin-Heavy Chain-β/Slow Content
3.3. Activin A-Induced Myotube Atrophy Is Associated with a Downregulation of MEF2C Expression and Activity
3.4. MEF2C Is Required to Maintain MyHC-β/Slow Gene Expression and Protein Content in Differentiated Myotubes
3.5. The Activin A-Induced Myotube Atrophy Is Not Associated with Increased Classical E3 Ubiquitin Ligases
3.6. Cancer Cachexia Is Associated with Downregulation of Muscle MEF2C Expression and Activity Which Is Blunted by Inhibition of Activin A
4. Discussion
4.1. Activin A Causes Human Muscle Cell Atrophy by Altering Myogenesis and MyHC-β/Slow Synthesis
4.2. Activin A Inhibits MyHC-β/Slow Synthesis by Downregulating MEF2C
4.3. Activin A Targets miR-1 Expression, a Positive Regulator of MEF2C Expression and Activity
4.4. Activin A Does Not Upregulate Classical E3 Ubiquitin Ligases Targeting Myosin-Heavy Chain
4.5. The Downregulation of MEF2C Expression and Activity in Animal Models of Cancer Cachexia Is Reversed by an Activin A Antagonist
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bloise, E.; Ciarmela, P.; Dela Cruz, C.; Luisi, S.; Petraglia, F.; Reis, F.M. Activin A in Mammalian Physiology. Physiol. Rev. 2019, 99, 739–780. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimek, M.E.B.; Aydogdu, T.; Link, M.J.; Pons, M.; Koniaris, L.G.; Zimmers, T.A. Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem. Biophys. Res. Commun. 2010, 391, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Busquets, S.; Toledo, M.; Orpí, M.; Massa, D.; Porta, M.; Capdevila, E.; Padilla, N.; Frailis, V.; López-Soriano, F.J.; Han, H.Q.; et al. Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J. Cachexia Sarcopenia Muscle 2012, 3, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Levolger, S.; Wiemer, E.A.C.; van Vugt, J.L.A.; Huisman, S.A.; van Vledder, M.G.; van Damme-van Engel, S.; Ambagtsheer, G.; IJzermans, J.N.M.; de Bruin, R.W.F. Inhibition of activin-like kinase 4/5 attenuates cancer cachexia associated muscle wasting. Sci. Rep. 2019, 9, 9826. [Google Scholar] [CrossRef]
- Nissinen, T.A.; Hentilä, J.; Penna, F.; Lampinen, A.; Lautaoja, J.H.; Fachada, V.; Holopainen, T.; Ritvos, O.; Kivelä, R.; Hulmi, J.J. Treating cachexia using soluble ACVR2B improves survival, alters mTOR localization, and attenuates liver and spleen responses. J. Cachexia Sarcopenia Muscle 2018, 9, 514–529. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Summermatter, S.; Jourdain, M.; Melly, S.; Minetti, G.C.; Lach-Trifilieff, E. ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti-cancer treatments. Skelet. Muscle 2016, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Han, H.Q.; Zhou, X.; Mitch, W.E.; Goldberg, A.L. Myostatin/activin pathway antagonism: Molecular basis and therapeutic potential. Int. J. Biochem. Cell Biol. 2013, 45, 2333–2347. [Google Scholar] [CrossRef]
- Wu, S.; Qi, Y.; Niu, L.M.; Xie, D.X.; Cui, X.L.; Liu, Z.H. Activin A as a novel biomarker for colorectal adenocarcinoma in humans. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4371–4378. [Google Scholar]
