
Preservation of Smooth Muscle Cell Integrity and Function: A Target for Limiting Abdominal Aortic Aneurysm Expansion?

 , , and
, , and 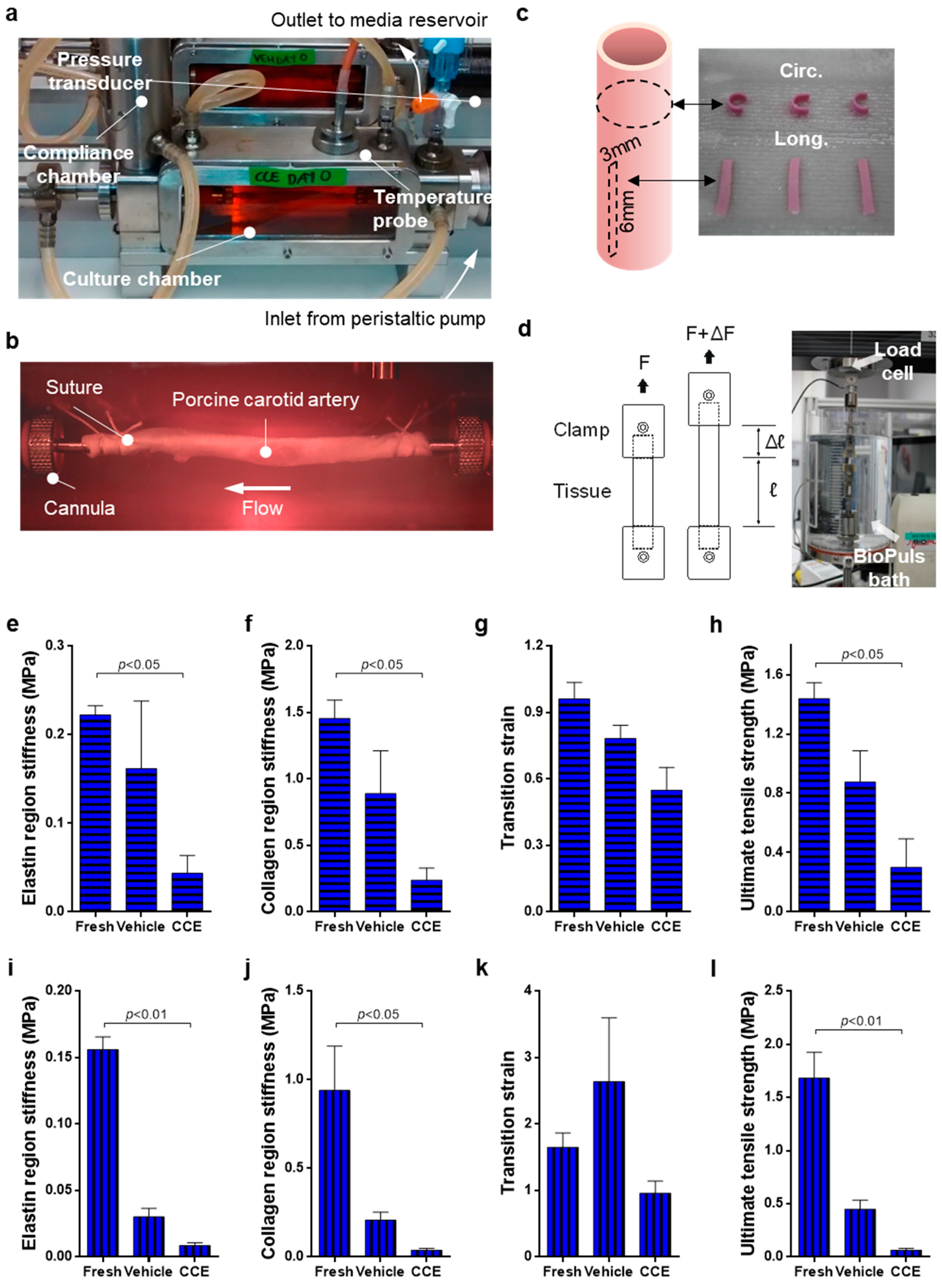{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
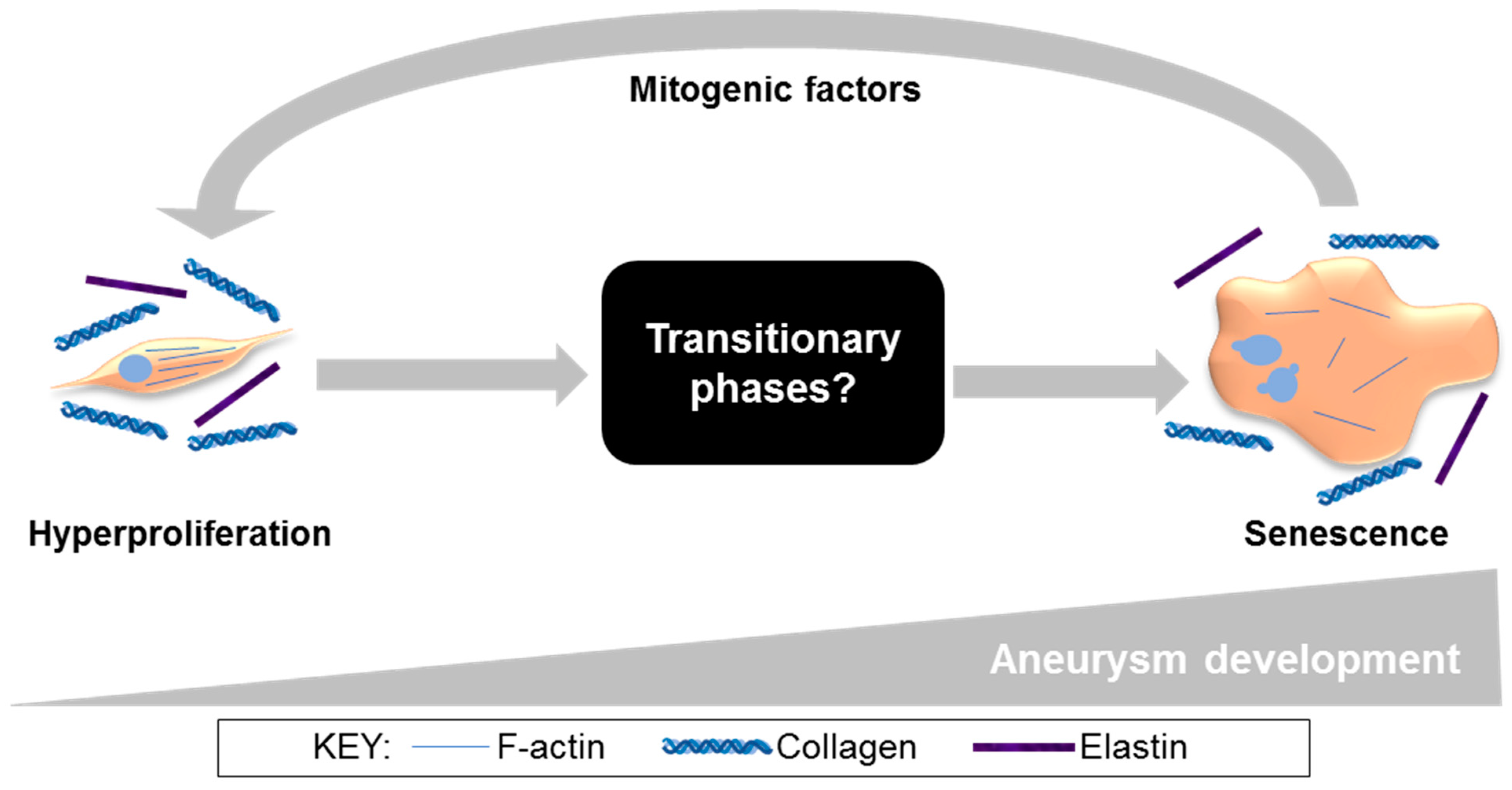{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bioreactor Model
2.2. Uniaxial Tensile Testing
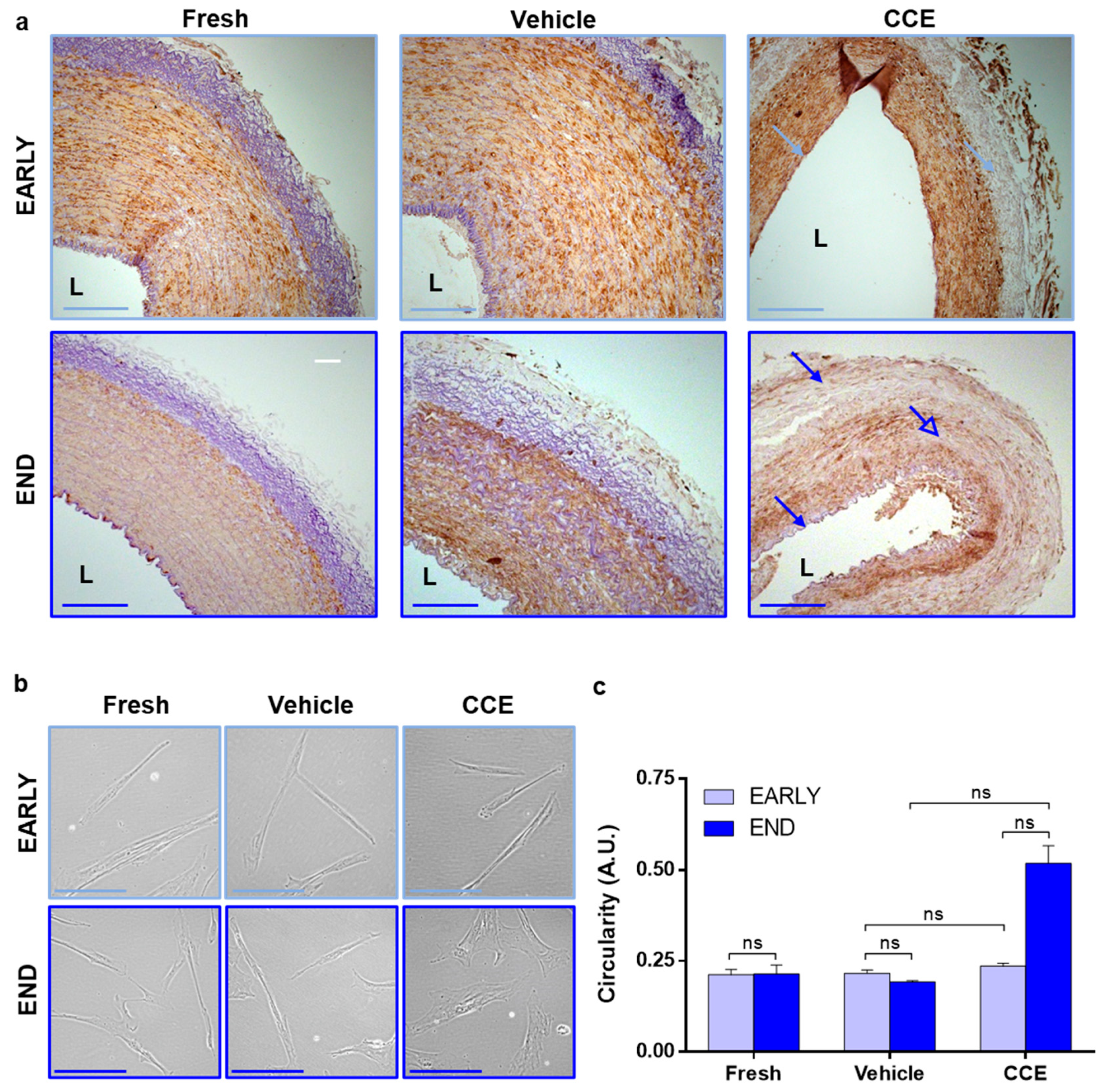
2.3. Histology
2.4. SMC Isolation and Culture
2.5. Morphometric Analyses
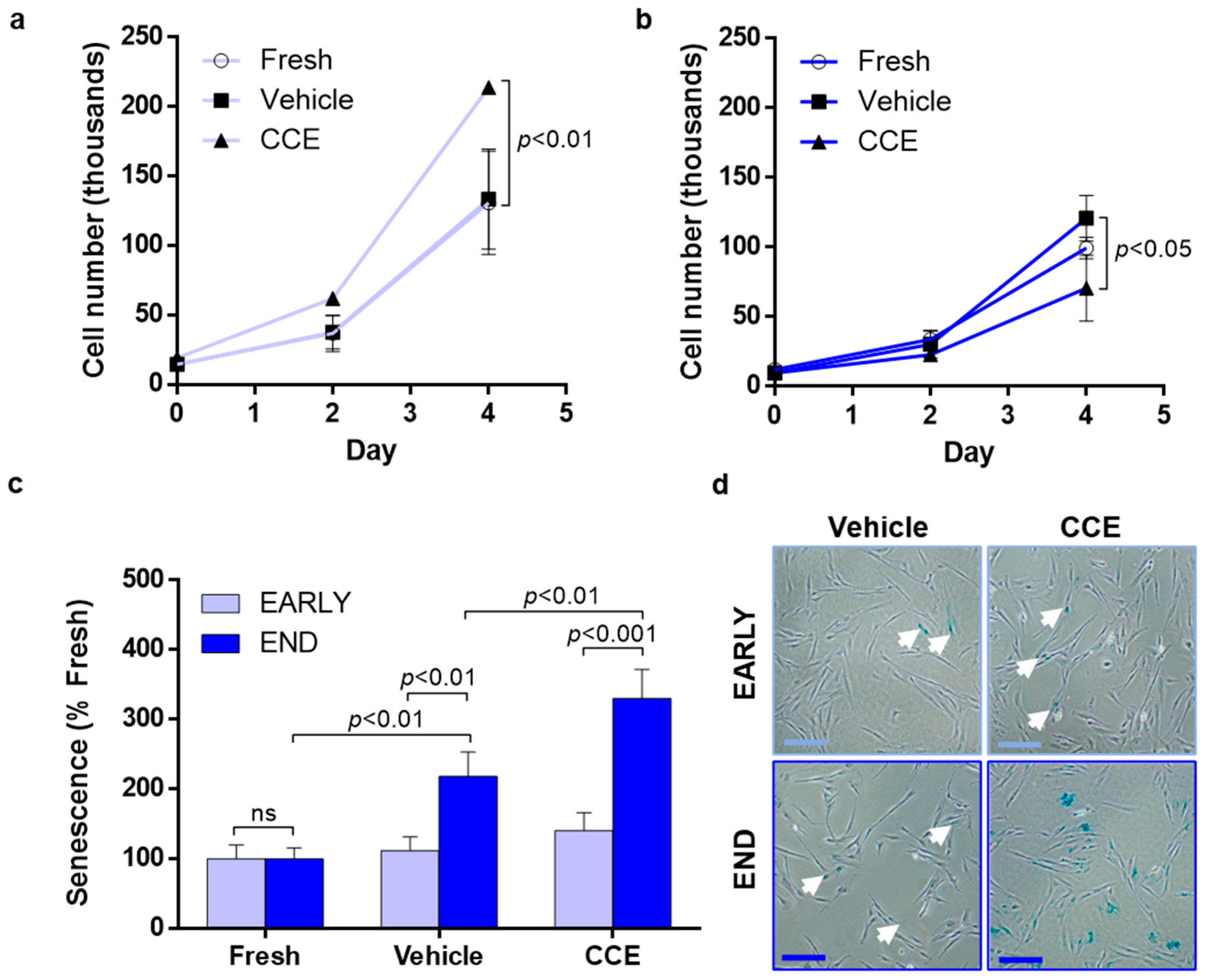
2.6. Proliferation
2.7. Senescence-Associated β-Galactosidase Assay
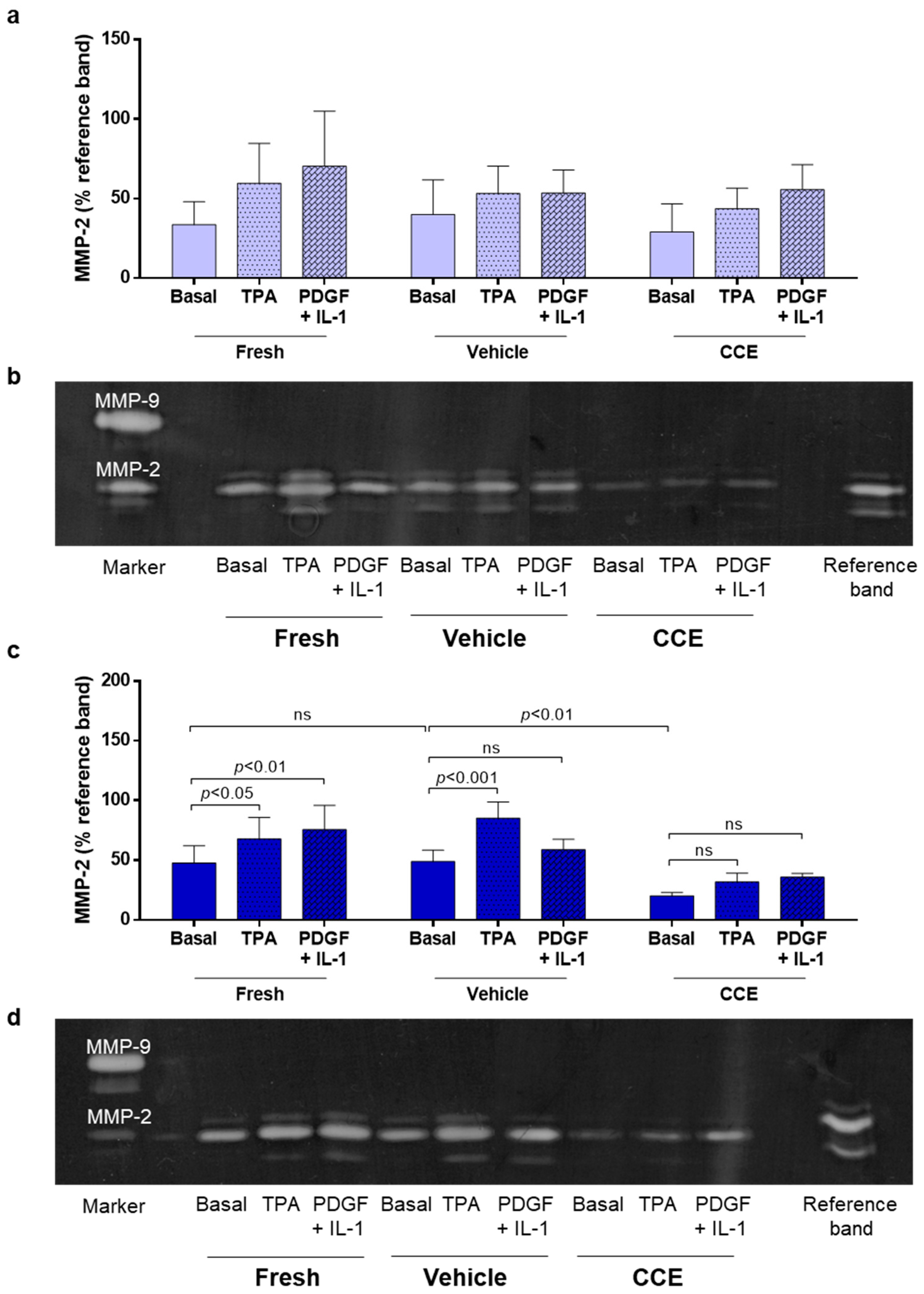
2.8. Gelatin Zymography
2.9. Statistical Analyses
3. Results
3.1. Biomechanics of END Stage CCE Tissue
3.2. Tissue and Cell Structure from EARLY and END Models
3.3. SMC Proliferation and Senescence
3.4. Matrix Remodeling
3.5. Secretion of Paracrine Factors That Propagate SMC Dysfunction
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jagadesham, V.P.; Scott, D.J.; Carding, S.R. Abdominal aortic aneurysms: An autoimmune disease? Trends Mol. Med. 2008, 14, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Jacomelli, J.; Summers, L.; Stevenson, A.; Lees, T.; Earnshaw, J.J. Impact of the first 5 years of a national abdominal aortic aneurysm screening programme. Br. J. Surg. 2016, 103, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.G.; Ashton, H.A.; Gao, L.; Scott, R.A. Screening men for abdominal aortic aneurysm: 10 year mortality and cost effectiveness results from the randomised Multicentre Aneurysm Screening Study. BMJ 2009, 338, b2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordon, I.M.; Hinchliffe, R.J.; Loftus, I.M.; Thompson, M.M. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat. Rev. Cardiol. 2011, 8, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Kent, K.C. Clinical practice. Abdominal aortic aneurysms. N. Engl. J. Med. 2014, 371, 2101–2108. [Google Scholar] [CrossRef] [PubMed]
