
KCNQ2 Selectivity Filter Mutations Cause Kv7.2 M-Current Dysfunction and Configuration Changes Manifesting as Epileptic Encephalopathies and Autistic Spectrum Disorders


Abstract
:1. Introduction
2. Materials
Participants
3. Methods
3.1. Computational Protein Analysis of SF Mutations and Their Phenotypes
3.2. Mutations of KCNQ2 in Corresponding to KCNQ2 Functional Domains and Phenotypes
3.3. Expression in HEK293 Cells
3.4. Transfecting Variants to HEK293 Cells
3.5. Whole-Cell Patch-Clamp Analysis
3.6. Cytoplasmic and Membranous Protein Separation
3.7. Western Blotting
3.8. Ethics
3.9. Statistics
4. Results
4.1. Clinical Presentations in 3 Patients with KCNQ2 Mutations in SF Domain
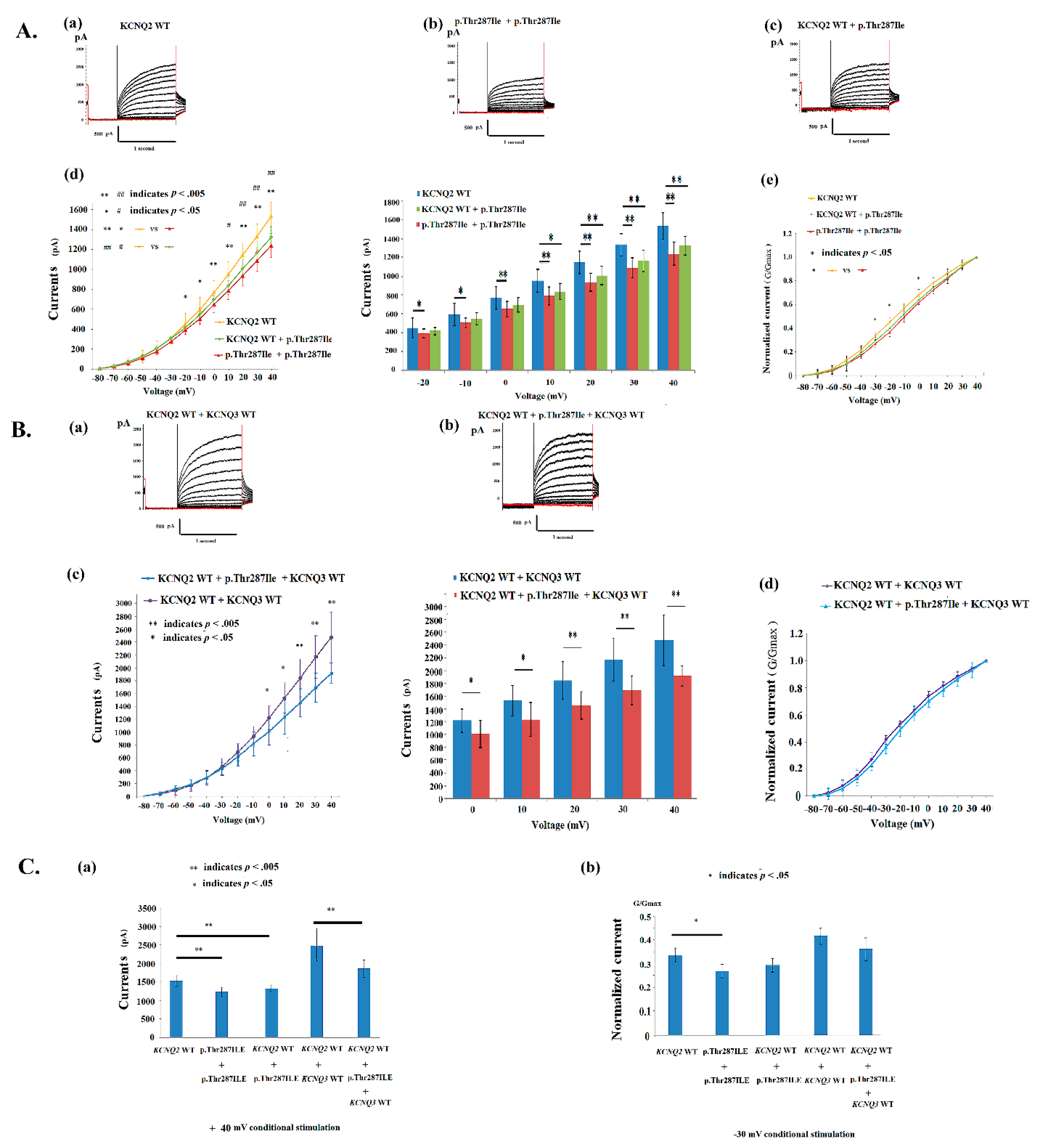
4.2. Electrophysiological Properties of p.Thr287Ile in KCNQ2 Mutations
4.3. Electrophysiological Properties of p.Gly281Glu in KCNQ2 Mutations
4.4. Conductance–Current Curves in p.Thr287Ile, p.Gly281Glu, p.Pro285Thr and KCNQ2 WT
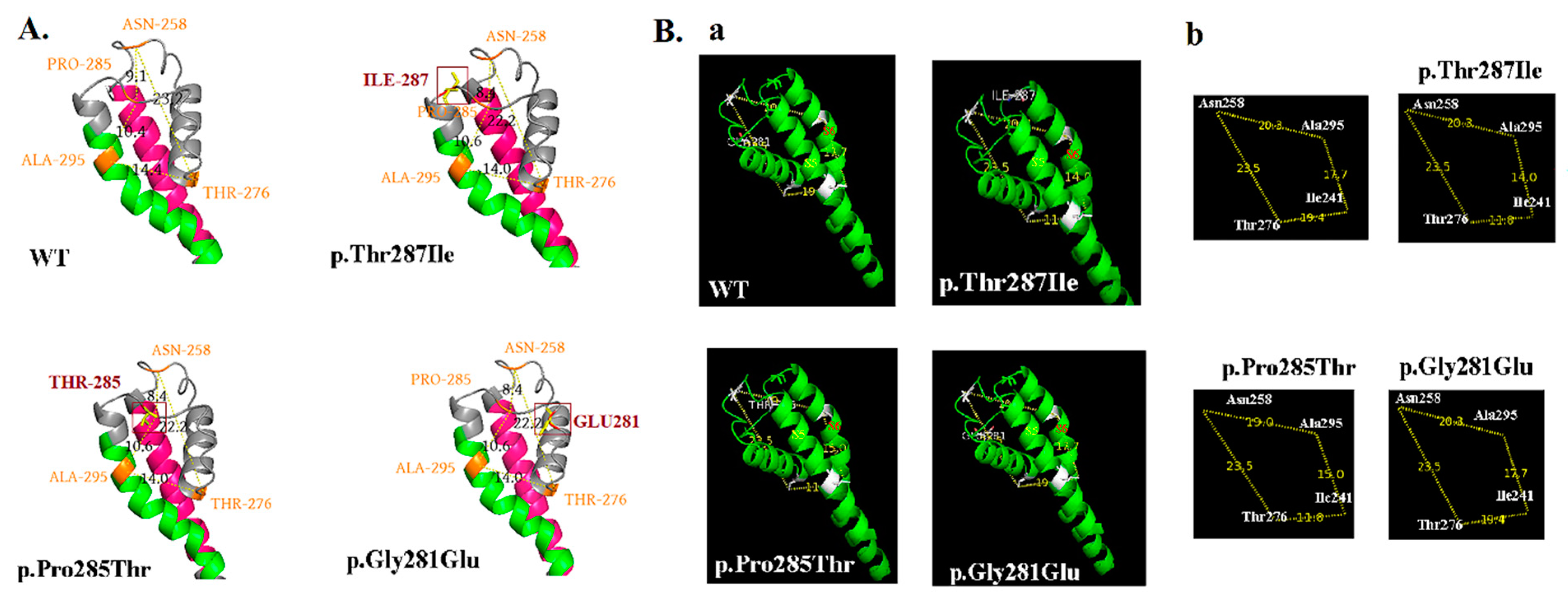
4.5. Phenotypes, KCNQ2 Protein Expression, and Configuration Change on Cell Membranes
4.6. Neurodevelopmental Outcomes Related to Mutations in the SF of KCNQ2
4.7. Neurodevelopmental Outcomes Related to Mutations in the SF of KCNQ2
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
References
- Leppert, M.; Anderson, V.E.; Quattlebaum, T.; Stauffer, D.; O’Connell, P.; Nakamura, Y.; White, R. Benign familial neonatal convulsions linked to genetic markers on chromosome 20. Nature 1989, 337, 647–648. [Google Scholar] [CrossRef] [PubMed]
- Biervert, C.; Schroeder, B.C.; Kubisch, C.; Berkovic, S.F.; Propping, P.; Jentsch, T.J.; Steinlein, O.K. A potassium channel mutation in neonatal human epilepsy. Science 1998, 279, 403–406. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.A.; Charlier, C.; Stauffer, D.; DuPont, B.R.; Leach, R.J.; Melis, R.; Leppert, M. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998, 18, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.S.; Pan, Z.; Shi, W.; Brown, B.S.; Wymore, R.S.; Cohen, I.S.; McKinnon, D. KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science 1998, 282, 1890–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerche, H.; Biervert, C.; Alekov, A.K.; Schleithoff, L.; Lindner, M.; Klinger, W.; Steinlein, O.K. A reduced K+ current due to a novel mutation in KCNQ2 causes neonatal convulsions. Ann. Neurol. 1999, 46, 305–312. [Google Scholar] [CrossRef]
- Kato, M.; Yamagata, T.; Kubota, M.; Arai, H.; Yamashita, S.; Nakagawa, T.; Saitsu, H. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia 2013, 54, 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Weckhuysen, S.; Mandelstam, S.; Suls, A.; Audenaert, D.; Deconinck, T.; Claes, L.R.; de Jonghe, P. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012, 71, 15–25. [Google Scholar] [CrossRef]
