
Oxidative Stress in Human Pathology and Aging: Molecular Mechanisms and Perspectives

 , , , and
, , , and 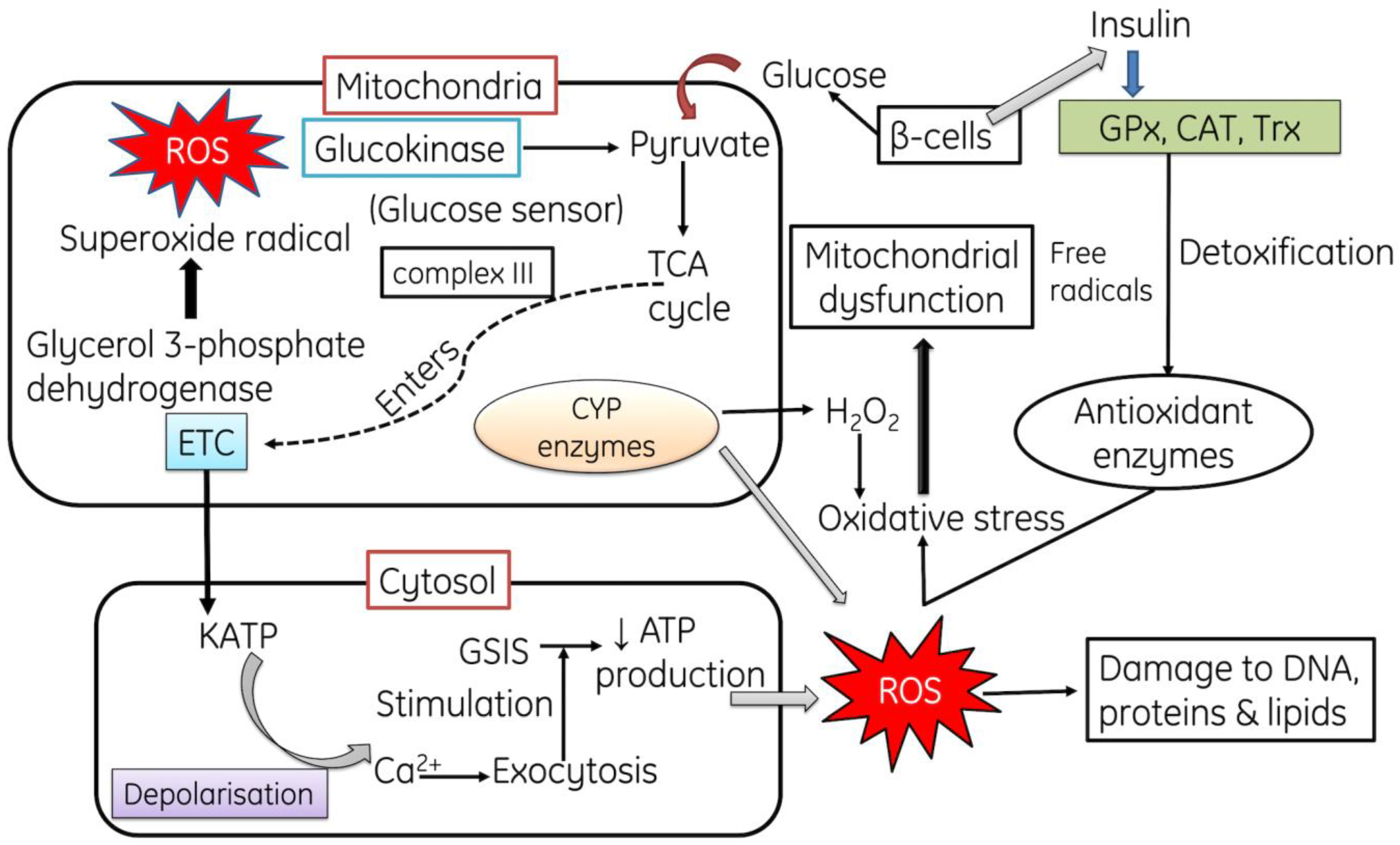{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Mechanism of ROS