- Leto, G.; Incorvaia, L.; Badalamenti, G.; Tumminello, F.M.; Gebbia, N.; Flandina, C.; Crescimanno, M.; Rini, G. Activin A circulating levels in patients with bone metastasis from breast or prostate cancer. Clin. Exp. Metastasis 2006, 23, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Yoshinaga, K.; Mimori, K.; Yamashita, K.; Utsunomiya, T.; Inoue, H.; Mori, M. Clinical significance of the expression of activin A in esophageal carcinoma. Int. J. Oncol. 2003, 22, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Hofland, J.; van Weerden, W.M.; Steenbergen, J.; Dits, N.F.; Jenster, G.; de Jong, F.H. Activin A stimulates AKR1C3 expression and growth in human prostate cancer. Endocrinology 2012, 153, 5726–5734. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.P.; Kao, H.K.; Liang, Y.; Cheng, M.H.; Chang, Y.L.; Liu, S.C.; Lin, Y.C.; Ko, T.Y.; Lee, Y.S.; Tsai, C.L.; et al. Overexpression of activin A in oral squamous cell carcinoma: Association with poor prognosis and tumor progression. Ann. Surg. Oncol. 2010, 17, 1945–1956. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Pons, M.; Poirier, C.; Jiang, Y.; Liu, J.; Sandusky, G.E.; Shahda, S.; Nakeeb, A.; Schmidt, C.M.; House, M.G.; et al. The systemic activin response to pancreatic cancer: Implications for effective cancer cachexia therapy. J. Cachexia Sarcopenia Muscle 2019, 10, 1083–1101. [Google Scholar] [CrossRef] [Green Version]
- Hoda, M.A.; Rozsas, A.; Lang, E.; Klikovits, T.; Lohinai, Z.; Torok, S.; Berta, J.; Bendek, M.; Berger, W.; Hegedus, B.; et al. High circulating activin A level is associated with tumor progression and predicts poor prognosis in lung adenocarcinoma. Oncotarget 2016, 7, 13388–13399. [Google Scholar] [CrossRef] [Green Version]
- Paajanen, J.; Ilonen, I.; Lauri, H.; Jarvinen, T.; Sutinen, E.; Ollila, H.; Rouvinen, E.; Lemstrom, K.; Rasanen, J.; Ritvos, O.; et al. Elevated Circulating Activin A Levels in Patients With Malignant Pleural Mesothelioma Are Related to Cancer Cachexia and Reduced Response to Platinum-based Chemotherapy. Clin. Lung Cancer 2020, 21, e142–e150. [Google Scholar] [CrossRef] [Green Version]
- Togashi, Y.; Kogita, A.; Sakamoto, H.; Hayashi, H.; Terashima, M.; de Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Kitano, M.; et al. Activin signal promotes cancer progression and is involved in cachexia in a subset of pancreatic cancer. Cancer Lett. 2015, 356, 819–827. [Google Scholar] [CrossRef]
- Loumaye, A.; de Barsy, M.; Nachit, M.; Lause, P.; Frateur, L.; van Maanen, A.; Trefois, P.; Gruson, D.; Thissen, J.P. Role of Activin A and myostatin in human cancer cachexia. J. Clin. Endocrinol. Metab. 2015, 100, 2030–2038. [Google Scholar] [CrossRef] [Green Version]
- Lerner, L.; Tao, J.; Liu, Q.; Nicoletti, R.; Feng, B.; Krieger, B.; Mazsa, E.; Siddiquee, Z.; Wang, R.; Huang, L.; et al. MAP3K11/GDF15 axis is a critical driver of cancer cachexia. J. Cachexia Sarcopenia Muscle 2015, 7, 467–482. [Google Scholar] [CrossRef]
- Lerner, L.; Hayes, T.G.; Tao, N.; Krieger, B.; Feng, B.; Wu, Z.; Nicoletti, R.; Chiu, M.I.; Gyuris, J.; Garcia, J.M. Plasma growth differentiation factor 15 is associated with weight loss and mortality in cancer patients. J. Cachexia Sarcopenia Muscle 2015, 6, 317–324. [Google Scholar] [CrossRef]