- Weckhuysen, S.; Ivanovic, V.; Hendrickx, R.; Van Coster, R.; Hjalgrim, H.; Moller, R.S.; De Jonghe, P. Extending the KCNQ2 encephalopathy spectrum: Clinical and neuroimaging findings in 17 patients. Neurology 2013, 81, 1697–1703. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.J.; Mukai, J.; Kvajo, M.; Xu, B.; Diamantopoulou, A.; Pitychoutis, P.M.; Zhang, H. A Schizophrenia-Related Deletion Leads to KCNQ2-Dependent Abnormal Dopaminergic Modulation of Prefrontal Cortical Interneuron Activity. Cereb. Cortex 2018, 28, 2175–2191. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.H.; Yuen, R.K.; Jin, X.; Wang, M.; Chen, N.; Wu, X.; Scherer, S.W. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 2013, 93, 249–263. [Google Scholar] [CrossRef] [Green Version]
- Vilan, A.; Mendes Ribeiro, J.; Striano, P.; Weckhuysen, S.; Weeke, L.C.; Brilstra, E.; Cilio, M.R. A Distinctive Ictal Amplitude-Integrated Electroencephalography Pattern in Newborns with Neonatal Epilepsy Associated with KCNQ2 Mutations. Neonatology 2017, 112, 387–393. [Google Scholar] [CrossRef]
- Chen, D.Y.; Chowdhury, S.; Farnaes, L.; Friedman, J.R.; Honold, J.; Dimmock, D.P.; Gold, O. Rapid Diagnosis of KCNQ2-Associated Early Infantile Epileptic Encephalopathy Improved Outcome. Pediatr. Neurol. 2018, 86, 69–70. [Google Scholar] [CrossRef]
- Lee, I.C.; Yang, J.J.; Wong, S.H.; Liou, Y.M.; Li, S.Y. Heteromeric Kv7.2 current changes caused by loss-of-function of KCNQ2 mutations are correlated with long-term neurodevelopmental outcomes. Sci. Rep. 2020, 10, 13375. [Google Scholar] [CrossRef] [PubMed]
- Miceli, F.; Soldovieri, M.V.; Ambrosino, P.; Barrese, V.; Migliore, M.; Cilio, M.R.; Taglialatela, M. Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proc. Natl. Acad. Sci. USA 2013, 110, 4386–4391. [Google Scholar] [CrossRef] [Green Version]
- Cooper, E.C.; Harrington, E.; Jan, Y.N.; Jan, L.Y. M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J. Neurosci. 2001, 21, 9529–9540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devaux, J.; Dhifallah, S.; De Maria, M.; Stuart-Lopez, G.; Becq, H.; Milh, M.; Aniksztejn, L. A possible link between KCNQ2- and STXBP1-related encephalopathies: STXBP1 reduces the inhibitory impact of syntaxin-1A on M current. Epilepsia 2017, 58, 2073–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soldovieri, M.V.; Ambrosino, P.; Mosca, I.; De Maria, M.; Moretto, E.; Miceli, F.; Taglialatela, M. Early-onset epileptic encephalopathy caused by a reduced sensitivity of Kv7.2 potassium channels to phosphatidylinositol 4,5-bisphosphate. Sci. Rep. 2016, 6, 38167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Cooper, E.C. An Ankyrin-G N-terminal Gate and Protein Kinase CK2 Dually Regulate Binding of Voltage-gated Sodium and KCNQ2/3 Potassium Channels. J. Biol. Chem. 2015, 290, 16619–16632. [Google Scholar] [CrossRef] [Green Version]
