
3D In Vitro Platform for Cell and Explant Culture in Liquid-like Solids

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Manufacturing, Assembly, and Sterilization of Injection-Molded Perfusion Plates
2.2. Human Cardiomyocyte AC16 Culture and Handling in LLS
2.3. Glucose and Lactate Assays
2.4. Mouse Colon Explant Culture
2.5. Microexplant Collection from LLS
2.6. Immunofluorescence Assay
2.7. Semi-Quantification Analysis of Mouse Colon Microexplant Viability
2.8. Flow Cytometry
2.9. Statistical Analysis
3. Results
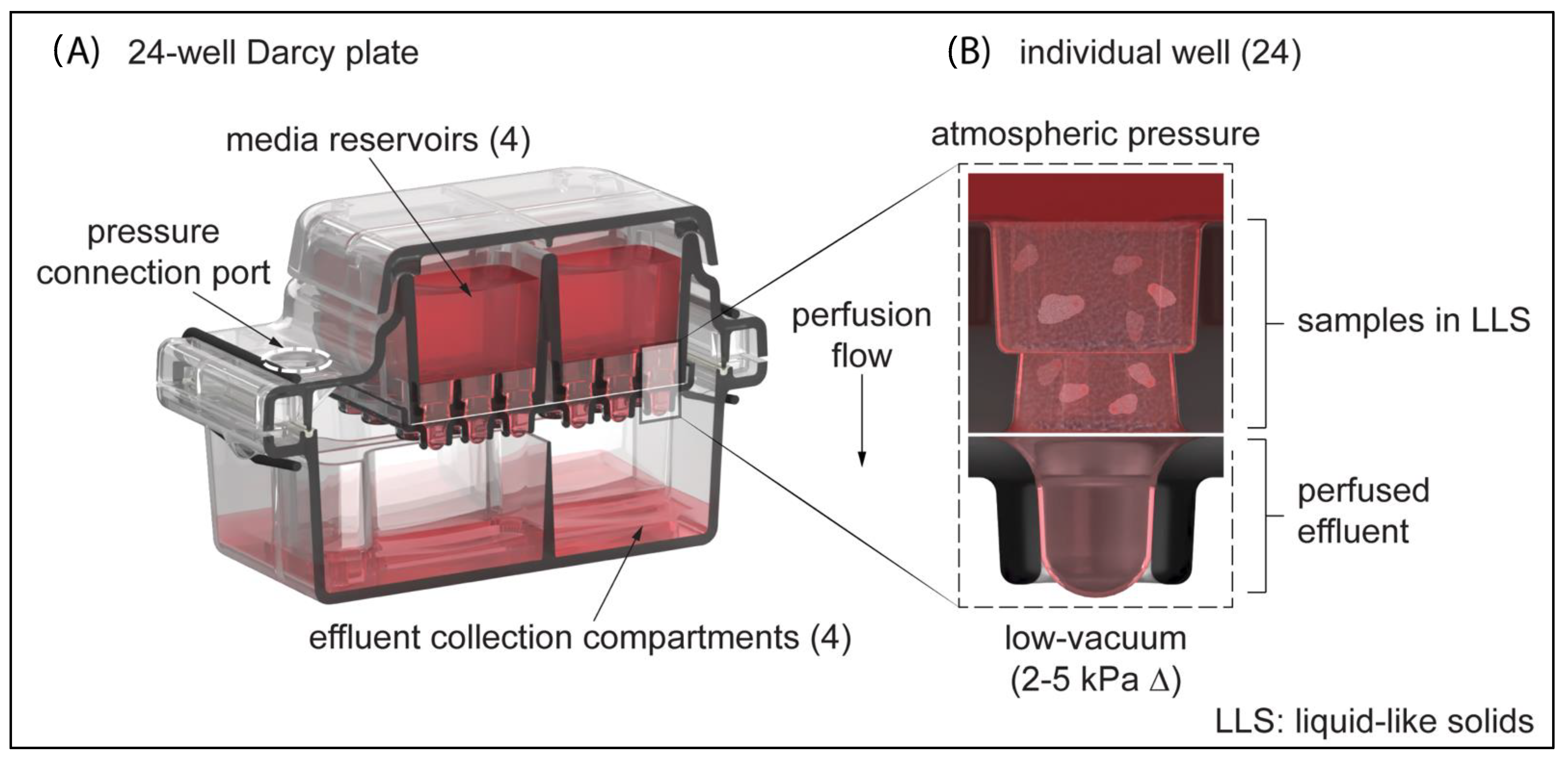
3.1. Three-Dimensional Cell Culture Platform
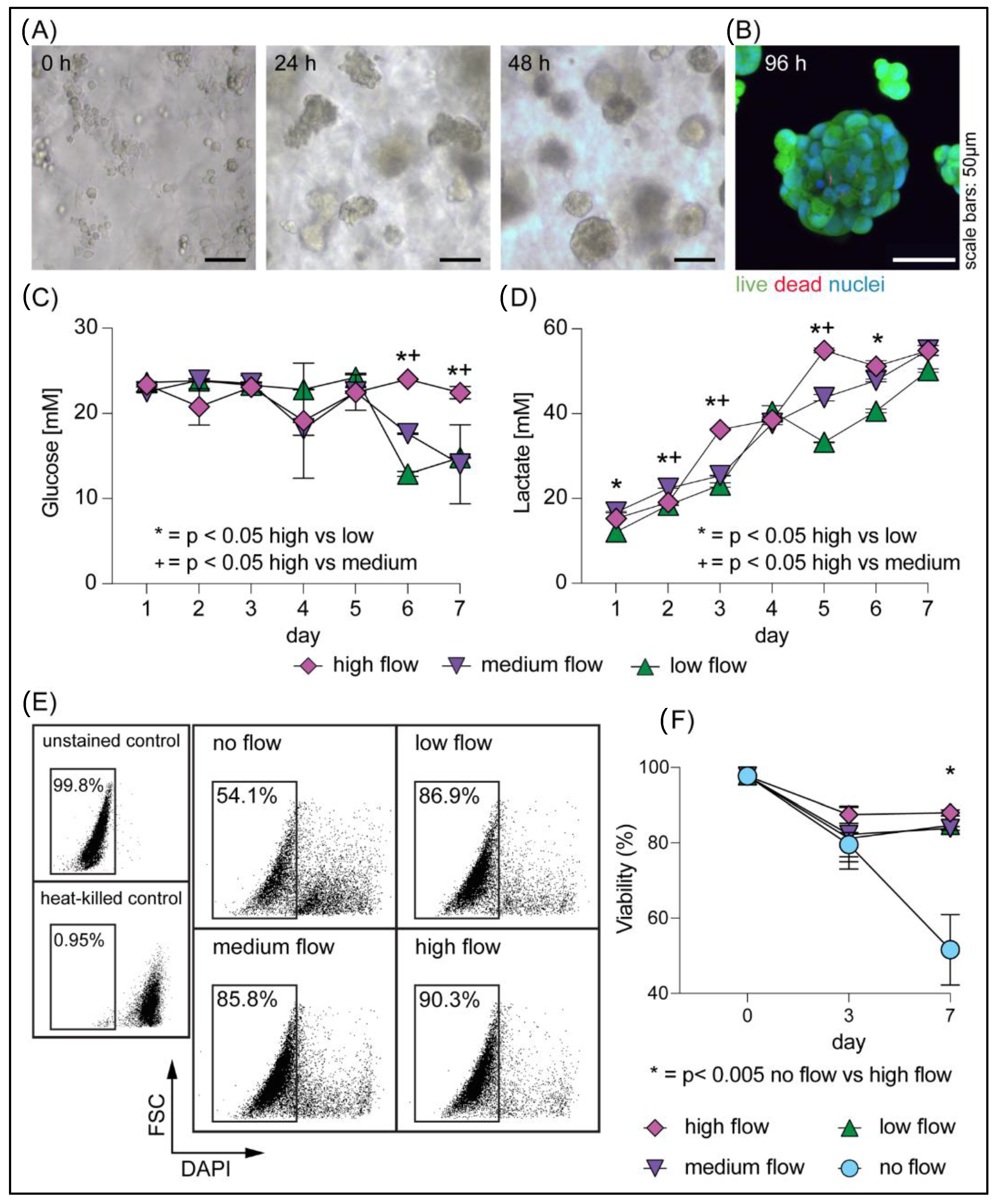
3.2. Rapid Generation of Cellular Spheroids in Perfusion Culture
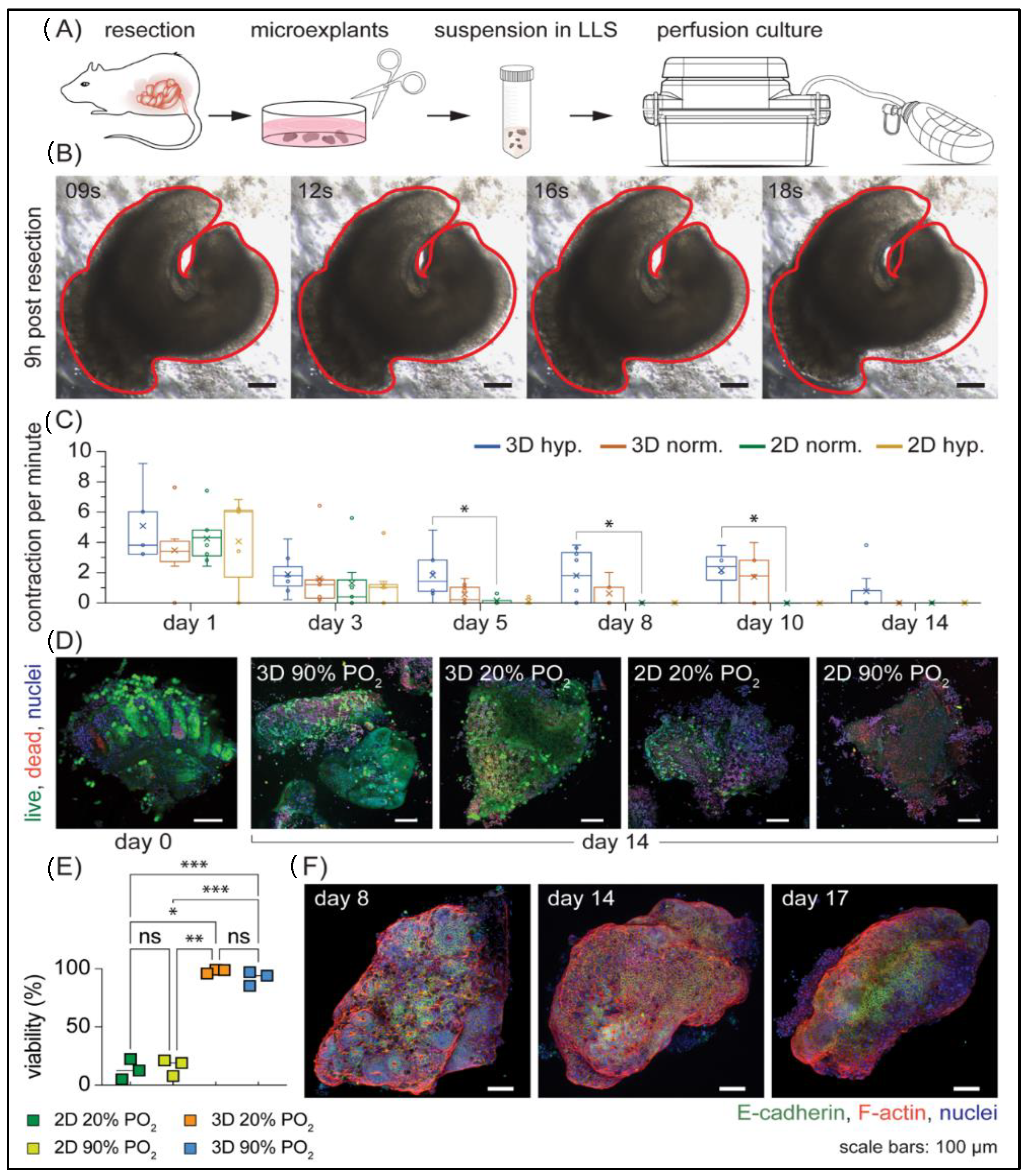
3.3. Long-Term Ex Vivo Culture of Microexplants in 3D
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clevers, H.; Tuveson, D.A. Organoid models for cancer research. Annu. Rev. Cancer Biol. 2019, 3, 223–234. [Google Scholar] [CrossRef]
- Powley, I.R.; Patel, M.; Miles, G.; Pringle, H.; Howells, L.; Thomas, A.; Kettleborough, C.; Bryans, J.; Hammonds, T.; Macfarlane, M.; et al. Patient-derived explants (PDEs) as a powerful preclinical platform for anti-cancer drug and biomarker discovery. Br. J. Cancer 2020, 122, 735–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunti, S.; Hoke, A.T.K.; Vu, K.; London, N.R., Jr. Organoid and spheroid tumor models: Techniques and applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef]
- Antoni, D.; Burckel, H.; Josset, E.; Noel, G. Three-dimensional cell culture: A breakthrough in vivo. Int. J. Mol. Sci. 2015, 16, 5517–5527. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarczyk, J.A.; Roberts, R.R.; Luke, B.T.; Chan, K.C.; Van Wagoner, C.M.; Felder, R.A.; Saul, R.G.; Simona, C.; Blonder, J. Comparative microsomal proteomics of a model lung cancer cell line NCI-H23 reveals distinct differences between molecular profiles of 3D and 2D cultured cells. Oncotarget 2021, 12, 2022–2038. [Google Scholar] [CrossRef] [PubMed]