- Kontopodis, N.; Pantidis, D.; Dedes, A.; Daskalakis, N.; Ioannou, C.V. The—Not So—Solid 5.5 cm Threshold for Abdominal Aortic Aneurysm Repair: Facts, Misinterpretations, and Future Directions. Front. Surg. 2016, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, K.; Mitchell, R.N.; Libby, P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arter. Thromb. Vasc. Biol. 2006, 26, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Tsuruda, T.; Kato, J.; Hatakeyama, K.; Kojima, K.; Yano, M.; Yano, Y.; Nakamura, K.; Nakamura-Uchiyama, F.; Matsushima, Y.; Imamura, T.; et al. Adventitial mast cells contribute to pathogenesis in the progression of abdominal aortic aneurysm. Circ. Res. 2008, 102, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.J.; McCarthy, W.J.; Dixit, S.N.; Lilly, M.P.; Shively, V.P.; Flinn, W.R.; Yao, J.S. Collagen types and matrix protein content in human abdominal aortic aneurysms. J. Vasc. Surg. 1989, 10, 365–373. [Google Scholar] [CrossRef] [Green Version]
- He, C.M.; Roach, M.R. The composition and mechanical properties of abdominal aortic aneurysms. J. Vasc. Surg. 1994, 20, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Carmo, M.; Colombo, L.; Bruno, A.; Corsi, F.R.; Roncoroni, L.; Cuttin, M.S.; Radice, F.; Mussini, E.; Settembrini, P.G. Alteration of elastin, collagen and their cross-links in abdominal aortic aneurysms. Eur. J. Vasc. Endovasc. Surg. 2002, 23, 543–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, E.L.; Geng, Y.J.; Sukhova, G.K.; Whittemore, A.D.; Knox, J.; Libby, P. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation 1999, 99, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Candales, A.; Holmes, D.R.; Liao, S.; Scott, M.J.; Wickline, S.A.; Thompson, R.W. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am. J. Pathol. 1997, 150, 993–1007. [Google Scholar] [PubMed]
- Thubrikar, M.J.; Labrosse, M.; Robicsek, F.; Al-Soudi, J.; Fowler, B. Mechanical properties of abdominal aortic aneurysm wall. J. Med. Eng. Technol. 2001, 25, 133–142. [Google Scholar] [CrossRef]