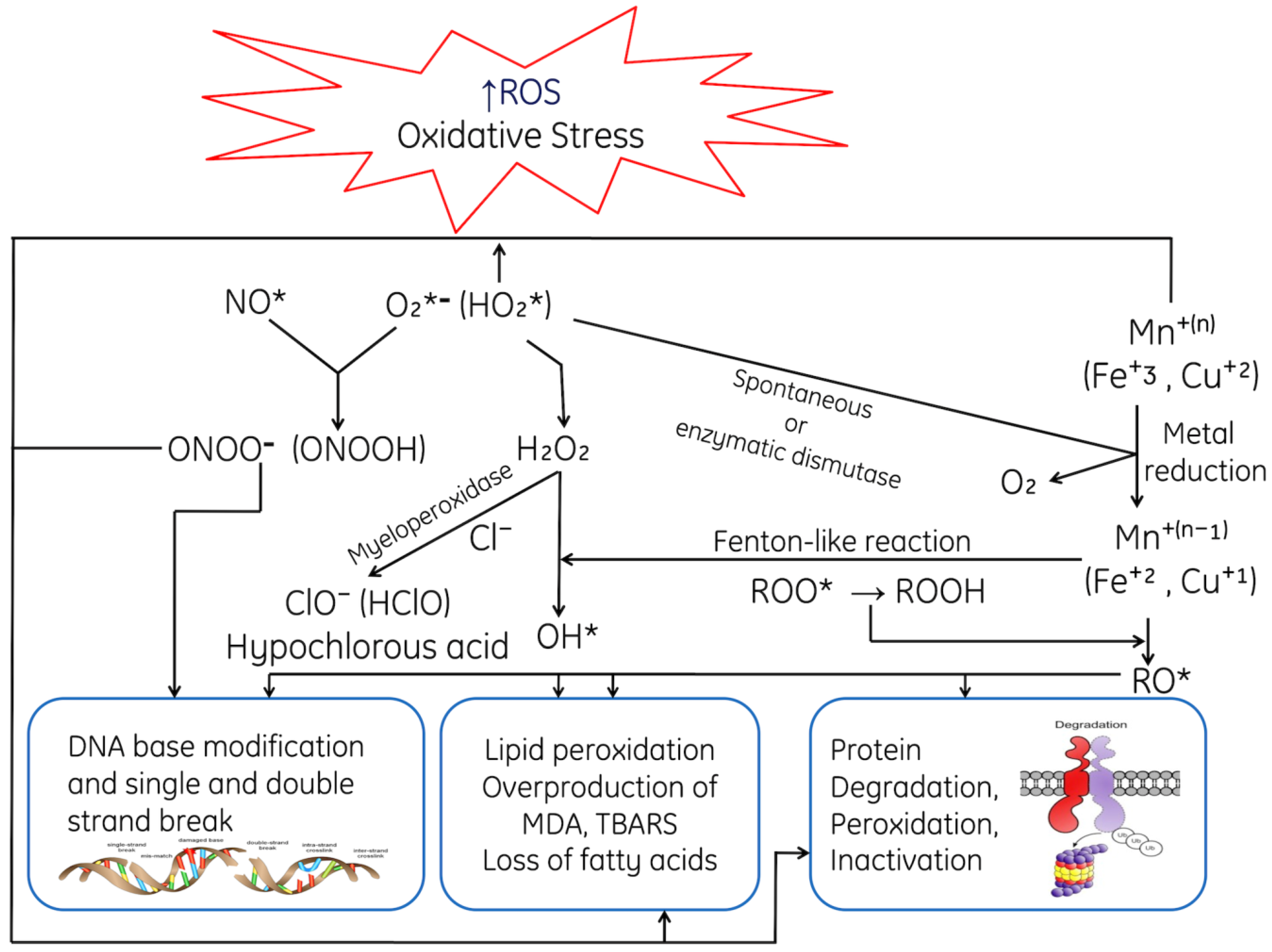
1.2. Mechanism of ROS-Mediated Toxicity
1.3. ROS as Second Messengers
1.4. ROS Induces Multifaceted Alterations
1.5. Physiological Impacts of ROS
2. Effect of ROS on Human Health
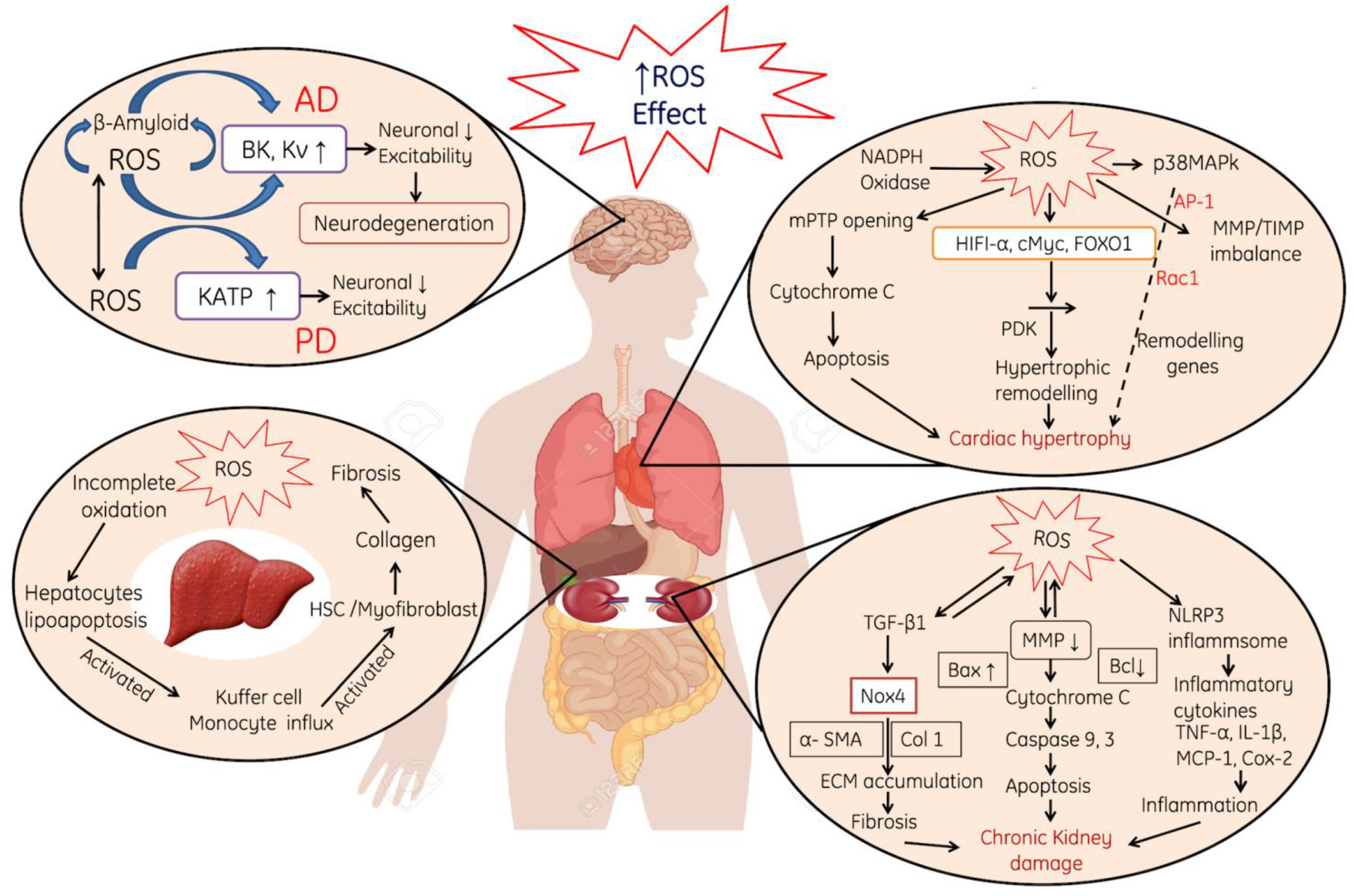
- ROS-induced neurotoxicity causes modulation in the neurons such as permeabilization of the cellular membrane, a decrease in the excitability property of neuronal membrane and the activation of the KATP pump (Figure 3).