- Zaydman, M.A.; Cui, J. PIP2 regulation of KCNQ channels: Biophysical and molecular mechanisms for lipid modulation of voltage-dependent gating. Front. Physiol. 2014, 5, 195. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Craciun, L.C.; Mirshahi, T.; Rohács, T.; Lopes, C.M.; Jin, T.; Logothetis, D.E. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 2003, 37, 963–975. [Google Scholar] [CrossRef] [Green Version]
- Ambrosino, P.; Alaimo, A.; Bartollino, S.; Manocchio, L.; De Maria, M.; Mosca, I.; Soldovieri, M.V. Epilepsy-causing mutations in Kv7.2 C-terminus affect binding and functional modulation by calmodulin. Biochim. Biophys. Acta 2015, 1852, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Choveau, F.S.; Shapiro, M.S. Regions of KCNQ K(+) channels controlling functional expression. Front. Physiol 2012, 3, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Zhuang, F.; Li, H.; Zheng, K.; Hong, Z.; Feng, W.; Chen, J. Calmodulin regulates KCNQ2 function in epilepsy. Am. J. Transl. Res. 2016, 8, 5610–5618. [Google Scholar] [PubMed]
- Wang, J.J.; Li, Y. KCNQ potassium channels in sensory system and neural circuits. Acta Pharmacol. Sin. 2016, 37, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Miceli, F.; Soldovieri, M.V.; Ambrosino, P.; De Maria, M.; Migliore, M.; Migliore, R.; Taglialatela, M. Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J. Neurosci. 2015, 35, 3782–3793. [Google Scholar] [CrossRef] [Green Version]
- Maljevic, S.; Naros, G.; Yalcin, O.; Blazevic, D.; Loeffler, H.; Caglayan, H.; Lerche, H. Temperature and pharmacological rescue of a folding-defective, dominant-negative KV 7.2 mutation associated with neonatal seizures. Hum. Mutat. 2011, 32, E2283–E2293. [Google Scholar] [CrossRef]
- Wuttke, T.V.; Penzien, J.; Fauler, M.; Seebohm, G.; Lehmann-Horn, F.; Lerche, H.; Jurkat-Rott, K. Neutralization of a negative charge in the S1-S2 region of the KV7.2 (KCNQ2) channel affects voltage-dependent activation in neonatal epilepsy. J. Physiol. 2008, 586, 545–555. [Google Scholar] [CrossRef]
- Orhan, G.; Bock, M.; Schepers, D.; Ilina, E.I.; Reichel, S.N.; Löffler, H.; Maljevic, S. Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann. Neurol. 2014, 75, 382–394. [Google Scholar] [CrossRef]
- Millichap, J.J.; Miceli, F.; De Maria, M.; Keator, C.; Joshi, N.; Tran, B.; Taglialatela, M. Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain-of-function variant. Epilepsia 2017, 58, e10–e15. [Google Scholar] [CrossRef] [Green Version]
- Devaux, J.; Abidi, A.; Roubertie, A.; Molinari, F.; Becq, H.; Lacoste, C.; Aniksztejn, L. A Kv7.2 mutation associated with early onset epileptic encephalopathy with suppression-burst enhances Kv7/M channel activity. Epilepsia 2016, 57, e87–e93. [Google Scholar] [CrossRef] [Green Version]
