
DHEA Protects Human Cholangiocytes and Hepatocytes against Apoptosis and Oxidative Stress


Abstract
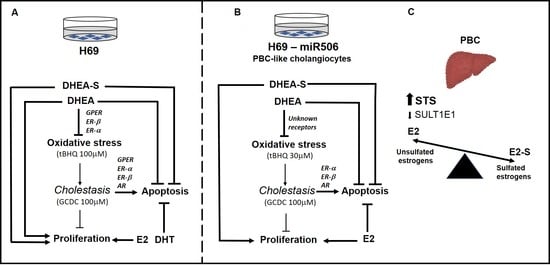
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Tissue Preparation
2.2. MTT Assay
2.3. Cell Proliferation and Apoptosis
2.4. SDS-PAGE and Immunoblotting
2.5. Immunohistochemical Analysis
2.6. Statistical Analysis
3. Results
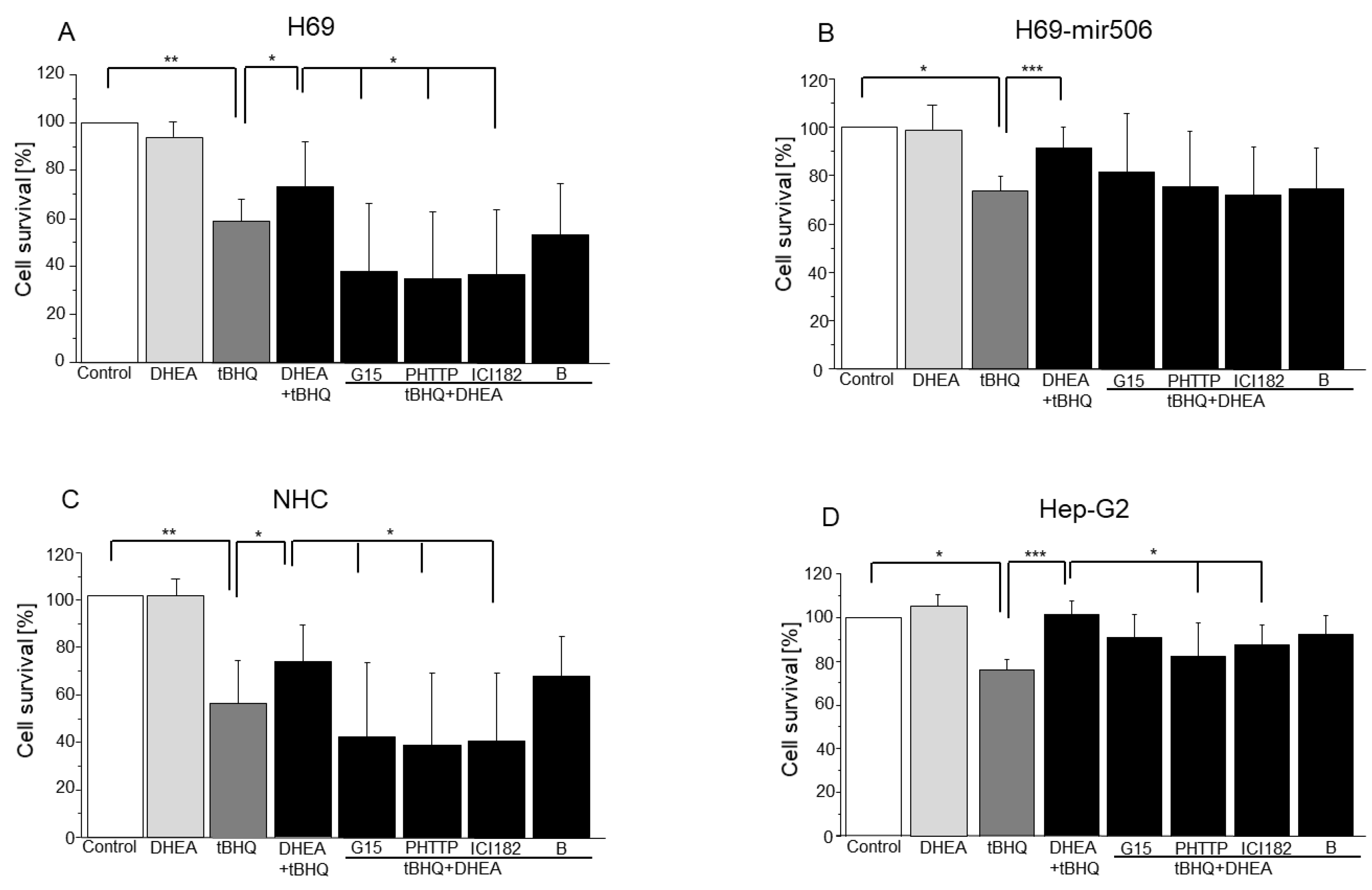
3.1. The Effect of DHEA and Its Metabolites on Cholangiocyte and Hepatocyte Proliferation and Apoptosis
3.2. Role of Estrogen and Androgen Receptors in Cholangiocyte and Hepatocyte Apoptosis Induced by GCDC
3.3. Involvement of Estrogen (ER-α, ER-β, and GPER) and Androgen Receptors in DHEA Protection against Mitochondrial Oxidative Stress Induced by tBHQ
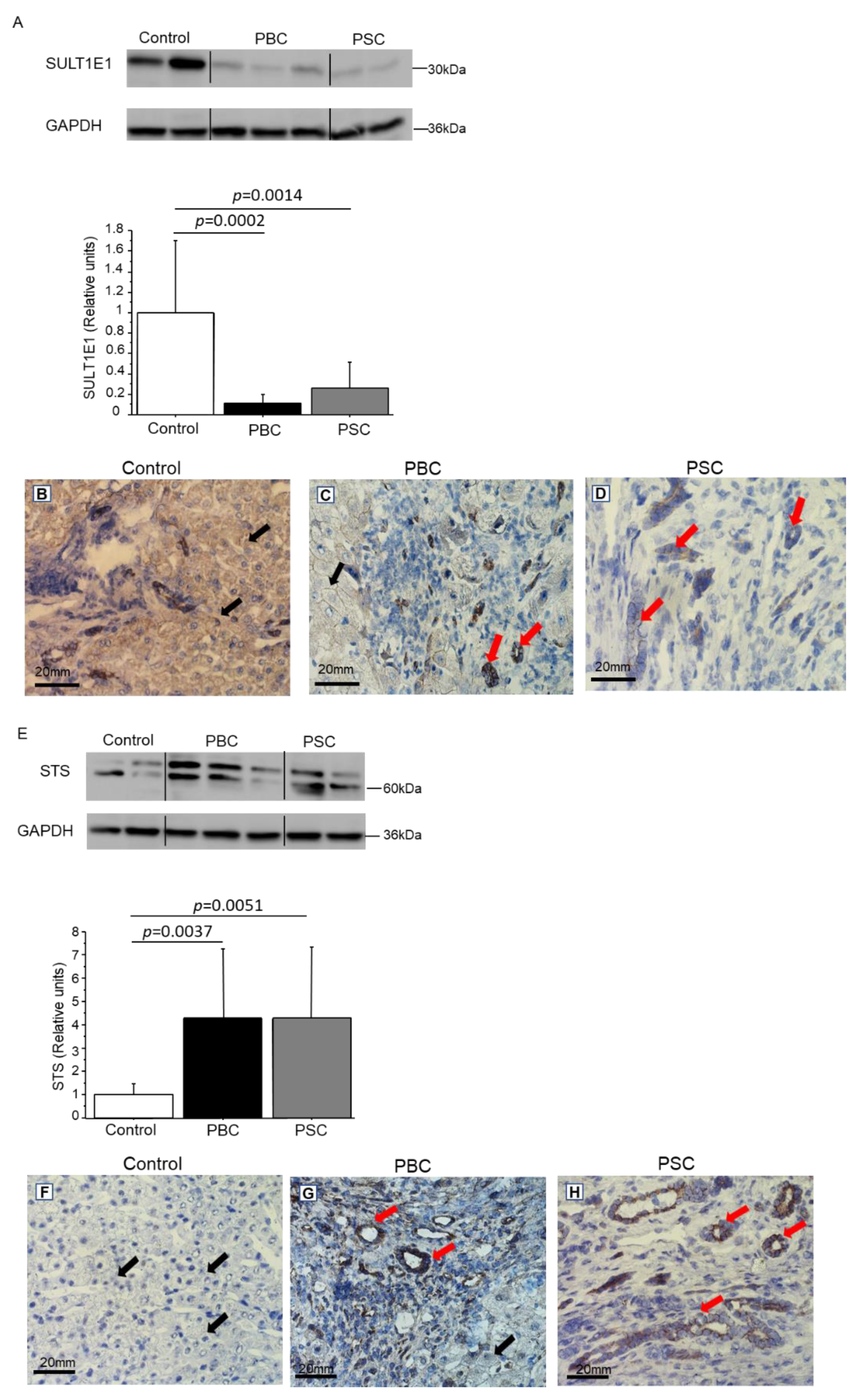
3.4. Expression of SULT1E1 and STS in Control and Cirrhotic Human Liver Tissues (PBC and PSC)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Alvaro, D.; Gigliozzi, A.; Attili, A.F. Regulation and deregulation of cholangiocyte proliferation. J. Hepatol. 2000, 33, 333–340. [Google Scholar] [CrossRef]
- Alvaro, D.; Invernizzi, P.; Onori, P.; Franchitto, A.; De Santis, A.; Crosignani, A.; Sferra, R.; Ginanni-Corradini, S.; Mancino, M.G.; Maggioni, M.; et al. Estrogen receptors in cholangiocytes and the progression of primary biliary cirrhosis. J. Hepatol. 2004, 41, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, Y.; Radde, B.N.; Litchfield, L.M.; Ivanova, M.M.; Prough, R.A.; Clark, B.J.; Doll, M.A.; Hein, D.W.; Klinge, C.M. Dehydroepiandrosterone Activation of G-protein-coupled Estrogen Receptor Rapidly Stimulates MicroRNA-21 Transcription in Human Hepatocellular Carcinoma Cells. J. Biol. Chem. 2015, 290, 15799–15811. [Google Scholar] [CrossRef] [Green Version]
- Van Erpecum, K.J.; Janssens, A.R.; Kreuning, J.; Ruiter, D.J.; Kroon, H.M.; Grond, A.J. Generalized peliosis hepatis and cirrhosis after long-term use of oral contraceptives. Am. J. Gastroenterol. 1988, 83, 572–575. [Google Scholar] [PubMed]
- Olsson, H.L.; Ingvar, C.; Bladstrom, A. Hormone replacement therapy containing progestins and given continuously increases breast carcinoma risk in Sweden. Cancer 2003, 97, 1387–1392. [Google Scholar] [CrossRef]
- Chen, J.; Robertson, G.; Field, J.; Liddle, C.; Farrell, G.C. Effects of bile duct ligation on hepatic expression of female-specific CYP2C12 in male and female rats. Hepatology 1998, 28, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Milkiewicz, P.; Roma, M.G.; Cardenas, R.; Mills, C.O.; Elias, E.; Coleman, R. Effect of tauroursodeoxycholate and S-adenosyl-L-methionine on 17beta-estradiol glucuronide-induced cholestasis. J. Hepatol. 2001, 34, 184–191. [Google Scholar] [CrossRef]