- Vande Geest, J.P.; Sacks, M.S.; Vorp, D.A. The effects of aneurysm on the biaxial mechanical behavior of human abdominal aorta. J. Biomech. 2006, 39, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Vorp, D.A.; Raghavan, M.L.; Muluk, S.C.; Makaroun, M.S.; Steed, D.L.; Shapiro, R.; Webster, M.W. Wall strength and stiffness of aneurysmal and nonaneurysmal abdominal aorta. Ann. N. Y. Acad. Sci. 1996, 800, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.E.; Riches, K. The vascular smooth muscle cell: A therapeutic target in Type 2 diabetes? Clin. Sci. 2013, 125, 167–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamley-Campbell, J.; Campbell, G.R.; Ross, R. The smooth muscle cell in culture. Physiol. Rev. 1979, 59, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef] [PubMed]
- Airhart, N.; Brownstein, B.H.; Cobb, J.P.; Schierding, W.; Arif, B.; Ennis, T.L.; Thompson, R.W.; Curci, J.A. Smooth muscle cells from abdominal aortic aneurysms are unique and can independently and synergistically degrade insoluble elastin. J. Vasc. Surg. 2014, 60, 1033–1041, discussion 1041–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, S.; Curci, J.A.; Kelley, B.J.; Sicard, G.A.; Thompson, R.W. Accelerated replicative senescence of medial smooth muscle cells derived from abdominal aortic aneurysms compared to the adjacent inferior mesenteric artery. J. Surg. Res. 2000, 92, 85–95. [Google Scholar] [CrossRef]
- Riches, K.; Angelini, T.G.; Mudhar, G.S.; Kaye, J.; Clark, E.; Bailey, M.A.; Sohrabi, S.; Korossis, S.; Walker, P.G.; Scott, D.J.; et al. Exploring smooth muscle phenotype and function in a bioreactor model of abdominal aortic aneurysm. J. Transl. Med. 2013, 11, 208. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.T.; Tromp, G.; Kuivaniemi, H.; Gretarsdottir, S.; Baas, A.F.; Giusti, B.; Strauss, E.; Van’t Hof, F.N.; Webb, T.R.; Erdman, R.; et al. Meta-Analysis of Genome-Wide Association Studies for Abdominal Aortic Aneurysm Identifies Four New Disease-Specific Risk Loci. Circ. Res. 2017, 120, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Zheng, C.; Liu, Y.; Ding, M.; Liu, P.; Xu, J.; Wang, W.; Wang, J. In vitro extracellular matrix deposition by vascular smooth muscle cells grown in fibroin scaffolds, and the regulation of TGF-β1. Mater. Des. 2021, 199, 109428. [Google Scholar] [CrossRef]