- Sands, T.T.; Miceli, F.; Lesca, G.; Beck, A.E.; Sadleir, L.G.; Arrington, D.K.; Cilio, M.R. Autism and developmental disability caused by KCNQ3 gain-of-function variants. Ann. Neurol. 2019, 86, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Abidi, A.; Devaux, J.J.; Molinari, F.; Alcaraz, G.; Michon, F.X.; Sutera-Sardo, J.; Aniksztejn, L. A recurrent KCNQ2 pore mutation causing early onset epileptic encephalopathy has a moderate effect on M current but alters subcellular localization of Kv7 channels. Neurobiol. Dis. 2015, 80, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kim, E.C.; Chen, C.; Procko, E.; Pant, S.; Lam, K.; Chung, H.J. Identifying mutation hotspots reveals pathogenetic mechanisms of KCNQ2 epileptic encephalopathy. Sci. Rep. 2020, 10, 4756. [Google Scholar] [CrossRef] [PubMed]
- Baculis, B.C.; Zhang, J.; Chung, H.J. The Role of K(v)7 Channels in Neural Plasticity and Behavior. Front. Physiol. 2020, 11, 568667. [Google Scholar] [CrossRef]
- Brown, D.A.; Passmore, G.M. Neural KCNQ (Kv7) channels. Br. J. Pharmacol. 2009, 156, 1185–1195. [Google Scholar] [CrossRef]
- Sun, J.; MacKinnon, R. Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell 2017, 169, 1042–1050.e1049. [Google Scholar] [CrossRef]
- Cui, J. Voltage-Dependent Gating: Novel Insights from KCNQ1 Channels. Biophys. J. 2016, 110, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Dhamija, R.; Goodkin, H.P.; Bailey, R.; Chambers, C.; Brenton, J.N. A Case of KCNQ2-Associated Movement Disorder Triggered by Fever. J. Child Neurol. 2017, 32, 1123–1124. [Google Scholar] [CrossRef]
- Blumkin, L.; Suls, A.; Deconinck, T.; De Jonghe, P.; Linder, I.; Kivity, S.; Lerman-Sagie, T. Neonatal seizures associated with a severe neonatal myoclonus like dyskinesia due to a familial KCNQ2 gene mutation. Eur. J. Paediatr. Neurol. 2012, 16, 356–360. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Volkers, L.; Rook, M.B.; Das, J.H.; Verbeek, N.E.; Groenewegen, W.A.; van Kempen, M.J.; Koeleman, B.P. Functional analysis of novel KCNQ2 mutations found in patients with Benign Familial Neonatal Convulsions. Neurosci. Lett. 2009, 462, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Selyanko, A.A.; Hadley, J.K.; Brown, D.A. Properties of single M-type KCNQ2/KCNQ3 potassium channels expressed in mammalian cells. J. Physiol. 2001, 534 Pt 1, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Stefani, E.; Toro, L.; Perozo, E.; Bezanilla, F. Gating of Shaker K+ channels: I. Ionic and gating currents. Biophys. J. 1994, 66, 996–1010. [Google Scholar] [CrossRef] [Green Version]
- Milh, M.; Lacoste, C.; Cacciagli, P.; Abidi, A.; Sutera-Sardo, J.; Tzelepis, I.; Villard, L. Variable clinical expression in patients with mosaicism for KCNQ2 mutations. Am. J. Med. Genet. A 2015, 167, 2314–2318. [Google Scholar] [CrossRef] [PubMed]