- A Tagle, D. The NIH microphysiological systems program: Developing in vitro tools for safety and efficacy in drug development. Curr. Opin. Pharmacol. 2019, 48, 146–154. [Google Scholar] [CrossRef]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Nii, T.; Makino, K.; Tabata, Y. Three-dimensional culture system of cancer cells combined with biomaterials for drug screening. Cancers 2020, 12, 2754. [Google Scholar] [CrossRef]
- Dillard, P.; Köksal, H.; Inderberg, E.-M.; Wälchli, S. a spheroid killing assay by CAR T cells. J. Vis. Exp. 2018, 2018, e58785. [Google Scholar] [CrossRef] [Green Version]
- Schnalzger, T.E.; De Groot, M.H.; Zhang, C.; Mosa, M.H.; Michels, B.E.; Röder, J.; Darvishi, T.; Wels, W.S.; Farin, H.F. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. 2019, 38, e100928. [Google Scholar] [CrossRef]
- Jacob, F.; Ming, G.-L.; Song, H. Generation and biobanking of patient-derived glioblastoma organoids and their application in CAR T cell testing. Nat. Protoc. 2020, 15, 4000–4033. [Google Scholar] [CrossRef] [PubMed]
- Browning, T.H.; Trier, J.S. Organ culture of mucosal biopsies of human small intestine. J. Clin. Investig. 1969, 48, 1423–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autrup, H.; Barrett, L.A.; Jackson, F.E.; Jesudason, M.L.; Stoner, G.; Phelps, P.; Trump, B.; Harris, C.C. Explant culture of human colon. Gastroenterology 1978, 74, 1248–1257. [Google Scholar] [CrossRef]
- Autrup, H.; Stoner, G.D.; Jackson, F.; Harris, C.C.; Shamsuddin, A.K.M.; Barrett, L.A.; Trump, B.F. Explant culture of rat colon: A model system for studying metabolism of chemical carcinogens. In Vitro 1978, 14, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Vis, M.A.M.; Ito, K.; Hofmann, S. Impact of Culture Medium on Cellular Interactions in in vitro Co-culture Systems. Front. Bioeng. Biotechnol. 2020, 8, 911. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, J.M.; Yu, W.-Y.; A Hornsey, M.; Tosh, D.; Slack, J.M. In vitroculture of embryonic mouse intestinal epithelium: Cell differentiation and introduction of reporter genes. BMC Dev. Biol. 2006, 6, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hearn, C.J.; Young, H.; Ciampoli, D.; Lomax, A.; Newgreen, D. Catenary cultures of embryonic gastrointestinal tract support organ morphogenesis, motility, neural crest cell migration, and cell differentiation. Dev. Dyn. 1999, 214, 239–247. [Google Scholar] [CrossRef]
