
Emerging Roles of Airway Epithelial Cells in Idiopathic Pulmonary Fibrosis

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: An Emerging Role of the Airway Epithelium in IPF Aetiology
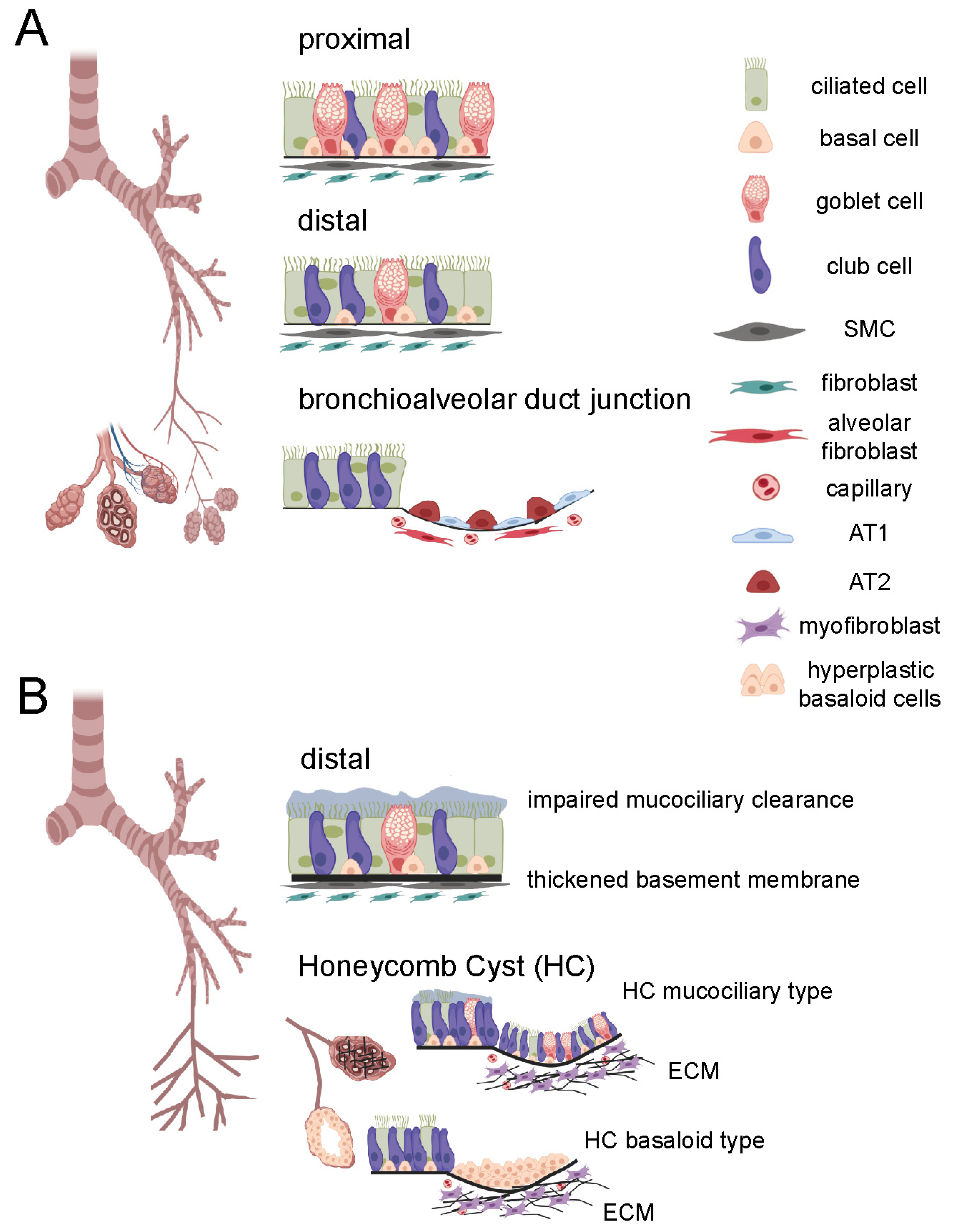
2. General Airway Structure
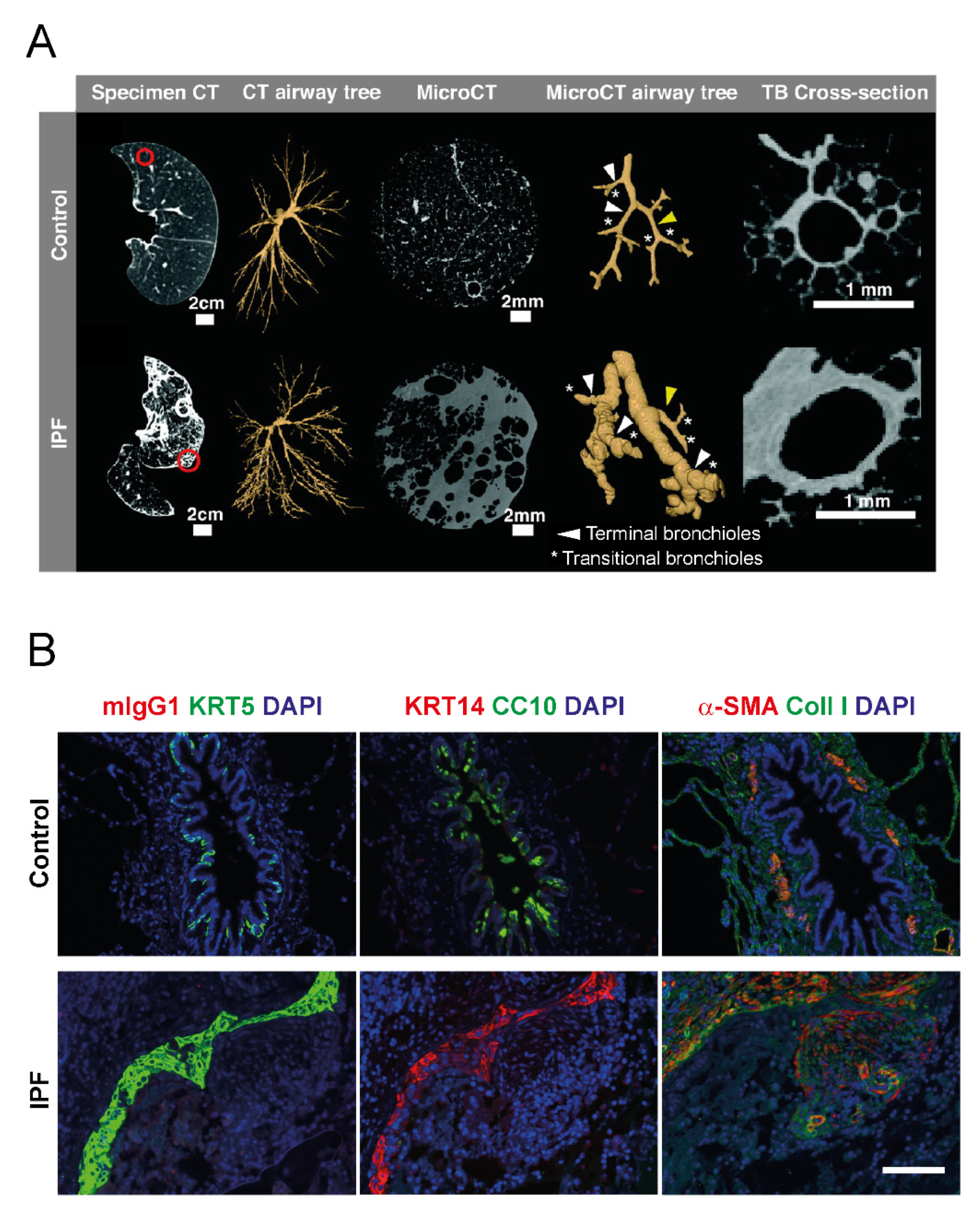
3. Changes in Airway Morphology in IPF
3.1. Airway Dilation
3.2. Increased Airway Wall Thickness (AWT)
3.3. Bronchiolar Abnormalities
3.4. Honeycomb Formation and Bronchiolization
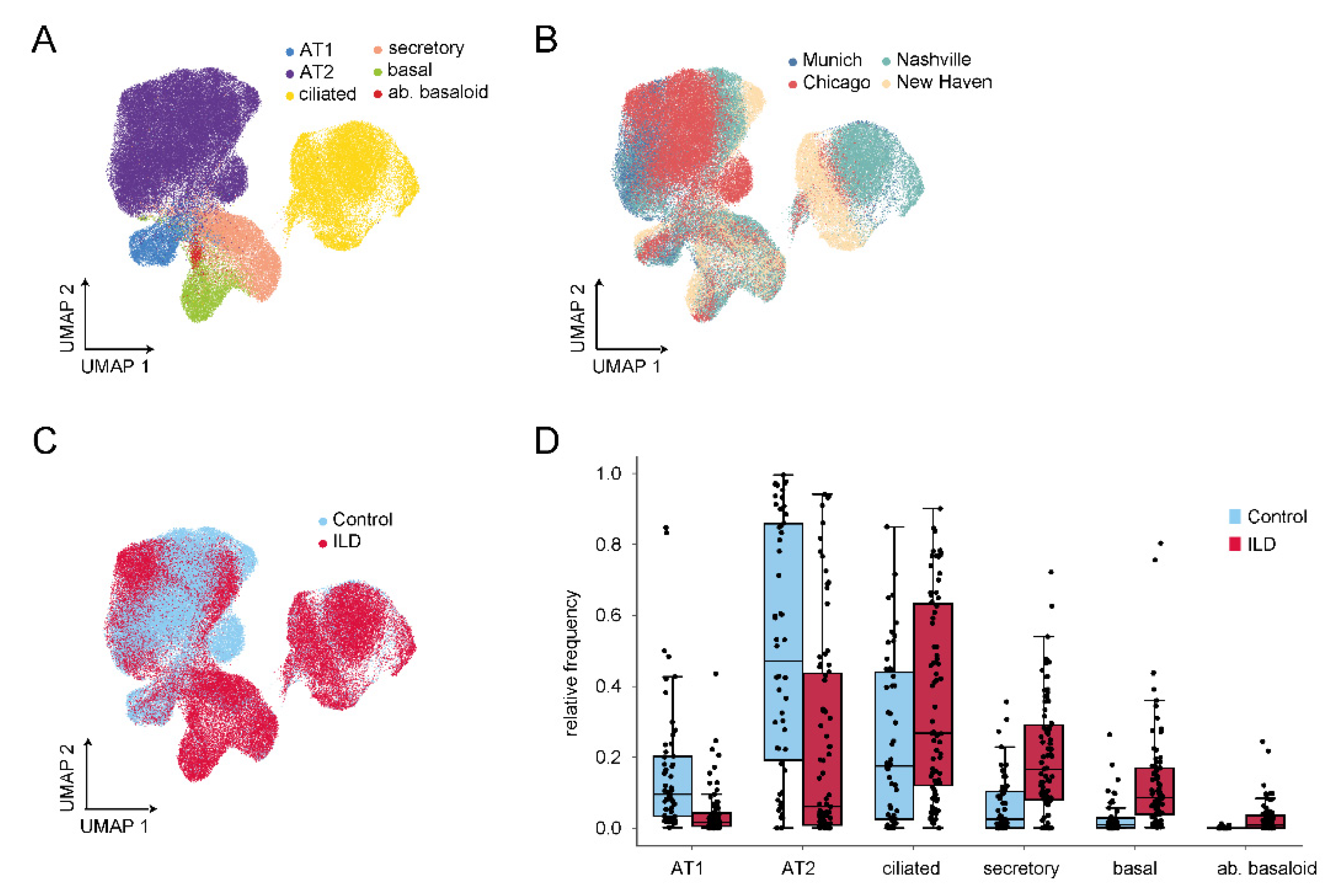
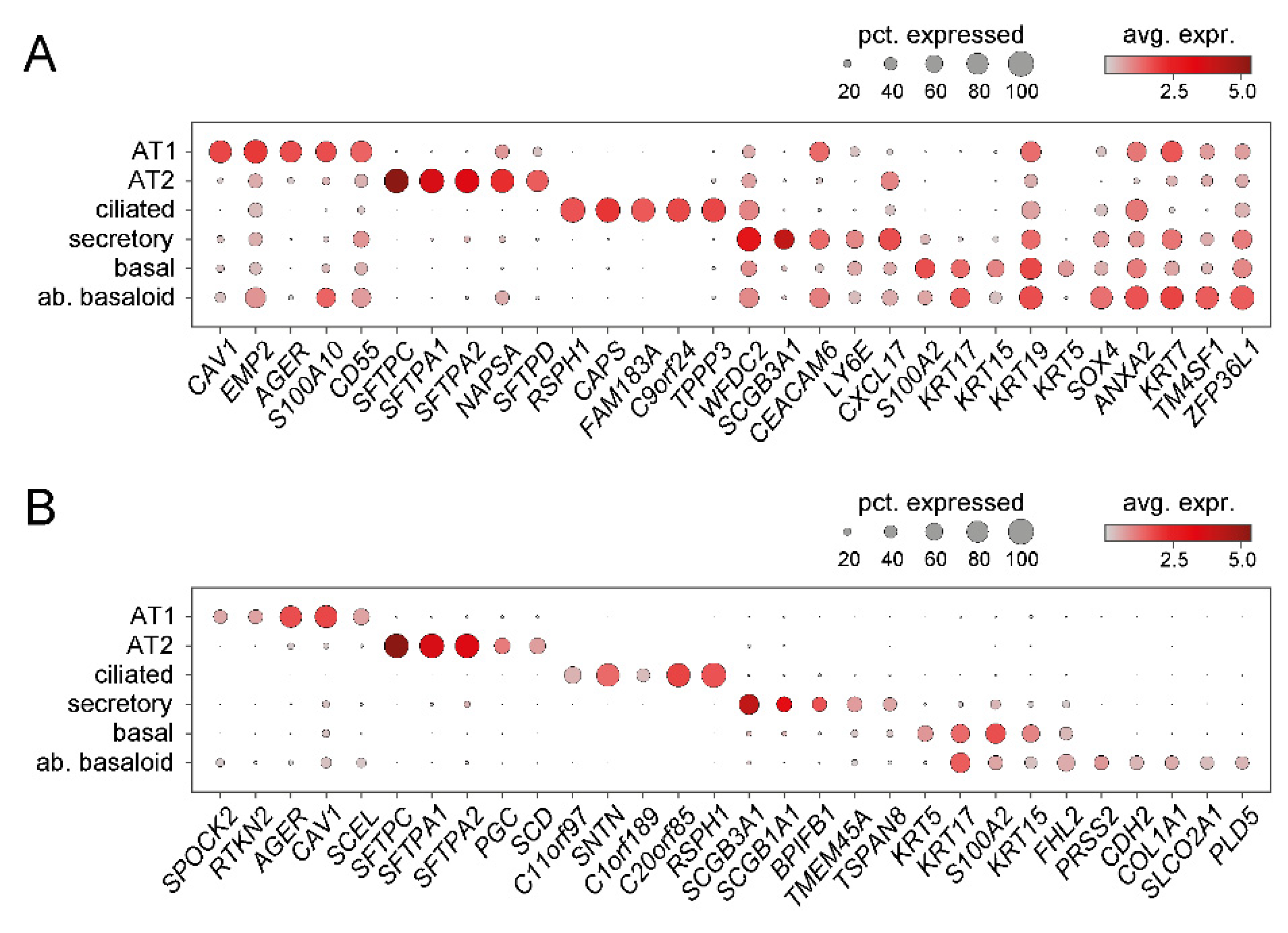
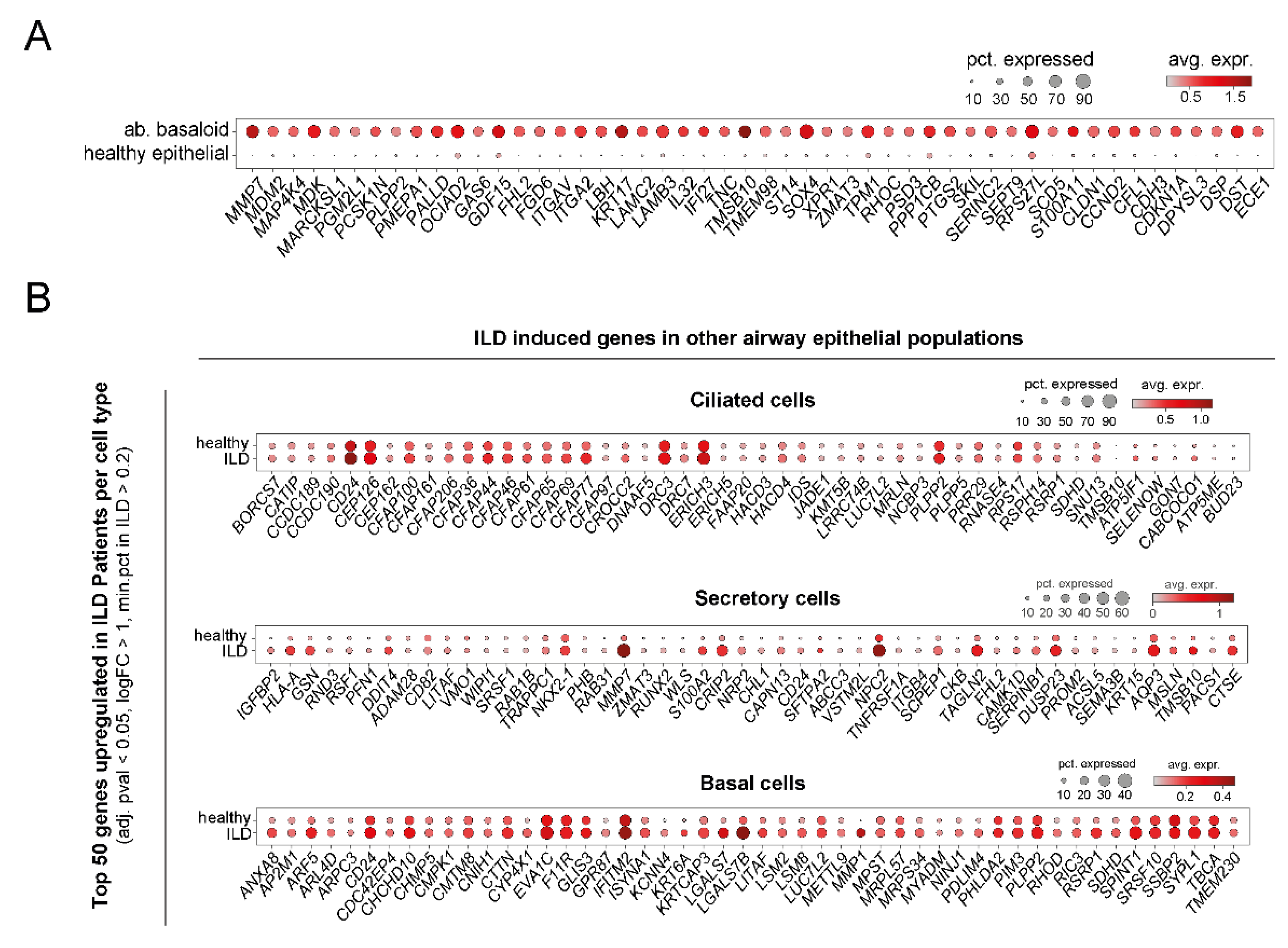
4. Recent Insights from Single Cell RNA-Sequencing (scRNA-Seq) Studies
5. Changes in Airway Function
5.1. Mucociliary Clearance
5.2. Epithelial Barrier Dysfunction in IPF Pathogenesis
5.3. Other Changes in Airway Function
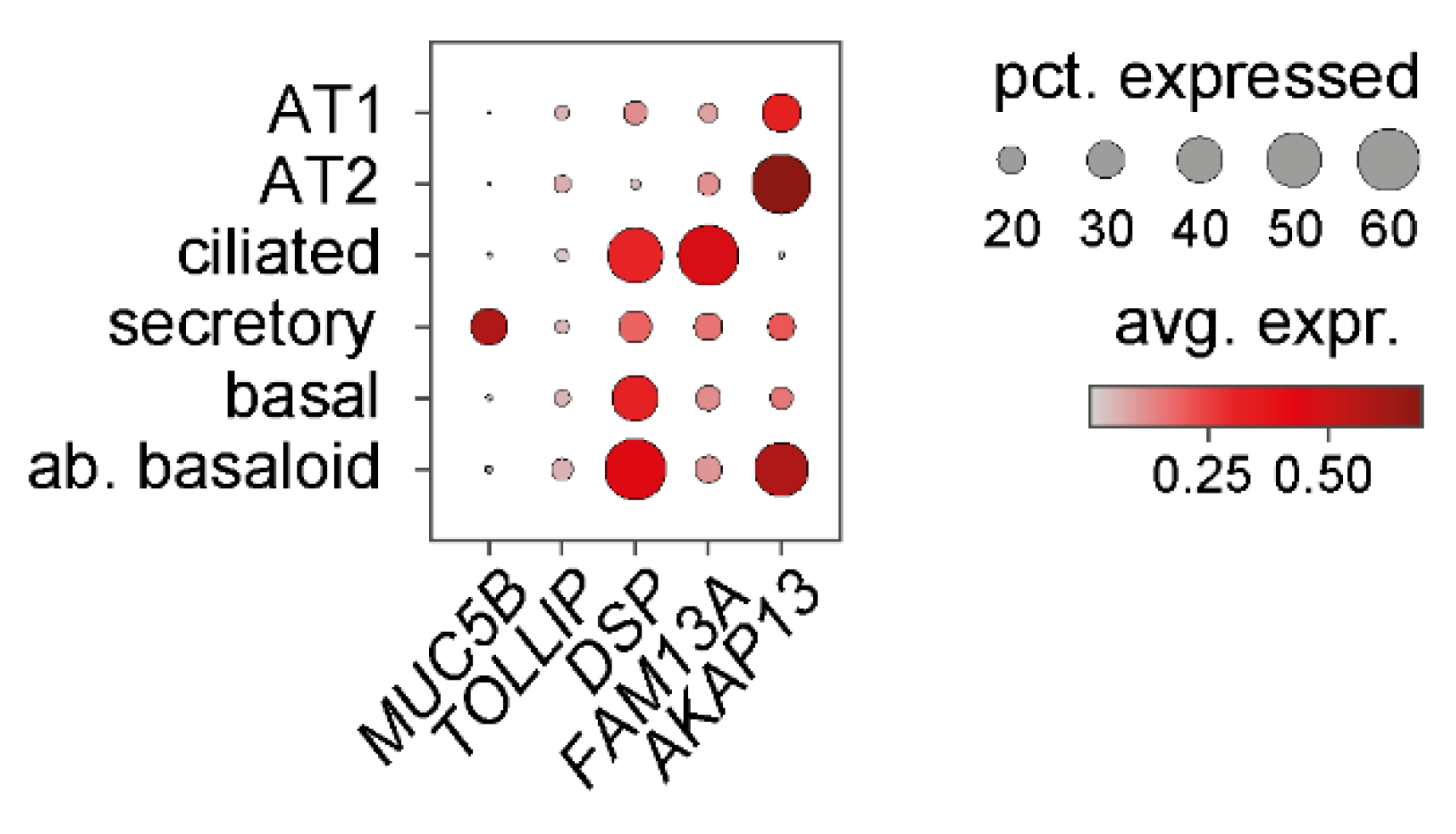
6. Genetic Evidence Indicating Involvement of Bronchial Epithelium in IPF
6.1. MUC5B
6.2. TOLLIP
6.3. DSP
6.4. FAM13A
6.5. AKAP13
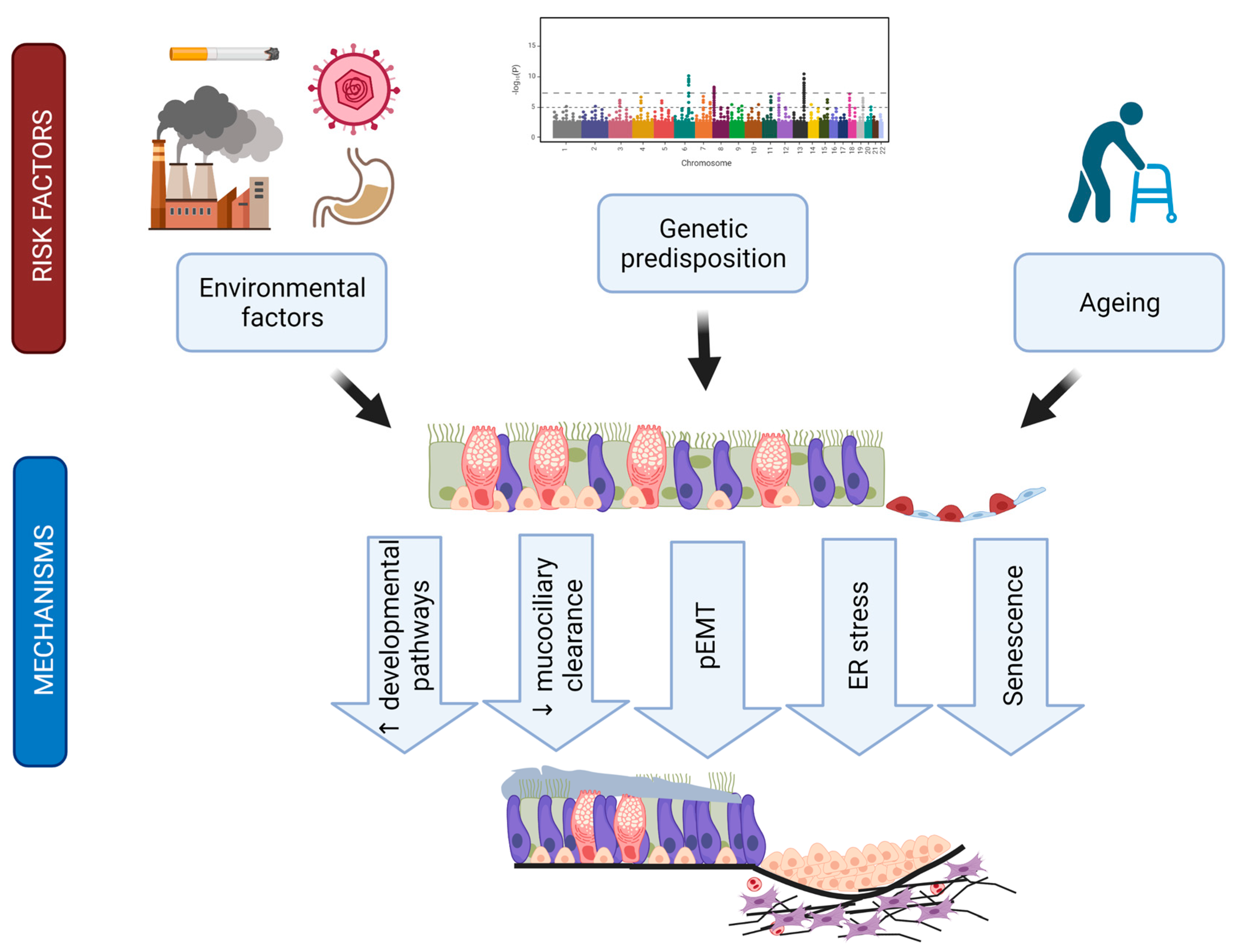
7. Implicated Mechanisms
7.1. Types of Epithelial Injury
7.2. Epithelial Apoptosis
7.3. Endoplasmic Reticulum (ER) Stress as Trigger for Epithelial Apoptosis
7.4. Ageing and Epithelial Senescence
7.5. Reactivation of Developmental Pathways
7.5.1. Transforming Growth Factor-β (TGF-β) Signalling
7.5.2. WNT Signalling Pathway
7.5.3. Sonic Hedgehog Signalling (SHH) Pathway
7.5.4. Notch Signalling Pathway
7.6. Epigenetic Mechanisms
7.7. Non-Coding RNAs
8. Summary, Conclusions, and Emerging Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 379, 797–798. [Google Scholar] [CrossRef] [PubMed]
- Crystal, R.G.; Randell, S.H.; Engelhardt, J.F.; Voynow, J.; Sunday, M.E. Airway epithelial cells: Current concepts and challenges. Proc. Am. Thorac. Soc. 2008, 5, 772–777. [Google Scholar] [CrossRef]
- Rock, J.R.; Randell, S.H.; Hogan, B.L.M. Airway basal stem cells: A perspective on their roles in epithelial homeostasis and remodeling. Dis. Model. Mech. 2010, 3, 545–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [Green Version]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, K.; Cui, G.; Huang, X.; Yao, S.; Guo, W.; Qin, Z.; Li, Y.; Yang, R.; Pu, W.; et al. Lung regeneration by multipotent stem cells residing at the bronchioalveolar-duct junction. Nat. Genet. 2019, 51, 728–738. [Google Scholar] [CrossRef]
- McLellan, T.; George, P.M.; Ford, P.; De Backer, J.; Van Holsbeke, C.; Mignot, B.; Screaton, N.J.; Ruggiero, A.; Thillai, M. Idiopathic pulmonary fibrosis: Airway volume measurement identifies progressive disease on computed tomography scans. ERJ Open Res. 2020, 6, 00290-2019. [Google Scholar] [CrossRef]
- Tanabe, N.; McDonough, J.E.; Vasilescu, D.M.; Ikezoe, K.; Verleden, S.E.; Xu, F.; Wuyts, W.A.; Vanaudenaerde, B.M.; Colby, T.V.; Hogg, J.C. Pathology of idiopathic pulmonary fibrosis assessed by a combination of microcomputed tomography, histology, and immunohistochemistry. Am. J. Pathol. 2020, 190, 2427–2435. [Google Scholar] [CrossRef]
- McDonough, J.E.; Verleden, S.E.; Verschakelen, J.; Wuyts, W.; Vanaudenaerde, B.M. The structural origin of honeycomb cysts in IPF. Am. J. Respir. Crit. Care Med. 2018, 197, A6388. [Google Scholar]
- Ikezoe, K.; Hackett, T.-L.; Peterson, S.; Prins, D.; Hague, C.J.; Murphy, D.; LeDoux, S.; Chu, F.; Xu, F.; Cooper, J.D.; et al. Small Airway Reduction and Fibrosis Is an Early Pathologic Feature of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 204, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Pastre, J.; Plantier, L.; Planes, C.; Borie, R.; Nunes, H.; Delclaux, C.; Israël-Biet, D. Different KCO and VA combinations exist for the same DLCO value in patients with diffuse parenchymal lung diseases. BMC Pulm. Med. 2015, 15, 100. [Google Scholar] [CrossRef] [Green Version]
- Plantier, L.; Cazes, A.; Dinh-Xuan, A.-T.; Bancal, C.; Marchand-Adam, S.; Crestani, B. Physiology of the lung in idiopathic pulmonary fibrosis. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2018, 27, 170062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, P.; Kohlha, M.; Meyer, T.; Selzer, T.; Heyder, J.; Häussinger, K.; Ha¨ußinger, K. Aerosol-derived airway morphometry and aerosol bolus dispersion in patients with lung fibrosis and lung emphysema. Chest 1999, 116, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Plantier, L.; Debray, M.-P.; Estellat, C.; Flamant, M.; Roy, C.; Bancal, C.; Borie, R.; Israel-Biet, D.; Mal, H.; Crestani, B.; et al. Increased volume of conducting airways in idiopathic pulmonary fibrosis is independent of disease severity: A volumetric capnography study. J. Breath Res. 2016, 10, 16005. [Google Scholar] [CrossRef] [PubMed]