- ROS-induced oxidative stress also leads to cardiac myopathy due to the mitochondrial damage, perforations in the mitochondria, the release of Cytochrome C and the initiation of apoptotic cascade (Figure 3).
- Liver and kidneys are the primary targets for ROS attack due to their direct involvement in metabolic and filtration processes. In the liver, ROS induce damages to the hepatocyte membranes and leads to deuteriation, which in turn leads to the deposition of collagen in the hepatocyte and finally cause liver fibrosis and cirrhosis. In addition, the incomplete oxidation of biomolecules causes lipoapoptosis in hepatic cells and induces immune reactions in the liver (Figure 3). In the kidneys, ROS-induced oxidative stress mainly initiates the production of various pro-inflammatory cytokines, which initiates nephrotic inflammation and finally affects the renal functions (Figure 3).
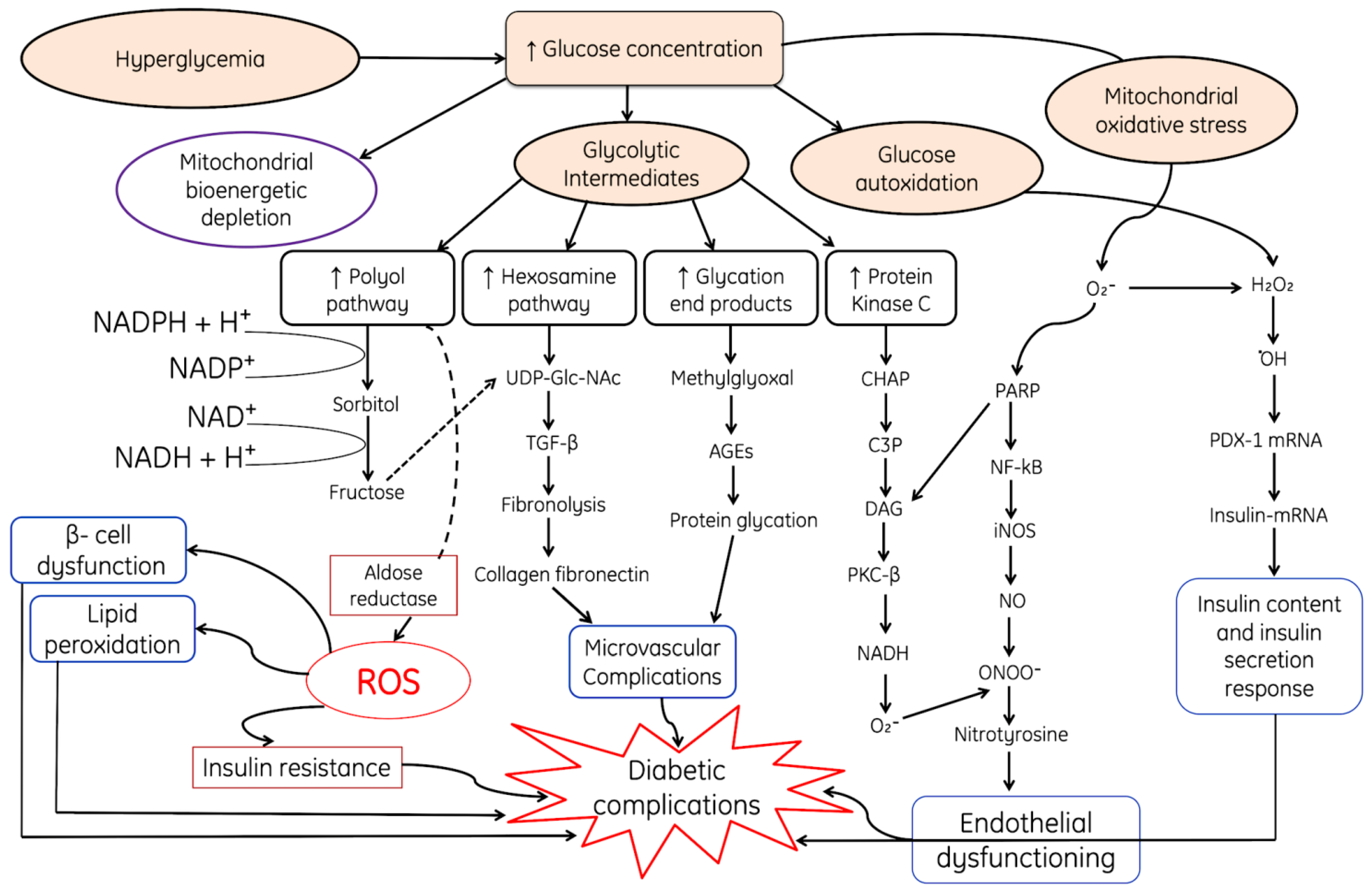
2.1. Role of ROS in Mitochondrial Dysfunction and in Diabetes Mellitus
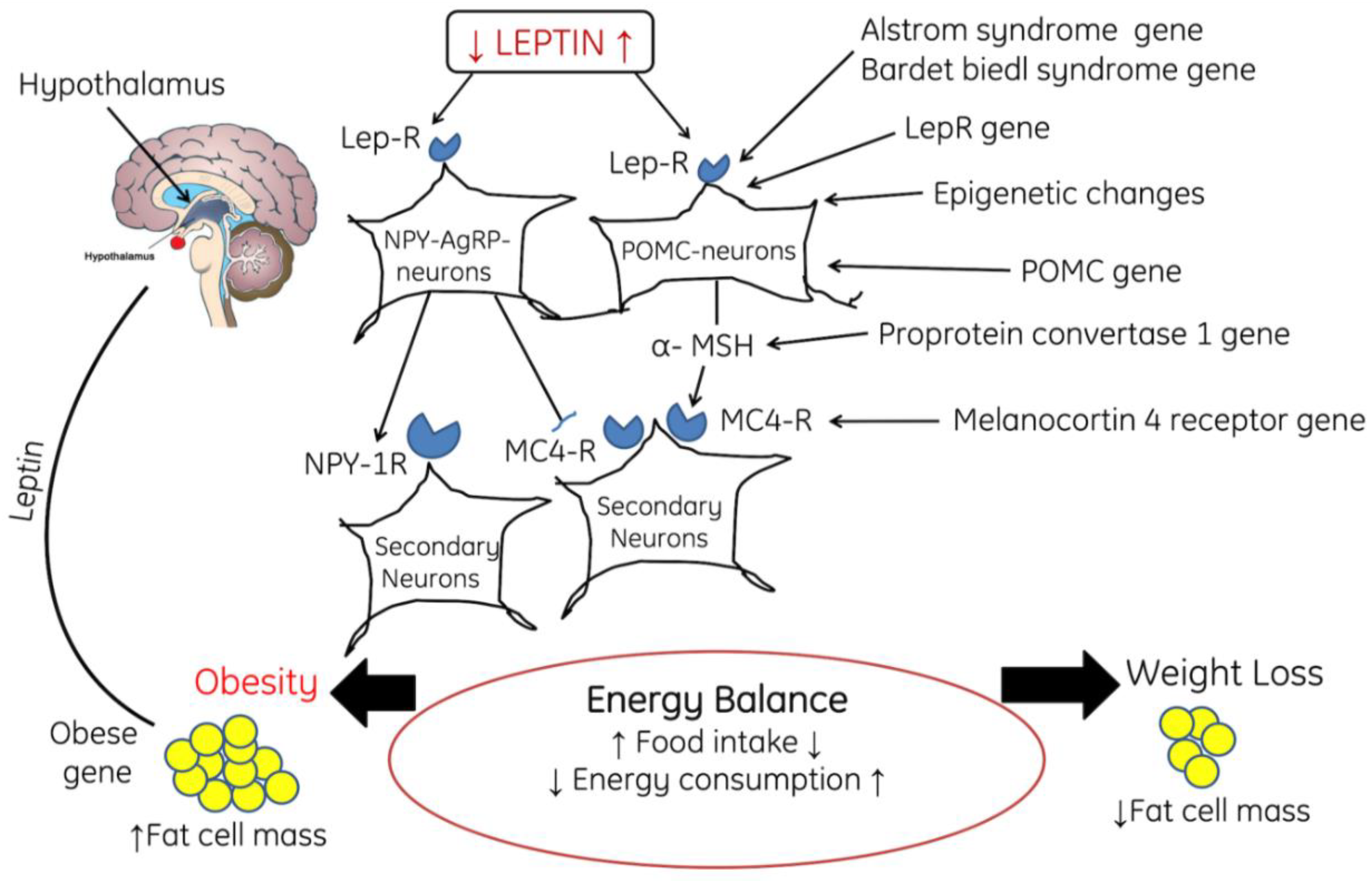
2.2. Role of ROS in Obesity and Associated Comorbidities
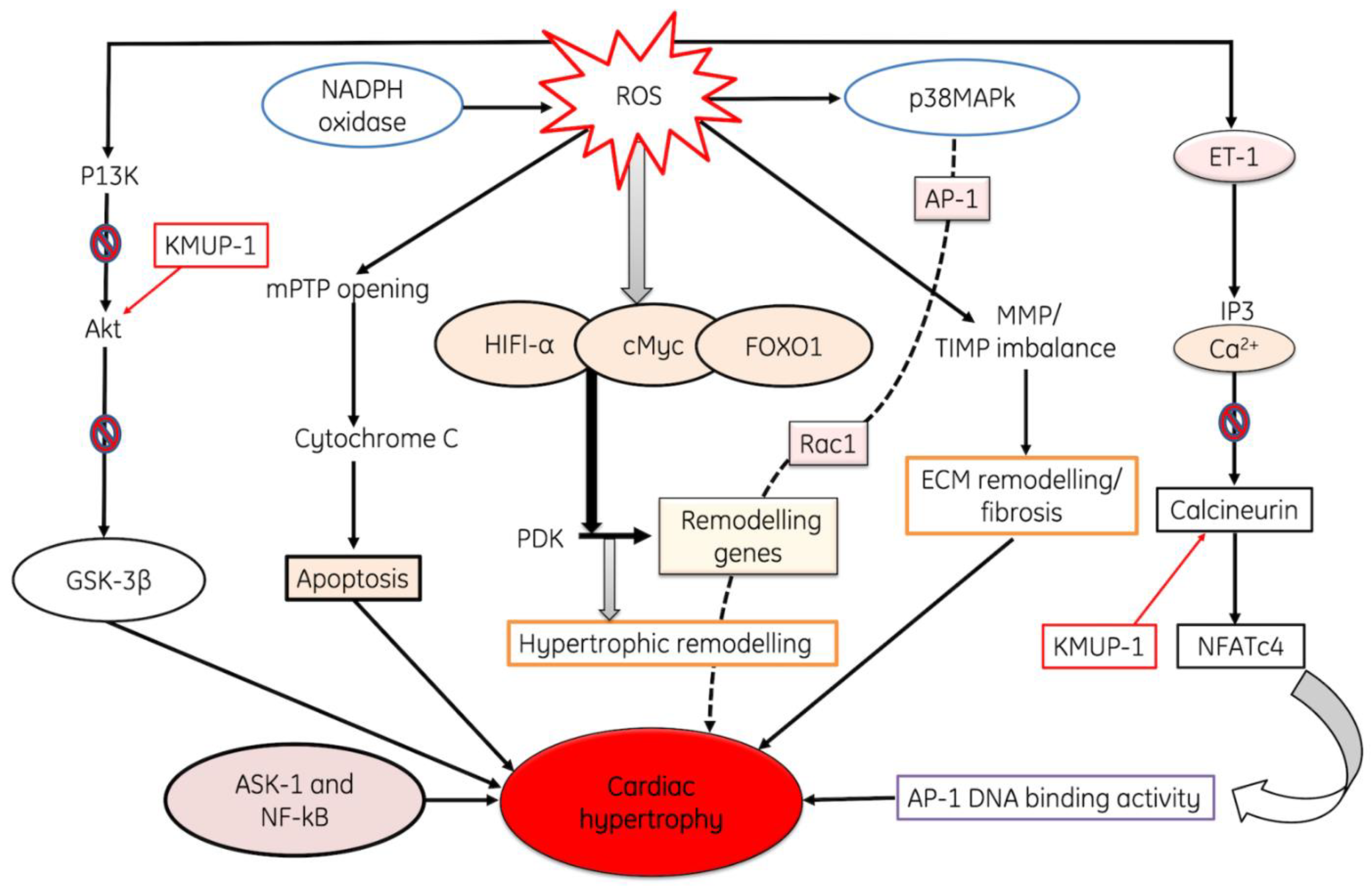