- Foty, R. A Simple hanging drop cell culture protocol for generation of 3D spheroids. J. Vis. Exp. 2011, 20, e2720. [Google Scholar] [CrossRef]
- Gupta, N.; Liu, J.; Patel, B.; Solomon, D.E.; Vaidya, B.; Gupta, V. Microfluidics-based 3D cell culture models: Utility in novel drug discovery and delivery research. Bioeng. Transl. Med. 2016, 1, 63–81. [Google Scholar] [CrossRef]
- van Helvert, S.; Friedl, P. Strain Stiffening of Fibrillar Collagen during Individual and Collective Cell Migration Identified by AFM Nanoindentation. ACS Appl. Mater. Interfaces 2016, 8, 21946–21955. [Google Scholar] [CrossRef]
- Porzionato, A.; Stocco, E.; Barbon, S.; Grandi, F.; Macchi, V.; De Caro, R. Tissue-engineered grafts from human decellularized extracellular matrices: A systematic review and future perspectives. Int. J. Mol. Sci. 2018, 19, 4117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Waele, J.; Reekmans, K.; Daans, J.; Goossens, H.; Berneman, Z.; Ponsaerts, P. 3D culture of murine neural stem cells on decellularized mouse brain sections. Biomaterials 2015, 41, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.S.; Postovit, L.M.; Lajoie, G.A. Matrigel: A complex protein mixture required for optimal growth of cell culture. Proteomics 2010, 10, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Bova, L.; Billi, F.; Cimetta, E. Mini-review: Advances in 3D bioprinting of vascularized constructs. Biol. Direct 2020, 15, 1–5. [Google Scholar] [CrossRef]
- Cao, X.; Maharjan, S.; Ashfaq, R.; Shin, J.; Zhang, Y.S. Bioprinting of Small-Diameter Blood Vessels. Engineering 2021, 7, 832–844. [Google Scholar] [CrossRef]
- Franco, C.; Gerhardt, H. Blood vessels on a chip. Nature 2012, 488, 465–466. [Google Scholar] [CrossRef]
- Schmid, J.; Schwarz, S.; Meier-Staude, R.; Sudhop, S.; Clausen-Schaumann, H.; Schieker, M.; Huber, R. A perfusion bioreactor system for cell seeding and oxygen-controlled cultivation of three-dimensional cell cultures. Tissue Eng.-Part C Methods 2018, 24, 585–595. [Google Scholar] [CrossRef]