- Westcott, J.L.; Cole, S.R. Traction bronchiectasis in end-stage pulmonary fibrosis. Radiology 1986, 161, 665–669. [Google Scholar] [CrossRef]
- Hino, T.; Lee, K.S.; Han, J.; Hata, A.; Ishigami, K.; Hatabu, H. Spectrum of pulmonary fibrosis from interstitial lung abnormality to usual interstitial pneumonia: Importance of identification and quantification of traction bronchiectasis in patient management. Korean J. Radiol. 2021, 22, 811–828. [Google Scholar] [CrossRef]
- Walsh, S.L.F.; Wells, A.U.; Sverzellati, N.; Devaraj, A.; von der Thusen, J.; Yousem, S.A.; Colby, T.V.; Nicholson, A.G.; Hansell, D.M. Relationship between fibroblastic foci profusion and high resolution CT morphology in fibrotic lung disease. BMC Med. 2015, 13, 1–8. [Google Scholar] [CrossRef]
- Piciucchi, S.; Tomassetti, S.; Ravaglia, C.; Gurioli, C.; Gurioli, C.; Dubini, A.; Carloni, A.; Chilosi, M.; Colby, T.V.; Poletti, V. From “traction bronchiectasis” to honeycombing in idiopathic pulmonary fibrosis: A spectrum of bronchiolar remodeling also in radiology? BMC Pulm. Med. 2016, 16, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Verleden, S.; Tanabe, N.; McDonough, J.; Vasilescu, D.M.; Xu, F.; A Wuyts, W.; Piloni, D.; De Sadeleer, L.; Willems, S.; Mai, C.; et al. Small airways pathology in idiopathic pulmonary fibrosis: A retrospective cohort study. Lancet Respir. Med. 2020, 8, 573–584. [Google Scholar] [CrossRef]
- Miller, E.R.; Putman, R.K.; Diaz, A.A.; Xu, H.; Estépar, R.S.J.; Araki, T.; Nishino, M.; De Frías, S.P.; Hida, T.; Ross, J.; et al. Increased airway wall thickness in interstitial lung abnormalities and idiopathic pulmonary fibrosis. Ann. Am. Thorac. Soc. 2019, 16, 447–454. [Google Scholar] [CrossRef]
- Chilosi, M.; Poletti, V.; Murer, B.; Lestani, M.; Cancellieri, A.; Montagna, L.; Piccoli, P.; Cangi, G.; Doglioni, C. Abnormal re-epithelialization and lung remodeling in idiopathic pulmonary fibrosis: The role of deltaN-P63. Lab. Investig. 2002, 82, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Zamò, A.; Doglioni, C.; Reghellin, D.; Lestani, M.; Montagna, L.; Pedron, S.; Ennas, M.G.; Cancellieri, A.; Murer, B.; et al. Migratory marker expression in fibroblast foci of idiopathic pulmonary fibrosis. Respir. Res. 2006, 7, 95. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, N.F.; Schamberger, A.C.; Nayakanti, S.; Hatz, R.; Behr, J.; Eickelberg, O. Detection and quantification of epithelial progenitor cell populations in human healthy and IPF lungs. Respir. Res. 2016, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Strickland, N.H.; Hughes, J.M.B.; Hart, D.A.; Myers, M.J.; Lavender, J.P. Cause of regional ventilation-perfusion mismatching in patients with idiopathic pulmonary fibrosis: A combined CT and scintigraphic study. Am. J. Roentgenol. 1993, 161, 719–725. [Google Scholar] [CrossRef] [Green Version]
- Hansell, D.M.; Bankier, A.A.; MacMahon, H.; McLoud, T.C.; Müller, N.L.; Remy, J. Fleischner society: Glossary of terms for thoracic imaging. Radiology 2008, 246, 697–722. [Google Scholar] [CrossRef] [Green Version]
- Akira, M. Radiographic differentiation of advanced fibrocystic lung diseases. Ann. Am. Thorac. Soc. 2017, 14, 432–440. [Google Scholar] [CrossRef]
- Evans, C.M.; Fingerlin, T.E.; Schwarz, M.I.; Lynch, D.; Kurche, J.; Warg, L.; Yang, I.V.; Schwartz, D.A. Idiopathic pulmonary fibrosis: A genetic disease that involves mucociliary dysfunction of the peripheral airways. Physiol. Rev. 2016, 96, 1567–1591. [Google Scholar] [CrossRef] [PubMed]
- Seibold, M.A.; Smith, R.W.; Urbanek, C.; Groshong, S.D.; Cosgrove, G.P.; Brown, K.K.; Schwarz, M.I.; Schwartz, D.A.; Reynolds, S.D. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS ONE 2013, 8, e58658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasse, A.; Binder, H.; Schupp, J.; Kayser, G.; Bargagli, E.; Jaeger, B.; Hess, M.; Rittinghausen, S.; Vuga, L.; Lynn, H.; et al. BAL cell gene expression is indicative of outcome and airway basal cell involvement in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Salwig, I.; Spitznagel, B.; Vazquez-Armendariz, A.I.; Khalooghi, K.; Guenther, S.; Herold, S.; Szibor, M.; Braun, T. Bronchioalveolar stem cells are a main source for regeneration of distal lung epithelia in vivo. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Plantier, L.; Crestani, B.; E Wert, S.; Dehoux, M.; Zweytick, B.; Guenther, A.; A Whitsett, J. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 2011, 66, 651–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.A.; Hu, Y.; Yamamoto, Y.; Hoe, N.B.; Wei, T.S.; Mu, D.; Sun, Y.; Joo, L.S.; Dagher, R.; Zielonka, E.M.; et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 2011, 147, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Zuo, W.; Zhang, T.; Wu, D.Z.; Guan, S.P.; Liew, A.-A.; Yamamoto, Y.; Wang, X.; Lim, S.J.; Vincent, M.; Lessard, M.; et al. p63(+)Krt5(+) distal airway stem cells are essential for lung regeneration. Nature 2014, 517, 616–620. [Google Scholar] [CrossRef]