- Lerner, L.; Gyuris, J.; Nicoletti, R.; Gifford, J.; Krieger, B.; Jatoi, A. Growth differentiating factor-15 (GDF-15): A potential biomarker and therapeutic target for cancer-associated weight loss. Oncol. Lett. 2016, 12, 4219–4223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talar-Wojnarowska, R.; Wozniak, M.; Borkowska, A.; Olakowski, M.; Malecka-Panas, E. Clinical significance of activin A and myostatin in patients with pancreatic adenocarcinoma and progressive weight loss. J. Physiol. Pharmacol. 2020, 71, 25. [Google Scholar] [CrossRef]
- Loumaye, A.; de Barsy, M.; Nachit, M.; Lause, P.; van Maanen, A.; Trefois, P.; Gruson, D.; Thissen, J.P. Circulating Activin A predicts survival in cancer patients. J. Cachexia Sarcopenia Muscle 2017, 8, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Matzuk, M.M.; Finegold, M.J.; Mather, J.P.; Krummen, L.; Lu, H.; Bradley, A. Development of cancer cachexia-like syndrome and adrenal tumors in inhibin-deficient mice. Proc. Natl. Acad. Sci. USA 1994, 91, 8817–8821. [Google Scholar] [CrossRef] [Green Version]
- Gilson, H.; Schakman, O.; Kalista, S.; Lause, P.; Tsuchida, K.; Thissen, J.P. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E157–E164. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.L.; Walton, K.L.; Winbanks, C.E.; Murphy, K.T.; Thomson, R.E.; Makanji, Y.; Qian, H.; Lynch, G.S.; Harrison, C.A.; Gregorevic, P. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J. 2014, 28, 1711–1723. [Google Scholar] [CrossRef]
- Chen, J.L.; Walton, K.L.; Qian, H.; Colgan, T.D.; Hagg, A.; Watt, M.J.; Harrison, C.A.; Gregorevic, P. Differential effects of interleukin-6 and activin A in the development of cancer-associated cachexia. Cancer Res. 2016, 76, 5372–5382. [Google Scholar] [CrossRef] [Green Version]
- Yaden, B.C.; Wang, Y.X.; Wilson, J.M.; Culver, A.E.; Milner, A.; Datta-Mannan, A.; Shetler, P.; Croy, J.E.; Dai, G.; Krishnan, V. Inhibition of activin A ameliorates skeletal muscle injury and rescues contractile properties by inducing efficient remodeling in female mice. Am. J. Pathol. 2014, 184, 1152–1166. [Google Scholar] [CrossRef]
- Walton, K.L.; Chen, J.L.; Arnold, Q.; Kelly, E.; La, M.; Lu, L.; Lovrecz, G.; Hagg, A.; Colgan, T.D.; Qian, H.; et al. Activin A-Induced Cachectic Wasting Is Attenuated by Systemic Delivery of Its Cognate Propeptide in Male Mice. Endocrinology 2019, 160, 2417–2426. [Google Scholar] [CrossRef]
- Trendelenburg, A.U.; Meyer, A.; Rohner, D.; Boyle, J.; Hatakeyama, S.; Glass, D.J. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am. J. Physiol.-Cell Physiol. 2009, 296, C1258–C1270. [Google Scholar] [CrossRef] [Green Version]