- Sekiya, S.; Kikuchi, T.; Shimizu, T. Perfusion culture maintained with an air-liquid interface to stimulate epithelial cell organization in renal organoids in vitro. BMC Biomed. Eng. 2019, 1, 15. [Google Scholar] [CrossRef] [Green Version]
- Jun, Y.; Lee, J.; Choi, S.; Yang, J.H.; Sander, M.; Chung, S.; Lee, S.-H. In vivo–mimicking microfluidic perfusion culture of pancreatic islet spheroids. Sci. Adv. 2019, 5, eaax4520. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, L.; Armstrong, J.P.K.; Chen, Q.; Lin, Y.; Stevens, M.M. Void-Free 3D Bioprinting for In Situ Endothelialization and Microfluidic Perfusion. Adv. Funct. Mater. 2020, 30, 1908349. [Google Scholar] [CrossRef]
- Priyadarshini, B.M.; Dikshit, V.; Zhang, Y. 3D-printed bioreactors for in vitro modeling and analysis. Int. J. Bioprint. 2020, 6, 267. [Google Scholar] [CrossRef]
- Fendler, C.; Denker, C.; Harberts, J.; Bayat, P.; Zierold, R.; Loers, G.; Münzenberg, M.; Blick, R.H. Microscaffolds by Direct Laser Writing for Neurite Guidance Leading to Tailor-Made Neuronal Networks. Adv. Biosyst. 2019, 3, e1800329. [Google Scholar] [CrossRef] [PubMed]
- Gale, B.K.; Jafek, A.R.; Lambert, C.J.; Goenner, B.L.; Moghimifam, H.; Nze, U.C.; Kamarapu, S.K. A review of current methods in microfluidic device fabrication and future commercialization prospects. Inventions 2018, 3, 60. [Google Scholar] [CrossRef] [Green Version]
- Convery, N.; Gadegaard, N. 30 years of microfluidics. Micro Nano Eng. 2019, 2, 76–91. [Google Scholar] [CrossRef]
- Eduati, F.; Utharala, R.; Madhavan, D.; Neumann, U.P.; Longerich, T.; Cramer, T.; Saez-Rodriguez, J.; Merten, C.A. A microfluidics platform for combinatorial drug screening on cancer biopsies. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Huh, D.; Leslie, D.C.; Matthews, B.D.; Fraser, J.P.; Jurek, S.; Hamilton, G.A.; Thorneloe, K.S.; McAlexander, M.A.; Ingber, D.E. A human disease model of drug toxicity–induced pulmonary edema in a lung-on-a-chip microdevice. Sci. Transl. Med. 2012, 4, 159ra147. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.W.; Lim, J.; Rhie, J.W.; Kim, D.S. Investigation of effective shear stress on endothelial differentiation of human adipose-derived stem cells with microfluidic screening device. Microelectron. Eng. 2017, 174, 24–27. [Google Scholar] [CrossRef]