- Milkiewicz, P.; Roma, M.G.; Elias, E.; Coleman, R. Pathobiology and experimental therapeutics in hepatocellular cholestasis: Lessons from the hepatocyte couplet model. Clin. Sci. 2002, 102, 603–614. [Google Scholar] [CrossRef]
- Alvaro, D.; Mancino, M.G.; Onori, P.; Franchitto, A.; Alpini, G.; Francis, H.; Glaser, S.; Gaudio, E. Estrogens and the pathophysiology of the biliary tree. World J. Gastroenterol. 2006, 12, 3537–3545. [Google Scholar] [CrossRef]
- Alvaro, D.; Alpini, G.; Onori, P.; Franchitto, A.; Glaser, S.S.; Le Sage, G.; Folli, F.; Attili, A.F.; Gaudio, E. Alfa and beta estrogen receptors and the biliary tree. Mol. Cell Endocrinol. 2002, 193, 105–108. [Google Scholar] [CrossRef]
- Secky, L.; Svoboda, M.; Klameth, L.; Bajna, E.; Hamilton, G.; Zeillinger, R.; Jager, W.; Thalhammer, T. The sulfatase pathway for estrogen formation: Targets for the treatment and diagnosis of hormone-associated tumors. J. Drug Deliv. 2013, 2013, 957605. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Negishi, M.; Lee, S.J. Estrogen Sulfotransferase (SULT1E1): Its Molecular Regulation, Polymorphisms, and Clinical Perspectives. J. Pers. Med. 2021, 11, 194. [Google Scholar] [CrossRef] [PubMed]
- Alnouti, Y. Bile Acid sulfation: A pathway of bile acid elimination and detoxification. Toxicol. Sci. 2009, 108, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Labrie, F. DHEA, important source of sex steroids in men and even more in women. Prog. Brain Res. 2010, 182, 97–148. [Google Scholar] [CrossRef] [PubMed]
- Ahboucha, S.; Pomier-Layrargues, G.; Vincent, C.; Hassoun, Z.; Tamaz, R.; Baker, G.; Butterworth, R.F. Reduced plasma dehydroepiandrosterone sulfate levels are significantly correlated with fatigue severity in patients with primary biliary cirrhosis. Neurochem. Int. 2008, 52, 569–574. [Google Scholar] [CrossRef]
- Prough, R.A.; Clark, B.J.; Klinge, C.M. Novel mechanisms for DHEA action. J. Mol. Endocrinol. 2016, 56, R139–R155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, S.J.; Geoghegan, T.E.; Prough, R.A.; Michael Miller, K.K. The biological actions of dehydroepiandrosterone involves multiple receptors. Drug Metab. Rev. 2006, 38, 89–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Iruthayanathan, M.; Homan, L.L.; Wang, Y.; Yang, L.; Wang, Y.; Dillon, J.S. Dehydroepiandrosterone stimulates endothelial proliferation and angiogenesis through extracellular signal-regulated kinase 1/2-mediated mechanisms. Endocrinology 