- Daugherty, A.; Cassis, L.A. Mouse models of abdominal aortic aneurysms. Arter. Thromb. Vasc. Biol. 2004, 24, 429–434. [Google Scholar] [CrossRef] [Green Version]
- Haskett, D.; Azhar, M.; Utzinger, U.; Vande Geest, J.P. Progressive alterations in microstructural organization and biomechanical response in the ApoE mouse model of aneurysm. Biomatter 2013, 3, e24648. [Google Scholar] [CrossRef] [Green Version]
- Anidjar, S.; Salzmann, J.L.; Gentric, D.; Lagneau, P.; Camilleri, J.P.; Michel, J.B. Elastase-induced experimental aneurysms in rats. Circulation 1990, 82, 973–981. [Google Scholar] [CrossRef] [Green Version]
- Cho, B.S.; Woodrum, D.T.; Roelofs, K.J.; Stanley, J.C.; Henke, P.K.; Upchurch, G.R., Jr. Differential regulation of aortic growth in male and female rodents is associated with AAA development. J. Surg. Res. 2009, 155, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Molacek, J.; Treska, V.; Kobr, J.; Certik, B.; Skalicky, T.; Kuntscher, V.; Krizkova, V. Optimization of the model of abdominal aortic aneurysm--experiment in an animal model. J. Vasc. Res. 2009, 46, 1–5. [Google Scholar] [CrossRef]
- Turnbull, I.C.; Hadri, L.; Rapti, K.; Sadek, M.; Liang, L.; Shin, H.J.; Costa, K.D.; Marin, M.L.; Hajjar, R.J.; Faries, P.L. Aortic implantation of mesenchymal stem cells after aneurysm injury in a porcine model. J. Surg. Res. 2011, 170, e179–e188. [Google Scholar] [CrossRef] [Green Version]
- Kloster, B.O.; Lund, L.; Lindholt, J.S. Induction of continuous expanding infrarenal aortic aneurysms in a large porcine animal model. Ann. Med. Surg. 2015, 4, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Riches, K.; Clark, E.; Helliwell, R.J.; Angelini, T.G.; Hemmings, K.E.; Bailey, M.A.; Bridge, K.I.; Scott, D.J.A.; Porter, K.E. Progressive Development of Aberrant Smooth Muscle Cell Phenotype in Abdominal Aortic Aneurysm Disease. J. Vasc. Res. 2018, 55, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.E.; Varty, K.; Jones, L.; Bell, P.R.; London, N.J. Human saphenous vein organ culture: A useful model of intimal