- Ko, J.; Ahn, J.; Kim, S.; Lee, Y.; Lee, J.; Park, D.; Jeon, N.L. Tumor spheroid-on-a-chip: A standardized microfluidic culture platform for investigating tumor angiogenesis. Lab Chip 2019, 19, 2822–2833. [Google Scholar] [CrossRef]
- Jeon, J.S.; Bersini, S.; Gilardi, M.; Dubini, G.; Charest, J.L.; Moretti, M.; Kamm, R.D. Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc. Natl. Acad. Sci. USA 2015, 112, 214–219, Erratum in Proc. Natl. Acad. Sci. USA 2015, 112, E818. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, T.; Gil, C.J.; Marshall, S.L.; Urueña, J.M.; O’Bryan, C.S.; Carstens, M.; Keselowsky, B.G.; Palmer, G.D.; Ghivizzani, S.; Gibbs, C.P.; et al. Liquid-like Solids Support Cells in 3D. ACS Biomater. Sci. Eng. 2016, 2, 1787–1795. [Google Scholar] [CrossRef]
- Davidson, M.M.; Nesti, C.; Palenzuela, L.; Walker, W.F.; Hernandez, E.; Protas, L.; Hirano, M.; Isaac, N.D. Novel cell lines derived from adult human ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2005, 39, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.A.; Sharma, Y.; Dupee, Z.; Nguyen, D.T.; Urueña, J.M.; Smolchek, R.A.; Loeb, J.C.; Machuca, T.N.; Lednicky, J.A.; Odde, D.J.; et al. Ex vivo SARS-CoV-2 infection of human lung reveals heterogeneous host defense and therapeutic responses. JCI Insight 2021, 6, e148003. [Google Scholar] [CrossRef] [PubMed]
- Poon, C. Measuring the density and viscosity of culture media for optimized computational fluid dynamics analysis of in vitro devices. bioRxiv 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.08.25.266221v1%0Ahttps://www.biorxiv.org/content/10.1101/2020.08.25.266221v1.abstract (accessed on 13 January 2022). [CrossRef] [PubMed]
- Chisti, Y. Hydrodynamic damage to animal cells. Crit. Rev. Biotechnol. 2001, 21, 67–110. [Google Scholar] [CrossRef]
- Natarajan, D.; Grigoriou, M.; Marcos-Gutierrez, C.; Atkins, C.; Pachnis, V. Multipotential progenitors of the mammalian enteric nervous system capable of colonising aganglionic bowel in organ culture. Development 1999, 126, 157–168. [Google Scholar] [CrossRef]