2008, 149, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Erice, O.; Munoz-Garrido, P.; Vaquero, J.; Perugorria, M.J.; Fernandez-Barrena, M.G.; Saez, E.; Santos-Laso, A.; Arbelaiz, A.; Jimenez-Aguero, R.; Fernandez-Irigoyen, J.; et al. MicroRNA-506 promotes primary biliary cholangitis-like features in cholangiocytes and immune activation. Hepatology 2018, 67, 1420–1440. [Google Scholar] [CrossRef]
- Kilanczyk, E.; Banales, J.M.; Wunsch, E.; Barbier, O.; Avila, M.A.; Mato, J.M.; Milkiewicz, M.; Milkiewicz, P. S-adenosyl-L-methionine (SAMe) halts the autoimmune response in patients with primary biliary cholangitis (PBC) via antioxidant and S-glutathionylation processes in cholangiocytes. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165895. [Google Scholar] [CrossRef]
- Banales, J.M.; Saez, E.; Uriz, M.; Sarvide, S.; Urribarri, A.D.; Splinter, P.; Tietz Bogert, P.S.; Bujanda, L.; Prieto, J.; Medina, J.F.; et al. Up-regulation of microRNA 506 leads to decreased Cl-/HCO3- anion exchanger 2 expression in biliary epithelium of patients with primary biliary cirrhosis. Hepatology 2012, 56, 687–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasik, U.; Milkiewicz, M.; Kempinska-Podhorodecka, A.; Milkiewicz, P. Protection against oxidative stress mediated by the Nrf2/Keap1 axis is impaired in Primary Biliary Cholangitis. Sci. Rep. 2017, 7, 44769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orentreich, N.; Brind, J.L.; Rizer, R.L.; Vogelman, J.H. Age changes and sex differences in serum dehydroepiandrosterone sulfate concentrations throughout adulthood. J. Clin. Endocrinol. Metab. 1984, 59, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Ansar Ahmed, S. The Immune System Is a Natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front. Immunol. 2015, 6, 635. [Google Scholar] [CrossRef] [Green Version]
- Klair, J.S.; Yang, J.D.; Abdelmalek, M.F.; Guy, C.D.; Gill, R.M.; Yates, K.; Unalp-Arida, A.; Lavine, J.E.; Clark, J.M.; Diehl, A.M.; et al. A longer duration of estrogen deficiency increases fibrosis risk among postmenopausal women with nonalcoholic fatty liver disease. Hepatology 2016, 64, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, Y.; Ding, Q.; Connelly, P.W.; Brunt, J.H.; Ban, M.R.; McIntyre, A.D.; Huff, M.W.; Gros, R.; Hegele, R.A.; Feldman, R.D. G-protein estrogen receptor as a regulator of low-density lipoprotein cholesterol metabolism: Cellular and population genetic studies. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.C.; Liu, Y.; Xiao, L.L.; Li, S.F.; Jiang, J.H.; Zhao, Y.; Qian, S.W.; Tang, Q.Q.; Li, X. Upregulation of miR-125b by estrogen protects against non-alcoholic fatty liver in female mice. J. Hepatol. 2015, 63, 1466–1475. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.C.; Johannsson, G.; Leong, G.M.; Ho, K.K. Estrogen regulation of growth hormone action. Endocr. Rev. 2004, 25, 693–721. [Google Scholar] [CrossRef]