- Yalçin, O.; Cağlayan, S.H.; Saltik, S.; Cokar, O.; Ağan, K.; Dervent, A.; Steinlein, O.K. A novel missense mutation (N258S) in the KCNQ2 gene in a Turkish family afflicted with benign familial neonatal convulsions (BFNC). Turk. J. Pediatr. 2007, 49, 385–389. [Google Scholar]
- Hortigüela, M.; Fernández-Marmiesse, A.; Cantarín, V.; Gouveia, S.; García-Peñas, J.J.; Fons, C.; Gutiérrez-Solana, L.G. Clinical and genetic features of 13 Spanish patients with KCNQ2 mutations. J. Hum. Genet. 2017, 62, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Saitsu, H.; Kato, M.; Koide, A.; Goto, T.; Fujita, T.; Nishiyama, K.; Matsumoto, N. Whole exome sequencing identifies KCNQ2 mutations in Ohtahara syndrome. Ann. Neurol. 2012, 72, 298–300. [Google Scholar] [CrossRef]
- Milh, M.; Boutry-Kryza, N.; Sutera-Sardo, J.; Mignot, C.; Auvin, S.; Lacoste, C.; Villard, L. Similar early characteristics but variable neurological outcome of patients with a de novo mutation of KCNQ2. Orphanet J. Rare Dis. 2013, 8, 80. [Google Scholar] [CrossRef]
- Pisano, T.; Numis, A.L.; Heavin, S.B.; Weckhuysen, S.; Angriman, M.; Suls, A.; Cilio, M.R. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia 2015, 56, 685–691. [Google Scholar] [CrossRef]
- Millichap, J.J.; Park, K.L.; Tsuchida, T.; Ben-Zeev, B.; Carmant, L.; Flamini, R.; Cooper, E.C. KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol. Genet. 2016, 2, e96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomis-Perez, C.; Urrutia, J.; Marce-Grau, A.; Malo, C.; Lopez-Laso, E.; Felipe-Rucian, A.; Villarroel, A. Homomeric Kv7.2 current suppression is a common feature in KCNQ2 epileptic encephalopathy. Epilepsia 2019, 60, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Ma, A.; Liu, X.; Huang, C.; Zhang, Y.; Shi, R.; Li, S. Infantile seizures and other epileptic phenotypes in a Chinese family with a missense mutation of KCNQ2. Eur. J. Pediatr. 2006, 165, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.C.; Kim, G.E.; Pagnamenta, A.T.; Murakami, Y.; Carvill, G.L.; Meyer, E.; Taylor, J.C. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum. Mol. Genet. 2014, 23, 3200–3211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Cheng, Y.; Li, H.; Jia, Q.; Zhang, H.; Zhao, S. Inhibition of Kv7/M Channel Currents by the Local Anesthetic Chloroprocaine. Pharmacology 2015, 96, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Yalcıntepe, S.; Ozguc Comlek, F.; Gurkan, H.; Demir, S.; Atlı, E.I.; Atlı, E.; Tutunculer Kokenli, F. The Application of Next Generation Sequencing Maturity Onset Diabetes of the Young Gene Panel in Turkish Patients from Trakya Region. J. Clin. Res. Pediatr. Endocrinol. 2021, 13, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Laccetta, G.; Fiori, S.; Giampietri, M.; Ferrari, A.; Cetica, V.; Bernardini, M.; Ghirri, P. A de novo KCNQ2 Gene Mutation Associated With Non-familial Early Onset Seizures: Case Report and Revision of Literature Data. Front. Pediatr. 2019, 7, 348. [Google Scholar] [CrossRef]