- Latres, E.; Mastaitis, J.; Fury, W.; Miloscio, L.; Trejos, J.; Pangilinan, J.; Okamoto, H.; Cavino, K.; Na, E.; Papatheodorou, A.; et al. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat. Commun. 2017, 8, 15153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, K. No longer going to waste. Nat. Biotechnol. 2016, 34, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, L.; Gandhi, R.; Warden, S.J.; Poiraudeau, S.; Myers, S.L.; Benson, C.T.; Hu, L.; Ahmad, Q.I.; Linnemeier, P.; Gomez, E.V.; et al. A Phase 2 Randomized Study Investigating the Efficacy and Safety of Myostatin Antibody LY2495655 versus Placebo in Patients Undergoing Elective Total Hip Arthroplasty. J. Frailty Aging 2016, 5, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Gueugneau, M.; d’Hose, D.; Barbe, C.; de Barsy, M.; Lause, P.; Maiter, D.; Bindels, L.B.; Delzenne, N.M.; Schaeffer, L.; Gangloff, Y.G.; et al. Increased Serpina3n release into circulation during glucocorticoid-mediated muscle atrophy. J. Cachexia Sarcopenia Muscle 2018, 9, 929–946. [Google Scholar] [CrossRef] [Green Version]
- Bindels, L.B.; Neyrinck, A.M.; Claus, S.P.; Le Roy, C.I.; Grangette, C.; Pot, B.; Martinez, I.; Walter, J.; Cani, P.D.; Delzenne, N.M. Synbiotic approach restores intestinal homeostasis and prolongs survival in leukaemic mice with cachexia. ISME J. 2016, 10, 1456–1470. [Google Scholar] [CrossRef]
- Ciciliot, S.; Rossi, A.C.; Dyar, K.A.; Blaauw, B.; Schiaffino, S. Muscle type and fiber type specificity in muscle wasting. Int. J. Biochem. Cell Biol. 2013, 45, 2191–2199. [Google Scholar] [CrossRef]
- Blaauw, B.; Schiaffino, S.; Reggiani, C. Mechanisms modulating skeletal muscle phenotype. Compr. Physiol. 2013, 3, 1645–1687. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [Green Version]
- Potthoff, M.J.; Arnold, M.A.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of skeletal muscle sarcomere integrity and postnatal muscle function by Mef2c. Mol. Cell. Biol. 2007, 27, 8143–8151. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Yang, X.Z.; Zhang, C.Y.; Liu, Y.Y.; Zhou, R.B.; Cheng, Q.D.; Yan, E.K.; Yin, D.C. Myocyte enhancer factor 2C and its directly-interacting proteins: A review. Prog. Biophys. Mol. Biol. 2017, 126, 22–30. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Calabria, E. Skeletal muscle mass is controlled by the MRF4-MEF2 axis. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Olson, E.N. MEF2: A central regulator of diverse developmental programs. Development 2007, 134, 4131–4140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, M. Protein breakdown in cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Costamagna, D.; Fanzani, A.; Bonelli, G.; Baccino, F.M.; Costelli, P. Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS ONE 2010, 5, e13604. [Google Scholar] [CrossRef]
- Penna, F.; Ballaro, R.; Beltra, M.; De Lucia, S.; Garcia Castillo, L.; Costelli, P. The Skeletal Muscle as an Active Player Against Cancer Cachexia. Front. Physiol. 2019, 10, 41. [Google Scholar] [CrossRef] [Green Version]
- Acharyya, S.; Butchbach, M.E.; Sahenk, Z.; Wang, H.; Saji, M.; Carathers, M.; Ringel, M.D.; Skipworth, R.J.; Fearon, K.C.; Hollingsworth, M.A.; et al. Dystrophin glycoprotein complex dysfunction: A regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 2005, 8, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Souza, T.A.; Chen, X.; Guo, Y.; Sava, P.; Zhang, J.; Hill, J.J.; Yaworsky, P.J.; Qiu, Y. Proteomic identification and functional validation of activins and bone morphogenetic protein 11 as candidate novel muscle mass regulators. Mol. Endocrinol. 2008, 22, 2689–2702. [Google Scholar] [CrossRef] [Green Version]