- Metzger, M.; Bareiss, P.M.; Nikolov, I.; Skutella, T.; Just, L. Three-dimensional slice cultures from murine fetal gut for investigations of the enteric nervous system. Dev. Dyn. 2006, 236, 128–133. [Google Scholar] [CrossRef]
- Baydoun, M.; Vanneste, S.B.; Creusy, C.; Guyot, K.; Gantois, N.; Chabe, M.; Delaire, B.; Mouray, A.; Baydoun, A.; Forzy, G.; et al. Three-dimensional (3D) culture of adult murine colon as an in vitro model of cryptosporidiosis: Proof of concept. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, P.S.; Elliott, J.; Grivel, J.-C.; Margolis, L.; Anton, P.; McGowan, I.; Shattock, R.J. Ex vivo culture of human colorectal tissue for the evaluation of candidate microbicides. Aids 2006, 20, 1237–1245. [Google Scholar] [CrossRef]
- Bareiss, P.M.; Metzger, M.; Sohn, K.; Rupp, S.; Frick, J.S.; Autenrieth, I.B.; Lang, F.; Schwarz, H.; Skutella, T.; Just, L. Organotypical tissue cultures from adult murine colon as an in vitro model of intestinal mucosa. Histochem. Cell Biol. 2008, 129, 795–804. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Platform | Culture Condition | Size | Duration | Note |
|---|---|---|---|---|
| Air–liquid interface | 37 °C, 5%CO2, 95% O2 | n/a | 24 h | The interchange of macromolecules in the culture medium was defined as an important factor [12]. |
| Rocker system | 37 °C, 5%CO2, 95% O2 | 5 × 5 mm | 3 months | The rocker system allowed the samples to be intermittently exposed to the air and prevented the accumulation of mucus, digestive enzyme, hormones, and growth factor by agitation. The technique allowed the culture of rat colon explants for up to 3 months [14], and human colon explants for up to 20 days [13]. |
| Rocker system | 37 °C, 5%CO2, 95% O2 | 5 × 5 mm | 20 days | |
| Catenary culture | 37 °C, 5%CO2, 95% Air | Whole section | 10 days | Embryonic gut segments were cultured in suspension while immersed in media. The cultured segments showed random contraction after day 6 [17]. |
| Free-floating culture | 37 °C, 5%CO2, 95% Air | Whole gut | 14 days | The whole embryonic mouse gut was cultured as a “free-floating organ”. Peristalsis was observed after 48 h [45]. |
| Air–liquid interface | 37 °C, 5%CO2, 95% Air | 2–3 mm3 | 12 days | The samples were cultured on a submerged gel foam raft. Human colon explants with epithelial and muscular mucosae were cultured for HIV-1 infection study [48]. |