- Avtanski, D.; Novaira, H.J.; Wu, S.; Romero, C.J.; Kineman, R.; Luque, R.M.; Wondisford, F.; Radovick, S. Both estrogen receptor alpha and beta stimulate pituitary GH gene expression. Mol. Endocrinol. 2014, 28, 40–52. [Google Scholar] [CrossRef]
- Lopez-Marure, R.; Contreras, P.G.; Dillon, J.S. Effects of dehydroepiandrosterone on proliferation, migration, and death of breast cancer cells. Eur. J. Pharmacol. 2011, 660, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kilanczyk, E.; Saraswat Ohri, S.; Whittemore, S.R.; Hetman, M. Antioxidant Protection of NADPH-Depleted Oligodendrocyte Precursor Cells Is Dependent on Supply of Reduced Glutathione. ASN Neuro 2016, 8, 1759091416660404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.R.; Dawood, T.; Ling, S.; Dai, A.; Lew, R.; Myles, K.; Funder, J.W.; Sudhir, K.; Komesaroff, P.A. Dehydroepiandrosterone increases endothelial cell proliferation in vitro and improves endothelial function in vivo by mechanisms independent of androgen and estrogen receptors. J. Clin. Endocrinol. Metab. 2004, 89, 4708–4715. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.; Zhang, G.; Sultana, H.; Yu, Y.; Wei, Z. The effects of 17-beta estradiol on enhancing proliferation of human bone marrow mesenchymal stromal cells in vitro. Stem Cells Dev. 2011, 20, 925–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charalampopoulos, I.; Tsatsanis, C.; Dermitzaki, E.; Alexaki, V.I.; Castanas, E.; Margioris, A.N.; Gravanis, A. Dehydroepiandrosterone and allopregnanolone protect sympathoadrenal medulla cells against apoptosis via antiapoptotic Bcl-2 proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 8209–8214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charalampopoulos, I.; Alexaki, V.I.; Tsatsanis, C.; Minas, V.; Dermitzaki, E.; Lasaridis, I.; Vardouli, L.; Stournaras, C.; Margioris, A.N.; Castanas, E.; et al. Neurosteroids as endogenous inhibitors of neuronal cell apoptosis in aging. Ann. N. Y. Acad. Sci. 2006, 1088, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Kousteni, S.; Han, L.; Chen, J.R.; Almeida, M.; Plotkin, L.I.; Bellido, T.; Manolagas, S.C. Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J. Clin. Investig. 