- Xi, Y.; Kim, T.; Brumwell, A.N.; Driver, I.; Wei, Y.; Tan, V.; Jackson, J.R.; Xu, J.; Lee, D.-K.; Gotts, J.E.; et al. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat. Cell Biol. 2017, 19, 904–914. [Google Scholar] [CrossRef]
- Ray, S.; Chiba, N.; Yao, C.; Guan, X.; McConnell, A.M.; Brockway, B.; Que, L.; McQualter, J.L.; Stripp, B.R. Rare SOX2+ airway progenitor cells generate KRT5+ cells that repopulate damaged alveolar parenchyma following influenza virus infection. Stem Cell Rep. 2016, 7, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Kathiriya, J.J.; Wang, C.; Zhou, M.; Brumwell, A.; Cassandras, M.; Le Saux, C.J.; Cohen, M.; Alysandratos, K.-D.; Wang, B.; Wolters, P.; et al. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5(+) basal cells. Nature 2021, 24, 10–23. [Google Scholar] [CrossRef]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.-K.T.; Funari, V.A.; Gokey, J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [Green Version]
- Danopoulos, S.; Alonso, I.; Thornton, M.E.; Grubbs, B.H.; Bellusci, S.; Warburton, D.; Al Alam, D. Human lung branching morphogenesis is orchestrated by the spatiotemporal distribution of ACTA2, SOX2, and SOX. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L144–L149. [Google Scholar] [CrossRef]
- Wasnick, R.M.; Shalashova, I.; Wilhelm, J.; Khadim, A.; Schmidt, N.; Hackstein, H.; Hecker, A.; Hoetzenecker, K.; Seeger, W.; Bellusci, S.; et al. Differential lysotracker uptake defines two populations of distal epithelial cells in idiopathic pulmonary fibrosis. Cells 2022, 11, 235. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.-I.; Ren, Z.; et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Mayr, C.H.; Simon, L.M.; Leuschner, G.; Ansari, M.; Schniering, J.; E Geyer, P.; Angelidis, I.; Strunz, M.; Singh, P.; Kneidinger, N.; et al. Integrative analysis of cell state changes in lung fibrosis with peripheral protein biomarkers. EMBO Mol. Med. 2021, 13, e12871. [Google Scholar] [CrossRef]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.-I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Behjati, S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience 2020, 9, giaa151. [Google Scholar] [CrossRef]
- Lun, A.T.; McCarthy, D.; Marioni, J. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research 2016, 5, 2122. [Google Scholar] [CrossRef] [PubMed]
- Polański, K.; Young, M.D.; Miao, Z.; Meyer, K.B.; A Teichmann, S.; Park, J.-E. BBKNN: Fast batch alignment of single cell transcriptomes. Bioinformatics 2019, 36, 964–965. [Google Scholar] [CrossRef]
- Schamberger, A.C.; Staab-Weijnitz, C.; Mise-Racek, N.; Eickelberg, O. Cigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci. Rep. 2015, 5, 8163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, W.-L.; Rostami, M.R.; Leblanc, M.; Kaner, R.J.; O’Beirne, S.L.; Mezey, J.G.; Leopold, P.L.; Quast, K.; Visvanathan, S.; Fine, J.S.; et al. Dysregulation of club cell biology in idiopathic pulmonary fibrosis. PLoS ONE 2020, 15, e0237529. [Google Scholar] [CrossRef] [PubMed]
- Jonsdottir, H.R.; Arason, A.J.; Palsson, R.; Franzdottir, S.R.; Gudbjartsson, T.; Isaksson, H.J.; Gudmundsson, G.; Gudjonsson, T.; Magnusson, M.K. Basal cells of the human airways acquire mesenchymal traits in idiopathic pulmonary fibrosis and in culture. Lab. Investig. 2015, 95, 1418–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carraro, G.; Mulay, A.; Yao, C.; Mizuno, T.; Konda, B.; Petrov, M.; Lafkas, D.; Arron, J.R.; Hogaboam, C.M.; Chen, P.; et al. Single cell reconstruction of human basal cell diversity in normal and idiopathic pulmonary fibrosis lungs. Am. J. Respir. Crit. Care Med. 2020, 202, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Seibold, M.A.; Anastasia, L.; Wise, L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, L.A.; Hennessy, C.E.; Solomon, G.M.; Dobrinskikh, E.; Estrella, A.; Hara, N.; Hill, D.B.; Kissner, W.J.; Markovetz, M.R.; Villalon, D.E.G.; et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat. Commun. 2018, 9, 5363. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.; Grubb, B.R.; Harkema, J.R.; O’Neal, W.K.; Boucher, R.C. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 2004, 10, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Duerr, J.; Leitz, D.H.W.; Szczygiel, M.; Dvornikov, D.; Fraumann, S.G.; Kreutz, C.; Zadora, P.K.; Agircan, A.S.; Konietzke, P.; Engelmann, T.A.; et al. Conditional deletion of Nedd4-2 in lung epithelial cells causes progressive pulmonary fibrosis in adult mice. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Comstock, A.T.; Sajjan, U.S. Barrier function of airway tract epithelium. Tissue Barriers 2013, 1, e24997. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Li, Y.; Yu, J.; Dong, L.; Husain, A.N.; Shen, L.; Weber, C.R. Idiopathic pulmonary fibrosis is associated with tight junction protein alterations. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183205. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.; Wang, G.; Hao, Q.; Wang, Q.J.; Tang, H. Protein kinase D promotes airway epithelial barrier dysfunction and permeability through down-regulation of claudin-1. J. Biol. Chem. 2013, 288, 37343–37354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, H.; McKenzie, R.; Hao, Q.; Idell, S.; Tang, H. Protein kinase d is increased and activated in lung epithelial cells and macrophages in idiopathic pulmonary fibrosis. PLoS ONE 2014, 9, e101983. [Google Scholar] [CrossRef] [PubMed]
- Hope-Gill, B.D.M.; Hilldrup, S.; Davies, C.; Newton, R.P.; Harrison, N.K. A study of the cough reflex in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Mathai, S.K.; Schwartz, D.A. Genetics in idiopathic pulmonary fibrosis pathogenesis, prognosis, and treatment. Front. Med. 2017, 4, 154. [Google Scholar] [CrossRef] [PubMed]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noth, I.; Zhang, Y.; Ma, S.-F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhao, Y.H.; Di, Y.-P.; Wu, R. Characterization of human mucin 5b gene expression in airway epithelium and the genomic clone of the amino-terminal and 5′-flanking region. Am. J. Respir. Cell Mol. Biol. 2001, 25, 542–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Goobie, G.C.; Gregory, A.D.; Kass, D.J.; Zhang, Y. Toll-Interacting Protein in Pulmonary Diseases. Abiding by the Goldilocks Principle. Am. J. Respir. Cell Mol. Biol. 2021, 64, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Michalski, J.E.; A Schwartz, D.A. Genetic risk factors for idiopathic pulmonary fibrosis: Insights into immunopathogenesis. J. Inflamm. Res. 2021, 13, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kim, S.E.; Chen, T.; Wang, J.; Yang, X.; Tabib, T.; Tan, J.; Guo, B.; Fung, S.; Zhao, J.; et al. Toll interacting protein protects bronchial epithelial cells from bleomycin-induced apoptosis. FASEB J. 2020, 34, 9884–9898. [Google Scholar] [CrossRef] [PubMed]
- Delva, E.; Tucker, D.K.; Kowalczyk, A.P. The desmosome. Cold Spring Harb. Perspect. Biol. 2009, 1, a002543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathai, S.K.; Pedersen, B.S.; Smith, K.; Russell, P.; Schwarz, M.I.; Brown, K.K.; Steele, M.P.; Loyd, J.; Crapo, J.D.; Silverman, E.K.; et al. Desmoplakin Variants Are Associated with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 1151–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; Bates, S.; Mou, H.; Yun, J.H.; Pham, B.; Liu, J.; Qiu, W.; Guo, F.; Morrow, J.D.; Hersh, C.P.; et al. Genome-wide association study: Functional variant rs2076295 regulates desmoplakin expression in airway epithelial cells. Am. J. Respir. Crit. Care Med. 2020, 202, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Corvol, H.; Hodges, C.A.; Drumm, M.L.; Guillot, L. Moving beyond genetics: Is FAM13A a major biological contributor in lung physiology and chronic lung diseases? J. Med. Genet. 2014, 51, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; de Vries, M.; Nwozor, K.O.; Noordhoek, J.A.; Brandsma, C.-A.; Boezen, H.M.; Heijink, I.H. A protective role of fam13a in human airway epithelial cells upon exposure to cigarette smoke extract. Front. Physiol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Lao, T.; Qiu, W.; Polverino, F.; Gupta, K.; Guo, F.; Mancini, J.D.; Naing, Z.Z.C.; Cho, M.H.; Castaldi, P.J.; et al. A chronic obstructive pulmonary disease susceptibility gene, fam13a, regulates protein stability of β-catenin. Am. J. Respir. Crit. Care Med. 2016, 194, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, B.D.; de Jong, K.; Lamontagne, M.; Bossé, M.L.Y.; Shrine, N.; Artigas, M.S.; Wain, L.V.; Hall, I.; Jackson, V.E.; Wyss, A.B.; et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat. Genet. 2017, 49, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Rahardini, E.P.; Ikeda, K.; Nugroho, D.B.; Hirata, K.-I.; Emoto, N. loss of family with sequence similarity 13, member a exacerbates pulmonary fibrosis potentially by promoting epithelial to mesenchymal transition. Kobe J. Med. Sci. 2020, 65, E100–E109. [Google Scholar] [PubMed]