- Rios, R.; Carneiro, I.; Arce, V.M.; Devesa, J. Myostatin is an inhibitor of myogenic differentiation. Am. J. Physiol.-Cell Physiol. 2002, 282, C993–C999. [Google Scholar] [CrossRef] [Green Version]
- Snijders, T.; Nederveen, J.P.; McKay, B.R.; Joanisse, S.; Verdijk, L.B.; van Loon, L.J.; Parise, G. Satellite cells in human skeletal muscle plasticity. Front. Physiol. 2015, 6, 283. [Google Scholar] [CrossRef] [Green Version]
- Trendelenburg, A.U.; Meyer, A.; Jacobi, C.; Feige, J.N.; Glass, D.J. TAK-1/p38/nNFkappaB signaling inhibits myoblast differentiation by increasing levels of Activin A. Skelet. Muscle 2012, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Lach-Trifilieff, E.; Minetti, G.C.; Sheppard, K.; Ibebunjo, C.; Feige, J.N.; Hartmann, S.; Brachat, S.; Rivet, H.; Koelbing, C.; Morvan, F.; et al. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol. Cell. Biol. 2014, 34, 606–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Nelson, B.R.; Bezprozvannaya, S.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proc. Natl. Acad. Sci. USA 2014, 111, 4109–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frontera, W.R.; Ochala, J. Skeletal muscle: A brief review of structure and function. Calcif. Tissue Int. 2015, 96, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Vanek, T.; Kohli, A. Biochemistry, Myoglobin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- De Angelis, L.; Borghi, S.; Melchionna, R.; Berghella, L.; Baccarani-Contri, M.; Parise, F.; Ferrari, S.; Cossu, G. Inhibition of myogenesis by transforming growth factor beta is density-dependent and related to the translocation of transcription factor MEF2 to the cytoplasm. Proc. Natl. Acad. Sci. USA 1998, 95, 12358–12363. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Kang, J.S.; Derynck, R. TGF-beta-activated Smad3 represses MEF2-dependent transcription in myogenic differentiation. EMBO J. 2004, 23, 1557–1566. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef]
- Graham, Z.A.; De Gasperi, R.; Bauman, W.A.; Cardozo, C.P. Recombinant myostatin reduces highly expressed microRNAs in differentiating C2C12 cells. Biochem. Biophys. Rep. 2017, 9, 273–280. [Google Scholar] [CrossRef]
- Sun, Y.; Ge, Y.; Drnevich, J.; Zhao, Y.; Band, M.; Chen, J. Mammalian target of rapamycin regulates miRNA-1 and follistatin in skeletal myogenesis. J. Cell Biol. 2010, 189, 1157–1169. [Google Scholar] [CrossRef] [Green Version]
- Shum, A.M.; Mahendradatta, T.; Taylor, R.J.; Painter, A.B.; Moore, M.M.; Tsoli, M.; Tan, T.C.; Clarke, S.J.; Robertson, G.R.; Polly, P. Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer-induced skeletal muscle wasting. Aging 2012, 4, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Judge, S.M.; Deyhle, M.R.; Neyroud, D.; Nosacka, R.L.; D’Lugos, A.C.; Cameron, M.E.; Vohra, R.S.; Smuder, A.J.; Roberts, B.M.; Callaway, C.S.; et al. MEF2c-Dependent Downregulation of Myocilin Mediates Cancer-Induced Muscle Wasting and Associates with Cachexia in Patients with Cancer. Cancer Res. 2020, 80, 1861–1874. [Google Scholar] [CrossRef] [Green Version]