| Immersion culture | 37 °C, 5%CO2, 95% Air | Whole section | 11 days | Mouse embryo intestinal explants were plated on fibronectin-coated coverslips and immersed in liquid media [16]. |
| Membrane insert | 37 °C, 5%CO2, 95% Air | 200 µm slices | 14 days | The contraction was maintained up to day 14. After 2 weeks of culture, treatment with 100 μM of serotonin and 10 μM of calcium channel blocker tetrodotoxin increased and reduced contraction, respectively [46]. |
| Membrane insert | 37 °C, 5%CO2, 95% Air | 2 mm2 | 14 days | Murine colon segments were cultured in liquid-cover media for up to 2 weeks [49]. |
| Membrane insert | 37 °C, 5%CO2, 95% Air | 12 mm2 | 35 days | Culture of colon segments from SCID mice by membrane insert technique [47]. |
| (a) | ||||
| Duration of Culture (Days) | 3-D | 2-D | ||
| PO2 (%) | PO2 (%) | |||
| 90% | 20% | 90% | 20% | |
| 3 | 1.88 | 1.60 | 1.14 | 1.29 |
| 5 | 1.83 | 0.57 | 0.07 | 0.17 |
| 8 | 1.78 | 0.60 | - | - |
| 14 | 0.77 | - | - | - |
| (b) | ||||
| Duration of Culture (Days) | 3-D Perfusion | 2-D | ||
| PO2 (%) | PO2 (%) | |||
| 90% | 20% | 90% | 20% | |
| 3 | 4.2 | 6.4 | 4.6 | 5.6 |
| 5 | 4.8 | 1.6 | 0.4 | 0.6 |
| 8 | 3.8 | 2 | - | - |
| 14 | 3.8 | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, D.T.; Famiglietti, J.E.; Smolchek, R.A.; Dupee, Z.; Diodati, N.; Pedro, D.I.; Urueña, J.M.; Schaller, M.A.; Sawyer, W.G. 3D In Vitro Platform for Cell and Explant Culture in Liquid-like Solids. Cells 2022, 11, 967. https://doi.org/10.3390/cells11060967
Nguyen DT, Famiglietti JE, Smolchek RA, Dupee Z, Diodati N, Pedro DI, Urueña JM, Schaller MA, Sawyer WG. 3D In Vitro Platform for Cell and Explant Culture in Liquid-like Solids. Cells. 2022; 11(6):967. https://doi.org/10.3390/cells11060967
Chicago/Turabian StyleNguyen, Duy T., Jack E. Famiglietti, Ryan A. Smolchek, Zadia Dupee, Nickolas Diodati, Diego I. Pedro, Juan M. Urueña, Matthew A. Schaller, and W. Gregory Sawyer. 2022. "3D In Vitro Platform for Cell and Explant Culture in Liquid-like Solids" Cells 11, no. 6: 967. https://doi.org/10.3390/cells11060967