- Schneider, A.L.; Myers, C.T.; Muir, A.M.; Calvert, S.; Basinger, A.; Perry, M.S.; Scheffer, I.E. FBXO28 causes developmental and epileptic encephalopathy with profound intellectual disability. Epilepsia 2021, 62, e13–e21. [Google Scholar] [CrossRef]
- Parrini, E.; Marini, C.; Mei, D.; Galuppi, A.; Cellini, E.; Pucatti, D.; Guerrini, R. Diagnostic Targeted Resequencing in 349 Patients with Drug-Resistant Pediatric Epilepsies Identifies Causative Mutations in 30 Different Genes. Hum. Mutat. 2017, 38, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Li, J.; Zhao, Y.; Bao, X.; Wei, L.; Wang, J. Gene mutation analysis of 175 Chinese patients with early-onset epileptic encephalopathy. Clin. Genet. 2017, 91, 717–724. [Google Scholar] [CrossRef]
- Cao, Y.; Bartolomé-Martín, D.; Rotem, N.; Rozas, C.; Dellal, S.S.; Chacon, M.A.; Faber, D.S. Rescue of homeostatic regulation of striatal excitability and locomotor activity in a mouse mode’l of Huntingtons disease. Proc. Natl. Acad. Sci. USA 2015, 112, 2239–2244. [Google Scholar] [CrossRef] [Green Version]
- Sander, S.E.; Lambrecht, C.; Richter, A. The K(V)7.2/3 preferring channel opener ICA 27243 attenuates L-DOPA-induced dyskinesia in hemiparkinsonian rats. Neurosci. Lett. 2013, 545, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Borgatti, R.; Zucca, C.; Cavallini, A.; Ferrario, M.; Panzeri, C.; Castaldo, P.; Bassi, M.T. A novel mutation in KCNQ2 associated with BFNC, drug resistant epilepsy, and mental retardation. Neurology 2004, 63, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Gururaj, X.; Palmer, E.E.; Sheehan, G.D.; Kandula, T.; Macintosh, R.; Bhattacharjee, A. A De Novo Mutation in the Sodium-Activated Potassium Channel KCNT2 Alters Ion Selectivity and Causes Epileptic Encephalopathy. Cell. Rep. 2017, 21, 926–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maljevic, S.; Wuttke, T.V.; Lerche, H. Nervous system KV7 disorders: Breakdown of a subthreshold brake. J. Physiol. 2008, 586, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.C.; Chang, M.Y.; Liang, J.S.; Chang, T.M. Ictal and interictal electroencephalographic findings can contribute to early diagnosis and prompt treatment in KCNQ2-associated epileptic encephalopathy. J. Formos. Med. Assoc. 2021, 120 Pt 3, 744–754. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Genotype in patients | p.Thr287Ile | p.Gly281Glu | p.Pro285Thr |
| Inheritance | De novo | De novo | De novo |
| Functional domain | Selectivity filter | Selectivity filter | Selectivity filter |
| Family history | No | No | No |
| First seizure day | Day 3 | Day 3 | Day 2 |