hyperplasia? Eur. J. Vasc. Endovasc. Surg. 1996, 11, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Goodall, S.; Porter, K.E.; Bell, P.R.; Thompson, M.M. Enhanced invasive properties exhibited by smooth muscle cells are associated with elevated production of MMP-2 in patients with aortic aneurysms. Eur. J. Vasc. Endovasc. Surg. 2002, 24, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Newman, K.M.; Jean-Claude, J.; Li, H.; Ramey, W.G.; Tilson, M.D. Cytokines that activate proteolysis are increased in abdominal aortic aneurysms. Circulation 1994, 90, II224–II227. [Google Scholar] [PubMed]
- Kanazawa, S.; Miyake, T.; Kakinuma, T.; Tanemoto, K.; Tsunoda, T.; Kikuchi, K. The expression of platelet-derived growth factor and connective tissue growth factor in different types of abdominal aortic aneurysms. J. Cardiovasc. Surg. 2005, 46, 271–278. [Google Scholar]
- Hemmings, K.E.; Riches-Suman, K.; Bailey, M.A.; O’Regan, D.J.; Turner, N.A.; Porter, K.E. Role of MicroRNA-145 in DNA Damage Signalling and Senescence in Vascular Smooth Muscle Cells of Type 2 Diabetic Patients. Cells 2021, 10, 919. [Google Scholar] [CrossRef]
- Bozzani, A.; Arici, V.; Franciscone, M.; Ticozzelli, G.; Sterpetti, A.V.; Ragni, F. COVID-19 patients with abdominal aortic aneurysm may be at higher risk for sudden enlargement and rupture. J. Vasc. Surg. 2022, 75, 387–388. [Google Scholar] [CrossRef] [PubMed]
- Allaire, E.; Muscatelli-Groux, B.; Mandet, C.; Guinault, A.M.; Bruneval, P.; Desgranges, P.; Clowes, A.; Melliere, D.; Becquemin, J.P. Paracrine effect of vascular smooth muscle cells in the prevention of aortic aneurysm formation. J. Vasc. Surg. 2002, 36, 1018–1026. [Google Scholar] [CrossRef] [Green Version]
- Allaire, E.; Muscatelli-Groux, B.; Guinault, A.M.; Pages, C.; Goussard, A.; Mandet, C.; Bruneval, P.; Melliere, D.; Becquemin, J.P. Vascular smooth muscle cell endovascular therapy stabilizes already developed aneurysms in a model of aortic injury elicited by inflammation and proteolysis. Ann. Surg. 2004, 239, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Losy, F.; Dai, J.; Pages, C.; Ginat, M.; Muscatelli-Groux, B.; Guinault, A.M.; Rousselle, E.; Smedile, G.; Loisance, D.; Becquemin, J.P.; et al. Paracrine secretion of transforming growth factor-beta1 in aneurysm healing and stabilization with endovascular smooth muscle cell therapy. J. Vasc. Surg. 2003, 37, 1301–1309. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, R.; Cawthorne, C.; Craggs, L.J.L.; Wright, J.D.; Domarkas, J.; He, P.; Koch-Paszkowski, J.; Shires, M.; Scarsbrook, A.F.; Archibald, S.J.; et al. Cell proliferation detected using [(18)F]FLT PET/CT as an early marker of abdominal aortic aneurysm. J. Nucl. Cardiol. 2021, 28, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Olson, S.L.; Panthofer, A.M.; Blackwelder, W.; Terrin, M.L.; Curci, J.A.; Baxter, B.T.; Weaver, F.A.; Matsumura, J.S.; Non-Invasive Treatment of Abdominal Aortic Aneurysm Clinical Trial Investigators. Role of volume in small abdominal aortic aneurysm surveillance. J. Vasc. Surg. 2021. [Google Scholar] [CrossRef]
- Shirasu, T.; Takagi, H.; Yasuhara, J.; Kuno, T.; Kent, K.C.; Clouse, W.D. Smaller size is more suitable for pharmacotherapy among undersized abdominal aortic aneurysm: A systematic review and meta-analysis. Vasc. Med. 2021, 1358863X211061603. [Google Scholar] [CrossRef]
- Ailawadi, G.; Moehle, C.W.; Pei, H.; Walton, S.P.; Yang, Z.; Kron, I.L.; Lau, C.L.; Owens, G.K. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2009, 138, 1392–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, M.; Johnston, W.F.; Woo, A.; Pope, N.H.; Su, G.; Upchurch, G.R., Jr.; Owens, G.K.; Ailawadi, G. KLF4 regulates abdominal aortic aneurysm morphology and deletion attenuates aneurysm formation. Circulation 2013, 128, S163–S174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rombouts, K.B.; van Merrienboer, T.A.R.; Ket, J.C.F.; Bogunovic, N.; van der Velden, J.; Yeung, K.K. The role of vascular smooth muscle cells in the development of aortic aneurysms and dissections. Eur. J. Clin. Investig. 2021, e13697. [Google Scholar] [CrossRef]