- Allen, R.J.; Porte, J.; Braybrooke, R.; Flores, C.; E Fingerlin, T.; Oldham, J.M.; Guillen-Guio, B.; Ma, S.-F.; Okamoto, T.; E John, A.; et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: A genome-wide association study. Lancet Respir. Med. 2017, 5, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Organ, L.; Porte, J.; John, A.; Jenkins, G. Investigating the role of AKAP13 on epithelial cell TGFß activation. Eur. Respir. J. 2019, 54, PA2429. [Google Scholar]
- Diviani, D.; Soderling, J.; Scott, J.D. AKAP-Lbc anchors protein kinase A and nucleates Galpha 12-selective Rho-mediated stress fiber formation. J. Biol. Chem. 2001, 276, 44247–44257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Ahn, C.; Kim, T.-H. Occupational and environmental risk factors of idiopathic pulmonary fibrosis: A systematic review and meta-analyses. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, S.; Harari, S.; Caminati, A.; Zanobetti, A.; Schwartz, J.D.; Bertazzi, P.A.; Cesana, G.; Madotto, F. The association between air pollution and the incidence of idiopathic pulmonary fibrosis in Northern Italy. Eur. Respir. J. 2018, 51, 1700397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.-M.; Seeger, W.; et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwano, K.; Kunitake, R.; Kawasaki, M.; Nomoto, Y.; Hagimoto, N.; Nakanishi, Y.; Hara, N. P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1996, 154, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Evans, I.C.; Bottoms, S.E.; Mercer, P.F.; Thorley, A.J.; Nicholson, A.G.; Laurent, G.J.; Tetley, T.D.; Chambers, R.C.; McAnulty, R.J. Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plataki, M.; Koutsopoulos, A.V.; Darivianaki, K.; Delides, G.; Siafakas, N.M.; Bouros, D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest 2005, 127, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-Y.; Hong, J.Y.; Lee, S.Y.; Lee, S.-H.; Kim, M.J.; Kim, S.Y.; Kim, K.W.; Shim, H.S.; Park, M.S.; Lee, C.G.; et al. Club cell-specific role of programmed cell death 5 in pulmonary fibrosis. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.-S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Cell. Mol. Physiol. 2008, 294, L1119–L1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, B.P.; McCormick, C. Herpesviruses and the unfolded protein response. Viruses 2019, 12, 17. [Google Scholar] [CrossRef] [Green Version]
- Tanjore, H.; Lawson, W.E.; Blackwell, T.S. Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim. Et. Biophys. Acta. BBA-Mol. Basis Dis. 2012, 1832, 940–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Ribeiro, C.M.P.; Sun, L.; Okuda, K.; Kato, T.; Glimore, R.C.; Martino, M.B.; Dang, H.; Abzhanove, A.; Lin, J.M.; et al. XBP1S regulates MUC5B in a promoter variant-dependent pathway in idiopathic pulmonary fibrosis airway epithelia. Am. J. Respir. Crit. Care Med. 2019, 200, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fuji, S.; Hara, H.; Yanagisawa, H.; Kobayashim, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, M.; Mutze, K.; Korfei, M.; Klee, S.; Wagner, D.; Costa, R.; Schiller, H.; Günther, A.; Königshoff, M. LSC-2017-Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir. J. 2017, 50, PA3471. [Google Scholar] [CrossRef]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L391–L401. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- DePianto, D.J.; Heiden, J.A.V.; Morshead, K.B.; Sun, K.-H.; Modrusan, Z.; Teng, G.; Wolters, P.J.; Arron, J.R. Molecular mapping of interstitial lung disease reveals a phenotypically distinct senescent basal epithelial cell population. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Meiners, S.; Lehmann, M. Senescent cells in IPF: Locked in repair? Front. Med. 2020, 7, 606330. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2018, 65, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-beta structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ask, K.; Bonniaud, P.; Maass, K.; Eickelberg, O.; Margetts, P.J.; Warburton, D.; Groffen, J.; Gauldie, J.; Kolb, M. Progressive pulmonary fibrosis is mediated by TGF-beta isoform 1 but not TGF-beta. Int. J. Biochem. Cell Biol. 2008, 40, 484–495. [Google Scholar] [PubMed] [Green Version]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Xaubet, A.; Marin-Arguedas, A.; Lario, S.; Ancochea, J.; Morell, F.; Ruiz-Manzano, J.; Rodriguez-Becerra, E.; Rodriguez-Arias, J.M.; Sanz, S.; Campistol, J.M.; et al. Transforming growth