| Seizure frequency before drug control | Daily | Daily | Daily |
| Age when seizure-free | Partial remission of seizures at 4 months, with recurrent febrile seizures | No remission of seizures | Partial remission of seizures after 1 year |
| MRI | Unremarkable at 4 years old | Thin corpus callosum, brain atrophy at 4 years old | Thin corpus callosum |
| Long-term neurodevelopmental outcomes | Lack of language production, can sit, inability to walk, severe cognitive disability at 5 years old. | Lack of language production, cannot sit without support, inability to walk, severe cognitive disability at 5 years old. | Lack of language, can sit, inability to walk, severe cognitive disability at 4 years old. |
| HEK293 Transfection | KCNQ2 WT | KCNQ2 WT + KCNQ3 WT | p.Gly281Glu | p.Thr287Ile | p.Pro285Thr |
|---|---|---|---|---|---|
| Homomeric variants (2 μg) | |||||
| V1/2 (mV) (mean ± SD) (N) (p) | −16.9 ± 2.0 (22) | −10.9 ± 1.8 ** (8) (p < 0.001) | −12.2 ± 1.9 ** (10) (p < 0.001) | −13.8 ± 3.2 * (10) (p = 0.012) | |
| K (mV/e-fold) (mean ± SD) (N) (p) | 9.5 ± 2.2 (22) | 8.0 ± 1.2 * (8) (p = 0.031) | 8.3 ± 1.3 * (10) (p = 0.015) | 7.9 ± 0.9 ** (10) (p = 0.009) | |
| Currents (pA) (mean ± SD) (N) (p) | 579.8 ± 46.0 (22) | 447.7 ± 63.7 ** (8) (p < 0.001) | 481.8 ± 56.9 * (10) (p = 0.015) | 436.0 ± 55.0 ** (10) (p <. 001) | |
| Heteromeric variants (KCNQ2 + mutants) (1 μg:1 μg) | |||||
| V1/2 (mV) (mean ± SD) (N) (p) | −13.0 ± 2.1 * (8) (p = 0.010) | −13.9 ± 1.5 * (10) (p = 0.011) | −14.8 ± 2.1 (10) (p = 0.073) | ||
| K (mV/e-fold) (mean ± SD) (N) (p) | 8.5 ± 1.6 (8) (p = 0.159) | 8.7 ± 1.1 (10) (p = 0.133) | 8.8 ± 1.5 (10) (p = 0.351) | ||
| Currents (pA) (mean ± SD) (N) (p) | 490.0 ± 38.0 * (8) (p = 0.016) | 519.4 ± 52.0 (10) (p = 0.056) | 518.0 ± 24.5 (10) (p = 0.74) | ||
| Heteromeric variants (KCNQ2 + mutants + KCNQ3) (0.5 μg:0.5 μg:1μg) | |||||
| V1/2 (mV) (mean ± SD) (N) (p) | −20.8 ± 1.6 (10) | −18.5 ± 2.0 (8) (p = 0.054) | −19.3 ± 1.4 (10) (p = 0.122) | −18.8 ± 2.2 (10) (p = 0.061) | |
| K (mV/e-fold) (mean ± SD) (N) (p) | 14.9 ± 3.0 (10) | 11.1 ± 0.7 ** (8) (p = 0.002) | 12.5 ± 2.7 (10) (p = 0.081) | 13.1 ± 1.7 (10) (p = 0.107) | |
| Currents (pA) (mean ± SD) (N) (p) | 813.9 ± 118.3 (10) | 705.0 ± 64.4 (8) (p = 0.051) | 753.3 ± 94.2 (10) (p = 0.053) | 715.2 ± 58.3 (10) (p = 0.054) |
| KCNQ2 Genotype # | WT | Weight of Amino Acid of KCNQ2 WT | Amino Acid Polarity | Amino Acid Change | Weight of Mutated Amino Acid | Mutated Amino Acid Polarity | Phenotype | References |
|---|---|---|---|---|---|---|---|---|
| Ala253Thr | Ala | 89 | N | Thr | 119 | Y | EE | [44] |
| Gly256Trp | Gly | 75 | N | Trp | 204 | Y | EE | [29] |
| Asn258Ser | Asn | 132 | Y | Ser | 105 | Y | BFNC | [45] |
| Asp259Tyr | Asp | 133 | Acid R group | Tyr | 181 | Y | BFNC | [46] |