- Lu, H.; Du, W.; Ren, L.; Hamblin, M.H.; Becker, R.C.; Chen, Y.E.; Fan, Y. Vascular Smooth Muscle Cells in Aortic Aneurysm: From Genetics to Mechanisms. J. Am. Heart Assoc. 2021, 10, e023601. [Google Scholar] [CrossRef] [PubMed]
- Goodall, S.; Crowther, M.; Hemingway, D.M.; Bell, P.R.; Thompson, M.M. Ubiquitous elevation of matrix metalloproteinase-2 expression in the vasculature of patients with abdominal aneurysms. Circulation 2001, 104, 304–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowther, M.; Goodall, S.; Jones, J.L.; Bell, P.R.; Thompson, M.M. Increased matrix metalloproteinase 2 expression in vascular smooth muscle cells cultured from abdominal aortic aneurysms. J. Vasc. Surg. 2000, 32, 575–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cione, E.; Piegari, E.; Gallelli, G.; Caroleo, M.C.; Lamirata, E.; Curcio, F.; Colosimo, F.; Cannataro, R.; Ielapi, N.; Colosimo, M.; et al. Expression of MMP-2, MMP-9, and NGAL in Tissue and Serum of Patients with Vascular Aneurysms and Their Modulation by Statin Treatment: A Pilot Study. Biomolecules 2020, 10, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, W.R.; Choke, E.C.; Dawson, J.; Loftus, I.M.; Thompson, M.M. Plasma matrix metalloproteinase levels do not predict tissue levels in abdominal aortic aneurysms suitable for elective repair. Vascular 2008, 16, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.O.; Lee, Y.K.; Kim, J.M.; Yoon, G. From cell senescence to age-related diseases: Differential mechanisms of action of senescence-associated secretory phenotypes. BMB Rep. 2015, 48, 549–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golledge, J.; Norman, P.E. Atherosclerosis and abdominal aortic aneurysm: Cause, response, or common risk factors? Arter. Thromb. Vasc. Biol. 2010, 30, 1075–1077. [Google Scholar] [CrossRef] [Green Version]
- Stojanovic, S.D.; Fuchs, M.; Kunz, M.; Xiao, K.; Just, A.; Pich, A.; Bauersachs, J.; Fiedler, J.; Sedding, D.; Thum, T. Inflammatory Drivers of Cardiovascular Disease: Molecular Characterization of Senescent Coronary Vascular Smooth Muscle Cells. Front. Physiol. 2020, 11, 520. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Pauli, J.; Winski, G.; Bleichert, S.; Chernogubova, E.; Metschl, S.; Winter, H.; Trenner, M.; Wiegering, A.; Otto, C.; et al. Lenvatinib halts aortic aneurysm growth by restoring smooth muscle cell contractility. JCI Insight 2021, 6, e140364. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Chen, S.; Gu, G.; Wang, W.; Ren, J.; Xu, F.; Li, F.; Wu, J.; Yang, D.; Zheng, Y. Discovery of potential biomarkers for human atherosclerotic abdominal aortic aneurysm through untargeted metabolomics and transcriptomics. J. Zhejiang Univ. Sci. B 2021, 22, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; He, X.; Li, J.; Guan, X.; Liu, X.; Wang, Y.; Niu, L.; Qiu, D.; Wu, X.; Wang, H. Profiling of the full-length transcriptome in abdominal aortic aneurysm using nanopore-based direct RNA sequencing. Open Biol. 2022, 12, 210172. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, Q.; Zhao, W.; Zhou, W. Circular RNA RBM33 contributes to extracellular matrix degradation via miR-4268/EPHB2 axis in abdominal aortic aneurysm. PeerJ 2021, 9, e12232. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clark, E.R.; Helliwell, R.J.; Bailey, M.A.; Hemmings, K.E.; Bridge, K.I.; Griffin, K.J.; Scott, D.J.A.; Jennings, L.M.; Riches-Suman, K.; Porter, K.E. Preservation of Smooth Muscle Cell Integrity and Function: A Target for Limiting Abdominal Aortic Aneurysm Expansion? Cells 2022, 11, 1043. https://doi.org/10.3390/cells11061043
Clark ER, Helliwell RJ, Bailey MA, Hemmings KE, Bridge KI, Griffin KJ, Scott DJA, Jennings LM, Riches-Suman K, Porter KE. Preservation of Smooth Muscle Cell Integrity and Function: A Target for Limiting Abdominal Aortic Aneurysm Expansion? Cells. 2022; 11(6):1043. https://doi.org/10.3390/cells11061043
Chicago/Turabian StyleClark, Emily R., Rebecca J. Helliwell, Marc A. Bailey, Karen E. Hemmings, Katherine I. Bridge, Kathryn J. Griffin, D. Julian A. Scott, Louise M. Jennings, Kirsten Riches-Suman, and Karen E. Porter. 2022. "Preservation of Smooth Muscle Cell Integrity and Function: A Target for Limiting Abdominal Aortic Aneurysm Expansion?" Cells 11, no. 6: 1043. https://doi.org/10.3390/cells11061043