2003, 111, 1651–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinge, C.M. Estrogens regulate life and death in mitochondria. J. Bioenerg. Biomembr. 2017, 49, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M. Estrogenic control of mitochondrial function and biogenesis. J. Cell Biochem. 2008, 105, 1342–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felty, Q.; Roy, D. Estrogen, mitochondria, and growth of cancer and non-cancer cells. J. Carcinog. 2005, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis-Wambi, J.S.; Jordan, V.C. Estrogen regulation of apoptosis: How can one hormone stimulate and inhibit? Breast Cancer Res. 2009, 11, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcredito, S.; Brusadelli, A.; Maggi, A. Estrogens, apoptosis and cells of neural origin. J. Neurocytol. 2000, 29, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, J.; Chen, S.; Irwin, R.W.; Iwamoto, S.; Brinton, R.D. Estrogen protects neuronal cells from amyloid beta-induced apoptosis via regulation of mitochondrial proteins and function. BMC Neurosci. 2006, 7, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.D.; Ma, L.; Gu, T.X.; Ding, W.Y.; Zhang, F.; Shen, Y.; Zhang, Y.Z.; Yang, D.L.; Zhang, D.; Sun, Y.P.; et al. 17beta-Estradiol protects against apoptosis induced by levofloxacin in rat nucleus pulposus cells by upregulating integrin alpha2beta1. Apoptosis 2014, 19, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Spyridopoulos, I.; Sullivan, A.B.; Kearney, M.; Isner, J.M.; Losordo, D.W. Estrogen-receptor-mediated inhibition of human endothelial cell apoptosis. Estradiol as a survival factor. Circulation 1997, 95, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Traish, A.M.; Kang, H.P.; Saad, F.; Guay, A.T. Dehydroepiandrosterone (DHEA)--a precursor steroid or an active hormone in human physiology. J. Sex. Med. 2011, 8, 2960–2982. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xie, W. The Role of Sulfotransferases in Liver Diseases. Drug Metab. Dispos. 2020, 48, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.C.S.; Feng, Y.; Yu, C.; Huang, M.; Xie, W. Estrogen sulfotransferase in the metabolism of estrogenic drugs and in the pathogenesis of diseases. Expert Opin. Drug Metab. Toxicol. 2019, 15, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Song, W.C. Biochemistry and reproductive endocrinology of estrogen sulfotransferase. Ann. N. Y. Acad. Sci. 2001, 948, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xue, R.; Yang, C.; Gu, J.; Chen, S.; Zhang, S. Cholestasis-induced bile acid elevates estrogen level via farnesoid X receptor-mediated suppression of the estrogen sulfotransferase SULT1E1. J. Biol. Chem. 2018, 293, 12759–12769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straub, R.H. The complex role of estrogens in inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.X.; Yin, X.M. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J. Cell Mol. Med. 2004, 8, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Klein, M.; Zanger, U.M.; Mohammad, M.K.; Cave, M.C.; Gaikwad, N.W.; Dias, N.J.; Selcer, K.W.; Guo, Y.; He, J.; et al. Inflammatory regulation of steroid sulfatase: A novel mechanism to control estrogen homeostasis and inflammation in chronic liver disease. J. Hepatol. 2016, 64, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Labrie, F.; Belanger, A.; Belanger, P.; Berube, R.; Martel, C.; Cusan, L.; Gomez, J.; Candas, B.; Chaussade, V.; Castiel, I.; et al. Metabolism of DHEA in postmenopausal women following percutaneous administration. J. Steroid. Biochem. Mol. Biol. 2007, 103, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Wronka, K.M.; Wunsch, E.; Kozlowska-Petriczko, K.; Wojcicki, M.; Kruk, B.; Milkiewicz, P. Dehydroepiandrosterone sulfate indicates decreased sulfation capacity and impaired quality of life in patients with primary sclerosing cholangitis. Pol. Arch. Intern. Med. 2021, 131, 790–796. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | PBC (n = 10) | PSC (n = 10) |
|---|---|---|
| Age (years) | 57 (45–69) | 46 (17–62) |
| Gender (M/F) | (1/9) | (7/3) |
| AST (IU/L; normal: 5–35) | 204 (78–652) | 199 (108–510) |
| ALP (IU/L; normal: 40–120) | 639 (234–1373) | 548 (171–984) |
| PLT (103 μl; normal: 150–400) | 124 (63–274) | 160 (56–321) |
| Bilirubin (μmol/L; normal: 3.4–20.6) | 113 (34–192) | 135 (36–316) |
| Albumin (g/dL; normal: 3.6–4.6) | 2.6 (1.7–3.1) | 2.2 (1.4–2.9) |
| INR (normal: 0.8–1.2) | 1.3 (1.1–1.7) | 1.3 (1.0–1.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kilanczyk, E.; Ruminkiewicz, D.; Banales, J.M.; Milkiewicz, P.; Milkiewicz, M. DHEA Protects Human Cholangiocytes and Hepatocytes against Apoptosis and Oxidative Stress. Cells 2022, 11, 1038. https://doi.org/10.3390/cells11061038
Kilanczyk E, Ruminkiewicz D, Banales JM, Milkiewicz P, Milkiewicz M. DHEA Protects Human Cholangiocytes and Hepatocytes against Apoptosis and Oxidative Stress. Cells. 2022; 11(6):1038. https://doi.org/10.3390/cells11061038
Chicago/Turabian StyleKilanczyk, Ewa, Dagmara Ruminkiewicz, Jesus M. Banales, Piotr Milkiewicz, and Małgorzata Milkiewicz. 2022. "DHEA Protects Human Cholangiocytes and Hepatocytes against Apoptosis and Oxidative Stress" Cells 11, no. 6: 1038. https://doi.org/10.3390/cells11061038