| Asp259Gly | Asp | 133 | Acid R group | Gly | 75 | N | BFNC | [47] |
| Asp259Glu | Asp | 133 | Acid R group | Glu | 147 | Acid R group | EE | [29] |
| Ala265Val | Ala | 89 | N | Val | 117 | N | EE | [48] |
| Ala265Pro | Ala | 89 | N | Pro | 115 | N | EE | [7] |
| Ala265Thr | Ala | 89 | N | Thr | 119 | Y | EE | [49] |
| Leu268Phe | Leu | 131 | N | Phe | 165 | N | EE | [50] |
| Trp269Leu | Trp | 204 | Y | Leu | 131 | N | EE | [51] |
| Trp270Arg | Trp | 204 | Y | Arg | 174 | Basic R group | EE | [52] |
| Gly271Val | Gly | 75 | N | Val | 117 | N | BFIC | [53] |
| Ile273Asn | Ile | 131 | N | Asn | 132 | Y | EE | [51] |
| Thr274Met | Thr | 119 | Y | Met | 149 | N | EE | [7] |
| Thr276Ile | Thr | 119 | Y | Ile | 131 | N | EE | [54] |
| Thr277Ile | Thr | 119 | Y | Ile | 131 | N | EE | [55] |
| Gly279Cys | Gly | 75 | N | Cys | 121 | Y | EE | [44] |
| Gly281 Trp | Gly | 75 | N | Trp | 204 | Y | EE | [50] |
| Gly281Arg | Gly | 75 | N | Arg | 174 | Basic R group | EE | [8] |
| Gly281Glu | Gly | 75 | N | Glu | 147 | Acid R group | EE | The study |
| Gly281Trp | Gly | 75 | N | Trp | 204 | Y | EE | [50] |
| Asp282Asn | Asp | 133 | Acid R group | Asn | 132 | Y | EE | [56] |
| Tyr284Asp | Tyr | 181 | Y | Asp | 133 | Acid R group | EE | [46] |
| Tyr284Cys | Tyr | 181 | Y | Cys | 121 | Y | BFNC | [3] |
| Pro285Thr | Pro | 115 | N | Thr | 119 | Y | EE | The study |
| Pro285Ser | Pro | 115 | N | Ser | 105 | Y | EE | [57] |
| Pro285His | Pro | 115 | N | His | 155 | Basic R group | EE | [6] |
| Thr287Ile | Thr | 119 | Y | Ile | 131 | N | EE | The study |
| Thr287Pro | Thr | 119 | Y | Pro | 115 | N | EE | [58] |
| Thr287Asn | Thr | 119 | Y | Asn | 132 | Y | EE | [49] |
| Gly290Asp | Gly | 75 | N | Asp | 133 | Acid R group | EE | [7] |
| Gly290Ser | Gly | 75 | N | Ser | 105 | Y | EE | [49] |
| Arg291Ser | Arg | 174 | Basic R group | Ser | 105 | Y | EE | [59] |
| Arg291Gly | Arg | 174 | Basic R group | Gly | 75 | N | EE | [60] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, I.-C.; Yang, J.-J.; Liou, Y.-M.; Wong, S.-H. KCNQ2 Selectivity Filter Mutations Cause Kv7.2 M-Current Dysfunction and Configuration Changes Manifesting as Epileptic Encephalopathies and Autistic Spectrum Disorders. Cells 2022, 11, 894. https://doi.org/10.3390/cells11050894
Lee I-C, Yang J-J, Liou Y-M, Wong S-H. KCNQ2 Selectivity Filter Mutations Cause Kv7.2 M-Current Dysfunction and Configuration Changes Manifesting as Epileptic Encephalopathies and Autistic Spectrum Disorders. Cells. 2022; 11(5):894. https://doi.org/10.3390/cells11050894
Chicago/Turabian StyleLee, Inn-Chi, Jiann-Jou Yang, Ying-Ming Liou, and Swee-Hee Wong. 2022. "KCNQ2 Selectivity Filter Mutations Cause Kv7.2 M-Current Dysfunction and Configuration Changes Manifesting as Epileptic Encephalopathies and Autistic Spectrum Disorders" Cells 11, no. 5: 894. https://doi.org/10.3390/cells11050894