factor-beta1 gene polymorphisms are associated with disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Lodyga, M.; Hinz, B. TGF-beta1-A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020, 101, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G. The role of proteases in transforming growth factor-beta activation. Int. J. Biochem. Cell Biol. 2008, 40, 1068–1078. [Google Scholar] [PubMed]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-beta and TGF-beta-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Coker, R.K.; Laurent, G.J.; Shahzeidi, S.; Hernandez-Rodriguez, N.A.; Pantelidis, P.; du Bois, R.M.; Jeffery, P.K.; McAnulty, R.J. Diverse cellular TGF-beta 1 and TGF-beta 3 gene expression in normal human and murine lung. Eur. Respir. J. 1996, 9, 2501–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Prentice, M.A.; Mariano, J.M.; Davarya, S.; Linnoila, R.I.; Moody, T.W.; Wakefield, L.M.; Jakowlew, S.B. Transforming growth factor-beta 1 and its receptors in human lung cancer and mouse lung carcinogenesis. Exp. Lung Res. 2000, 26, 685–707. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D. Functions of pulmonary epithelial integrins: From development to disease. Physiol. Rev. 2003, 83, 673–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, V.J.; Polverino, F.; Laucho-Contreras, M.E.; Shi, Y.; Liu, Y.; Osorio, J.C.; Tesfaigzi, Y.; Pinto-Plata, V.; Gochuico, B.R.; Rosas, I.O.; et al. Mononuclear phagocytes and airway epithelial cells: Novel sources of matrix metalloproteinase-8 (MMP-8) in patients with idiopathic pulmonary fibrosis. PLoS ONE 2014, 9, e97485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Câmara, J.; Jarai, G. Epithelial-mesenchymal transition in primary human bronchial epithelial cells is Smad-dependent and enhanced by fibronectin and TNF-α. Fibrogenesis Tissue Repair 2010, 3, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackett, T.-L.; Warner, S.M.; Stefanowicz, D.; Shaheen, F.; Pechkovsky, D.V.; Murray, L.A.; Argentieri, R.; Kicic, A.; Sick, S.M.; Bai, T.R.; et al. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta. Am. J. Respir. Crit. Care Med. 2009, 180, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Gabasa, M.; Duch, P.; Jorba, I.; Giménez, A.; Lugo, R.; Pavelescu, I.; Rodriguez-Pascual, F.; Molina, M.M.; Xaubet, A.; Pereda, J.; et al. Epithelial contribution to the profibrotic stiff microenvironment and myofibroblast population in lung fibrosis. Mol. Biol. Cell 2017, 28, 3741–3755. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Neilson, E.G. Origin and functional heterogeneity of fibroblasts. FASEB J. 2020, 34, 3519–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aros, C.J.; Pantoja, C.J.; Gomperts, B.N. Wnt signaling in lung development, regeneration, and disease progression. Commun. Biol. 2021, 4, 1–13. [Google Scholar] [CrossRef]
- Herr, P.; Hausmann, G.; Basler, K. WNT secretion and signalling in human disease. Trends Mol. Med. 2012. 18, 483–493. [CrossRef]
- Baarsma, H.A.; Königshoff, M. ‘WNT-er is coming’: WNT signalling in chronic lung diseases. Thorax 2017, 72, 746–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Königshoff, M.; Balsara, N.; Pfaff, E.-M.; Kramer, M.; Chrobak, I.; Seeger, W.; Eickelberg, O. Functional wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS ONE 2008, 3, e2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaff, E.-M.; Becker, S.; Gunther, A.; Konigshoff, M. Dickkopf proteins influence lung epithelial cell proliferation in idiopathic pulmonary fibrosis. Eur. Respir. J. 2010, 37, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Aros, C.J.; Paul, M.; Pantoja, C.J.; Bisht, B.; Meneses, K.; Vijayaraj, P.; Sandlin, J.M.; France, B.; Tse, J.A.; Chen, M.W.; et al. High-throughput drug screening identifies a potent wnt inhibitor that promotes airway basal stem cell homeostasis. Cell Rep. 2020, 30, 2055–2064.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aros, C.J.; Vijayaraj, P.; Pantoja, C.J.; Bisht, B.; Meneses, L.K.; Sandlin, J.M.; Tse, J.A.; Chen, M.W.; Purkayastha, A.; Shia, D.W.; et al. Distinct spatiotemporally dynamic wnt-secreting niches regulate proximal airway regeneration and aging. Cell Stem Cell 2020, 27, 413–429. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Anderson, P.J.; Rotti, P.G.; Tyler, S.R.; Crooke, A.K.; Choi, S.H.; Montoro, D.T.; Montoro, C.L.; Silverman, C.L.; Shahin, W.; et al. Submucosal gland myoepithelial cells are reserve stem cells that can regenerate mouse tracheal epithelium. Cell Stem Cell 2018, 22, 779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, T.J.; Anderson, P.J.; Xie, W.; Crooke, A.K.; Liu, X.; Tyler, S.R.; Luo, M.; Kusner, D.M.; Zhang, Y.; Neff, T.; et al. Wnt signaling regulates airway epithelial stem cells in adult murine submucosal glands. Stem Cells 2016, 34, 2758–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, M.; Vázquez, G.J.L.; Sun, D.I.; Tran, H.T.; Brislinger, M.; Tasca, A.; Shomroni, O.; Vleminckx, K.; Walentek, P. DeltaN-Tp63 mediates wnt/beta-catenin-induced inhibition of differentiation in basal stem cells of mucociliary epithelia. Cell Rep. 2019, 28, 3338–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volckaert, T.; Yuan, T.; Chao, C.-M.; Bell, H.; Sitaula, A.; Szimmtenings, L.; El Agha, E.; Chanda, D.; Majka, S.; Bellusci, S.; et al. Fgf10-hippo epithelial-mesenchymal crosstalk maintains and recruits lung basal stem cells. Dev. Cell 2017, 43, 48–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, A.E.; Brumwell, A.N.; Xi, Y.; Gotts, J.E.; Brownfield, D.G.; Treutlein, B.; Tan, K.; Tan, V.; Liu, F.C.; Looney, M.R.; et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 2015, 517, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Kugler, M.C.; Joyner, A.L.; Loomis, C.A.; Munger, J.S. Sonic hedgehog signaling in the lung. From development to disease. Am. J. Respir. Cell Mol. Biol. 2015, 52, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kugler, M.C.; Loomis, C.A.; Zhao, Z.; Cushman, J.C.; Liu, L.; Munger, J.S. Sonic hedgehog signaling regulates myofibroblast function during alveolar septum formation in murine postnatal lung. Am. J. Respir. Cell Mol. Biol. 2017, 57, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Bolanos, A.L.; Milla, C.M.; Liram, J.C.; Ramírez, R.; Checa, M.; Barrera, L.; García-Alvarez, J.; Carbajal, V.; Becerril, C.; Gaxiola, M. Role of Sonic Hedgehog in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, 978–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassandras, M.; Wang, C.; Kathiriya, J.; Tsukui, T.; Matatia, P.; Matthay, M.; Wolters, P.; Molofsky, A.; Sheppard, D.; Chapman, H.; et al. Gli1+ mesenchymal stromal cells form a pathological niche to promote airway progenitor metaplasia in the fibrotic lung. Nat. Cell Biol. 2020, 22, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, M.; Liu, Z.; Cheng, H.-T.; Winters, N.; Bader, D.; Kopan, R. Canonical Notch signaling in the developing lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate. J. Cell Sci. 2010, 123, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, P.-N.; Vasconcelos, M.; Izvolsky, K.I.; Qian, J.; Lu, J.; Cardoso, W.V. Notch signaling controls the balance of ciliated and secretory cell fates in developing airways. Development 2009, 136, 2297–2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyagi-Ikeda, K.; Maeno, T.; Hiroki Matsui, H.; Ueno, M.; Hara, K.; Aoki, Y.; Aoki, F.; Shimizu, T.; Doi, H.; Kawai-Kowase, K.; et al. Notch induces myofibroblast differentiation of alveolar epithelial cells via transforming growth factor-{beta}-Smad3 pathway. Am. J. Respir. Cell Mol. Biol. 2011, 45, 136–144. [Google Scholar]
- Finn, J.; Sottoriva, K.; Pajcini, K.V.; Kitajewski, J.K.; Chen, C.; Zhang, W.; Malik, A.B.; Liu, Y. Dlk1-mediated temporal regulation of notch signaling is required for differentiation of alveolar type ii to type i cells during repair. Cell Rep. 2019, 26, 2942–2954. [Google Scholar] [CrossRef] [Green Version]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Schwartz, D.A. Epigenetics of idiopathic pulmonary fibrosis. Transl. Res. J. Lab. Clin. Med. 2015, 165, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Alhamwe, B.A.; Miethe, S.; Von Strandmann, E.P.; Potaczek, D.P.; Garn, H. Epigenetic regulation of airway epithelium immune functions in asthma. Front. Immunol. 2020, 11, 1747. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, N.; Alashkar Alhamwe, B.; Caraballo, L.; Ding, M.; Ferrante, A.; Garn, H.; Garssen, J.; Hii, C.S.; Irvine, J.; Llinás-Caballero, K.; et al. Perinatal and early-life nutrition, epigenetics, and allergy. Nutrients 2021, 13, 724. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Ambalavanan, N.; Halloran, B.; Zhang, X.; Liu, H.; Crossman, D.K.; Bray, M.; Zhang, K.; Thannickal, V.J.; Hagood, J.S. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Pedersen, B.S.; Rabinovich, E.; Hennessy, C.E.; Davidson, E.J.; Murphy, E.; Guardela, B.J.; Tedrow, J.R.; Zhang, Y.; Singh, M.K.; et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Korfei, M.; Skwarna, S.; Henneke, I.; MacKenzie, B.; Klymenko, O.; Saito, S.; Ruppert, C.; von der Beck, D.; Mahavadi, P.; Klepetko, W.; et al. Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax 2015, 70, 1022–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, B.; Platel, A.; Antherieu, S.; Alleman, L.; Hardy, E.; Perdrix, E.; Grova, N.; Riffault, V.; Appenzeller, B.; Happillon, M.; et al. Genetic and epigenetic alterations in normal and sensitive COPD-diseased human bronchial epithelial cells repeatedly exposed to air pollution-derived PM2.5. Environ. Pollut. 2017, 230, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Real, A.D.; Santurtún, A.; Zarrabeitia, M.T. Epigenetic related changes on air quality. Environ. Res. 2021, 197, 111155. [Google Scholar] [CrossRef] [PubMed]