- Rullman, E.; Fernandez-Gonzalo, R.; Mekjavic, I.B.; Gustafsson, T.; Eiken, O. MEF2 as upstream regulator of the transcriptome signature in human skeletal muscle during unloading. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R799–R809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, I.; Ciciliot, S.; Dyar, K.A.; Abraham, R.; Murgia, M.; Agatea, L.; Akimoto, T.; Bicciato, S.; Forcato, M.; Pierre, P.; et al. MRF4 negatively regulates adult skeletal muscle growth by repressing MEF2 activity. Nat. Commun. 2016, 7, 12397. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Freyssenet, D. Phenotypic features of cancer cachexia-related loss of skeletal muscle mass and function: Lessons from human and animal studies. J. Cachexia Sarcopenia Muscle 2021, 12, 252–273. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primers 5′-3′ | Accession No. | |
|---|---|---|---|
| Forward | Reverse | ||
| Human | |||
| MYH7 | GAGCAAGCCAACACCAACCT | TGTGGCAAAGCTACTCCTCCATT | NM_000257.3 |
| MYH1 | TCCACTTTAAGGTCGCATCTCT | GTTCTGGGCTTCAATTCGCTC | NM_005963.3 |
| MYH2 | AGCCCTTGGAATGAGGCTGA | GCTCCGCCACAAAGACAGAT | NM_017534.5 |
| MEF2C | TTCCAGTATGCCAGCACCG | GGCCCTTCTTTCTCAACGTCTC | NM_002397.4 |
| MB | AGATTAAGCCCCTGGCACAGT | GATGCATTCCGAGATGAACTC | NM_005368.2 |
| MYOM-1 | GCAGCCTCAGCCTACGATTA | TGACATGCTTTTGACGTCCTG | NM_003803.3 |
| PPARGC1A | CGGGATGATGGAGACAGCTA | CTTGGTGGAAGCAGGGTCAA | NM_001354825.1 |
| MyoD | CGACGGCATGATGGACTACA | GGCAGTCTAGGCTCGACAC | NM_002478.4 |
| MyoG | GCCATCCAGTACATCGAGCG | ATCTGTAGGGTCAGCCGTGA | NM_002479.5 |
| Myf5 | AACTACTATAGCCTGCCGGG | GATCCTGGAGAGGCAACCCA | NM_005593.2 |
| MRF4 | CTTGAGGGTGCGGATTTCCT | AAGCGCAGGCTCAGTTACTT | NM_002469.2 |
| HDAC4 | TTGGATGTCACAGACTCCGC | CCTTCTCGTGCCACAAGTCT | NM_006037.3 |
| TRIM63 | CATGTGCAAGGAGCACGAAG | GCCACCAGCATGGAGATACA | NM_032588.3 |
| Atrogin1 | TCACAGCTCACATCCCTGAG | AGACTTGCCGACTCTTTGGA | NM_058229.3 |
| MUSA1 | GTCATACTGCAGTGGGGGAAA | CGTGTCACACACATACATGGC | NM_032145.4 |
| GAPDH | CGCTGAGTACGTCGTGGAGTC | GCAGGAGGCATTGCTGATGA | |
| Mice | |||
| MYH7 | GGTGCCAAGGGCCTGAATGAGGAG | GGTCTGAGGGCTTCACGGGCAC | |
| MEF2C | GCTGTTCCAGTACGCCAGCAC | NM_025282.3 | |
| GAPDH | TGCACCACCAACTGCTTA | GGATGCAGGGATGATGTTC | NM_001289726.1 |
| Tbp | Mm01277042_m1 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loumaye, A.; Lause, P.; Zhong, X.; Zimmers, T.A.; Bindels, L.B.; Thissen, J.-P. Activin A Causes Muscle Atrophy through MEF2C-Dependent Impaired Myogenesis. Cells 2022, 11, 1119. https://doi.org/10.3390/cells11071119
Loumaye A, Lause P, Zhong X, Zimmers TA, Bindels LB, Thissen J-P. Activin A Causes Muscle Atrophy through MEF2C-Dependent Impaired Myogenesis. Cells. 2022; 11(7):1119. https://doi.org/10.3390/cells11071119
Chicago/Turabian StyleLoumaye, Audrey, Pascale Lause, Xiaoling Zhong, Teresa A. Zimmers, Laure B. Bindels, and Jean-Paul Thissen. 2022. "Activin A Causes Muscle Atrophy through MEF2C-Dependent Impaired Myogenesis" Cells 11, no. 7: 1119. https://doi.org/10.3390/cells11071119