- Vaz, M.; Hwang, S.Y.; Kagiampakis, I.; Phallen, J.; Patil, A.; O’Hagan, H.M.; Murphy, L.; Zahnow, C.A.; Gabrielson, E.; Velculescu, V.E.; et al. Chronic cigarette smoke-induced epigenomic changes precede sensitization of bronchial epithelial cells to single-step transformation by KRAS mutations. Cancer Cell 2017, 32, 360–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattick, J.S. Non-coding RNAs: The architects of eukaryotic complexity. EMBO Rep. 2001, 2, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulitsky, I. Interactions between short and long noncoding RNAs. FEBS Lett. 2018, 592, 2874–2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjicharalambous, M.R.; Lindsay, M.A. Idiopathic pulmonary fibrosis: Pathogenesis and the emerging role of long non-coding RNAs. Int. J. Mol. Sci. 2020, 21, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omote, N.; Sauler, M. Non-coding RNAs as regulators of cellular senescence in idiopathic pulmonary fibrosis and chronic obstructive pulmonary disease. Front. Med. 2020, 7, 603047. [Google Scholar] [CrossRef] [PubMed]
- Savary, G.; Savary, G.; Dewaeles, E.; Diazzi, S.; Buscot, M.; Nottet, N.; Fassy, J.; Courcot, E.; Henaoui, I.-S.; Lemaire, J.; et al. The long noncoding RNA DNM3OS is a reservoir of fibromirs with major functions in lung fibroblast response to TGF-beta and pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Gokey, J.; Snowball, J.; Sridharan, A.; Speth, J.P.; Black, K.E.; Hariri, L.P.; Perl, A.-K.T.; Xu, Y.; Whitsett, J.A. MEG3 is increased in idiopathic pulmonary fibrosis and regulates epithelial cell differentiation. JCI Insight 2018, 3, e122490. [Google Scholar] [CrossRef] [PubMed]
- Omote, N.; Sakamoto, K.; Li, Q.; Schupp, J.C.; Adams, T.; Ahangari, F.; Chioccioli, M.; DeIuliis, G.; Hashimoto, N.; Hasegawa, Y.; et al. Long noncoding RNA TINCR is a novel regulator of human bronchial epithelial cell differentiation state. Physiol. Rep. 2021, 9, e14727. [Google Scholar] [CrossRef] [PubMed]
- Omote, N.; Sakamoto, K.; Li, Q.; Schupp, J.; Adams, T.; Ahangari, F.; Chioccioli, M.; Xylourgidis, N.; Deiuliis, G.; Hashimoto, N.; et al. TINCR, a long intergenic noncoding RNA decreased in IPF, is a novel regulator of airway epithelial cell differentiation. Am. J. Respir. Crit. Care Med. 2019, 199, A5406. [Google Scholar] [CrossRef]
- Sun, H.; Chen, J.; Qian, W.; Kang, J.; Wang, J.; Jiang, L.; Qiao, L.; Chen, W.; Zhang, J. Integrated long non-coding RNA analyses identify novel regulators of epithelial-mesenchymal transition in the mouse model of pulmonary fibrosis. J. Cell. Mol. Med. 2016, 20, 1234–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cheng, Z.; Dai, L.; Jiang, T.; Jia, L.; Jing, X.; An, L.; Wang, H.; Liu, M. Knockdown of long noncoding RNA H19 represses the progress of pulmonary fibrosis through the transforming growth factor beta/smad3 pathway by regulating microRNA 140. Mol. Cell. Biol. 2019, 39, e00143-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, L.; Habiel, D.M.; Hansbro, P.; Kim, R.Y.; Gharib, S.A.; Edelman, J.D.; Koenigshoff, M.; Parimon, T.; Brauer, R.; Huang, Y.; et al. miR-323a-3p regulates lung fibrosis by targeting multiple profibrotic pathways. JCI Insight 2016, 1, e90301. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 2020, 22, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakraborty, A.; Mastalerz, M.; Ansari, M.; Schiller, H.B.; Staab-Weijnitz, C.A. Emerging Roles of Airway Epithelial Cells in Idiopathic Pulmonary Fibrosis. Cells 2022, 11, 1050. https://doi.org/10.3390/cells11061050
Chakraborty A, Mastalerz M, Ansari M, Schiller HB, Staab-Weijnitz CA. Emerging Roles of Airway Epithelial Cells in Idiopathic Pulmonary Fibrosis. Cells. 2022; 11(6):1050. https://doi.org/10.3390/cells11061050
Chicago/Turabian StyleChakraborty, Ashesh, Michal Mastalerz, Meshal Ansari, Herbert B. Schiller, and Claudia A. Staab-Weijnitz. 2022. "Emerging Roles of Airway Epithelial Cells in Idiopathic Pulmonary Fibrosis" Cells 11, no. 6: 1050. https://doi.org/10.3390/cells11061050