2.3. Role of ROS in Cardiac Hypertrophy
3. Oxidative Stress and Neurodegenerative Diseases
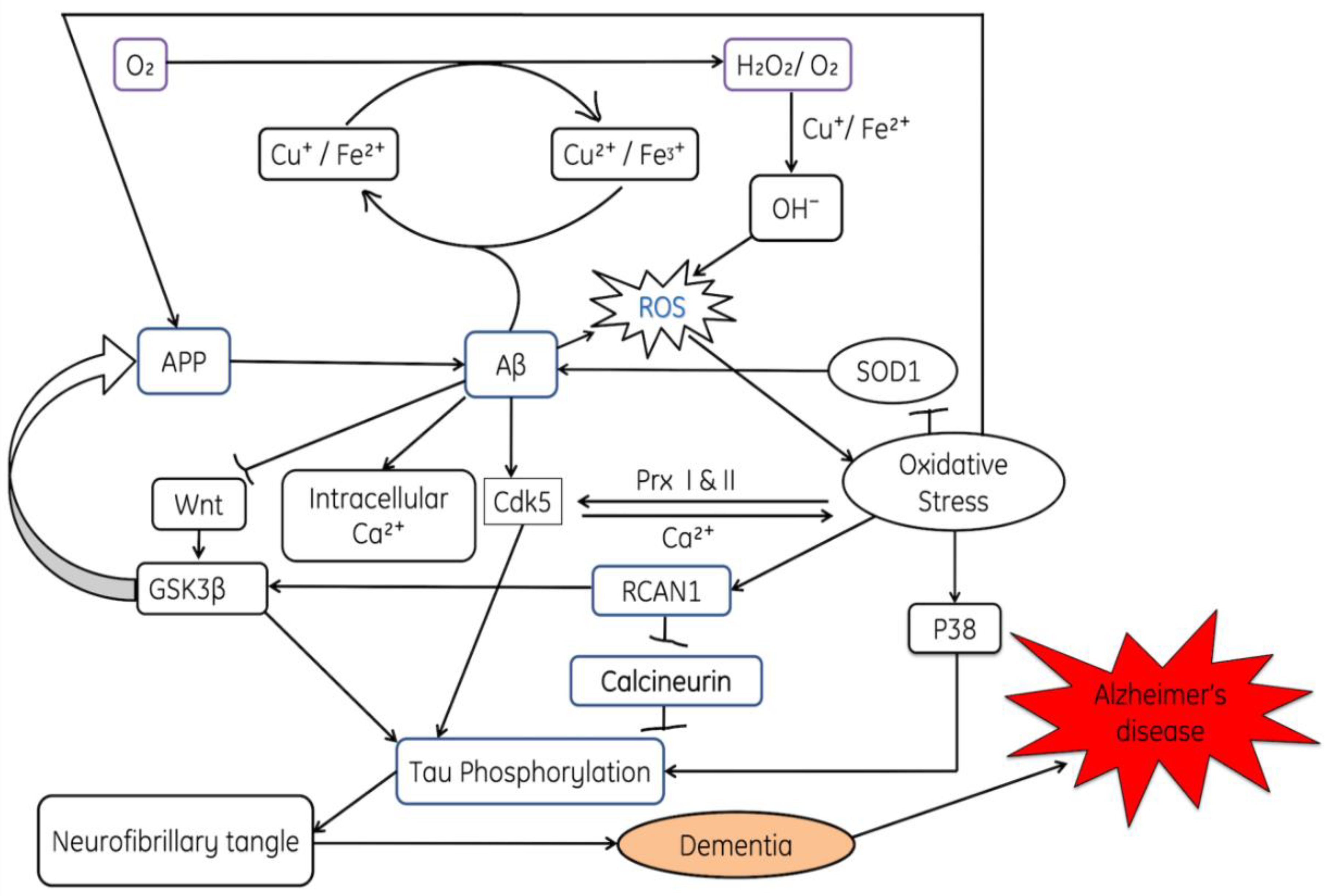
3.1. Alzheimer’s Disease
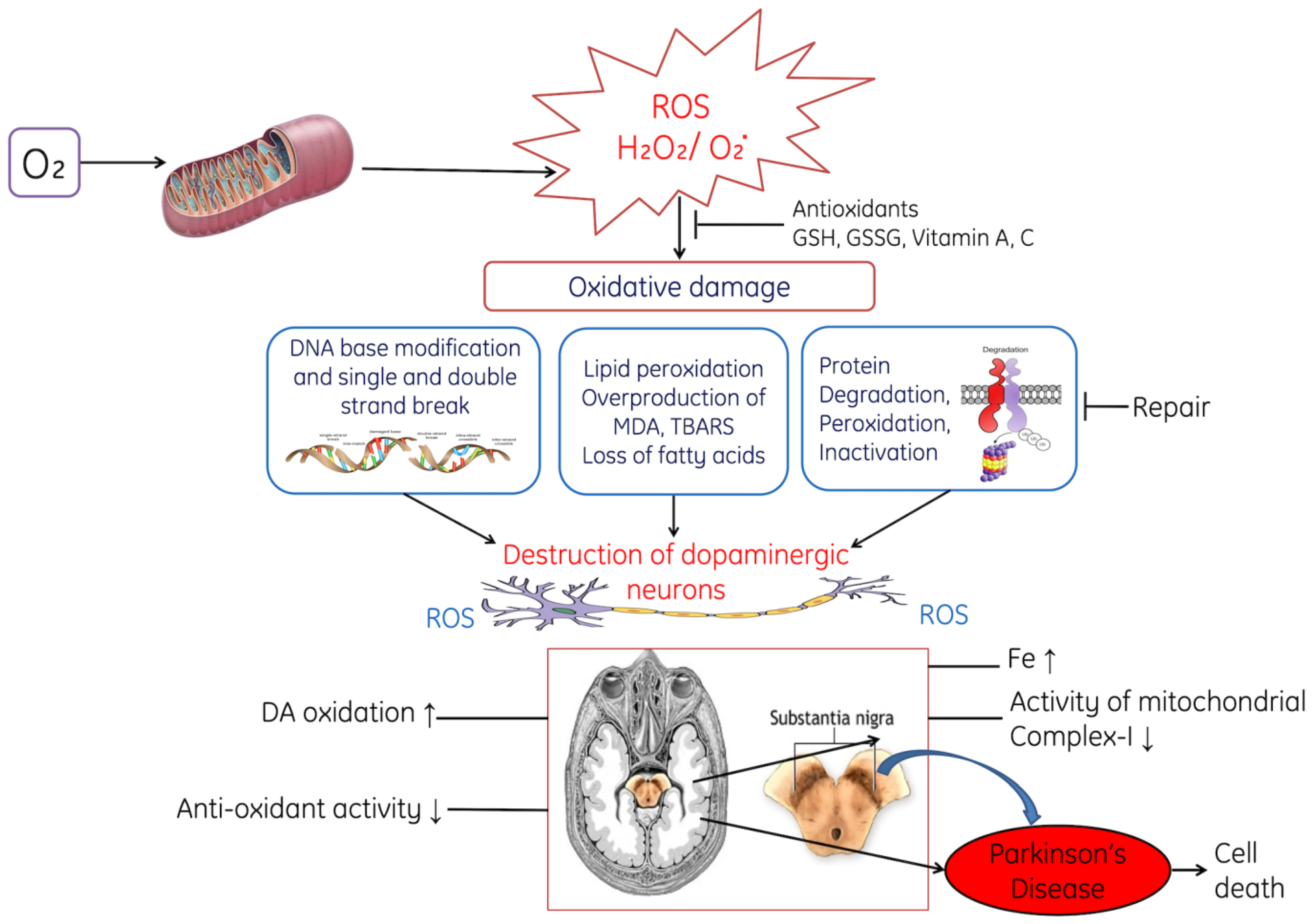
3.2. Parkinson’s Disease
4. Oxidative Stress and Chronic Kidney Diseases
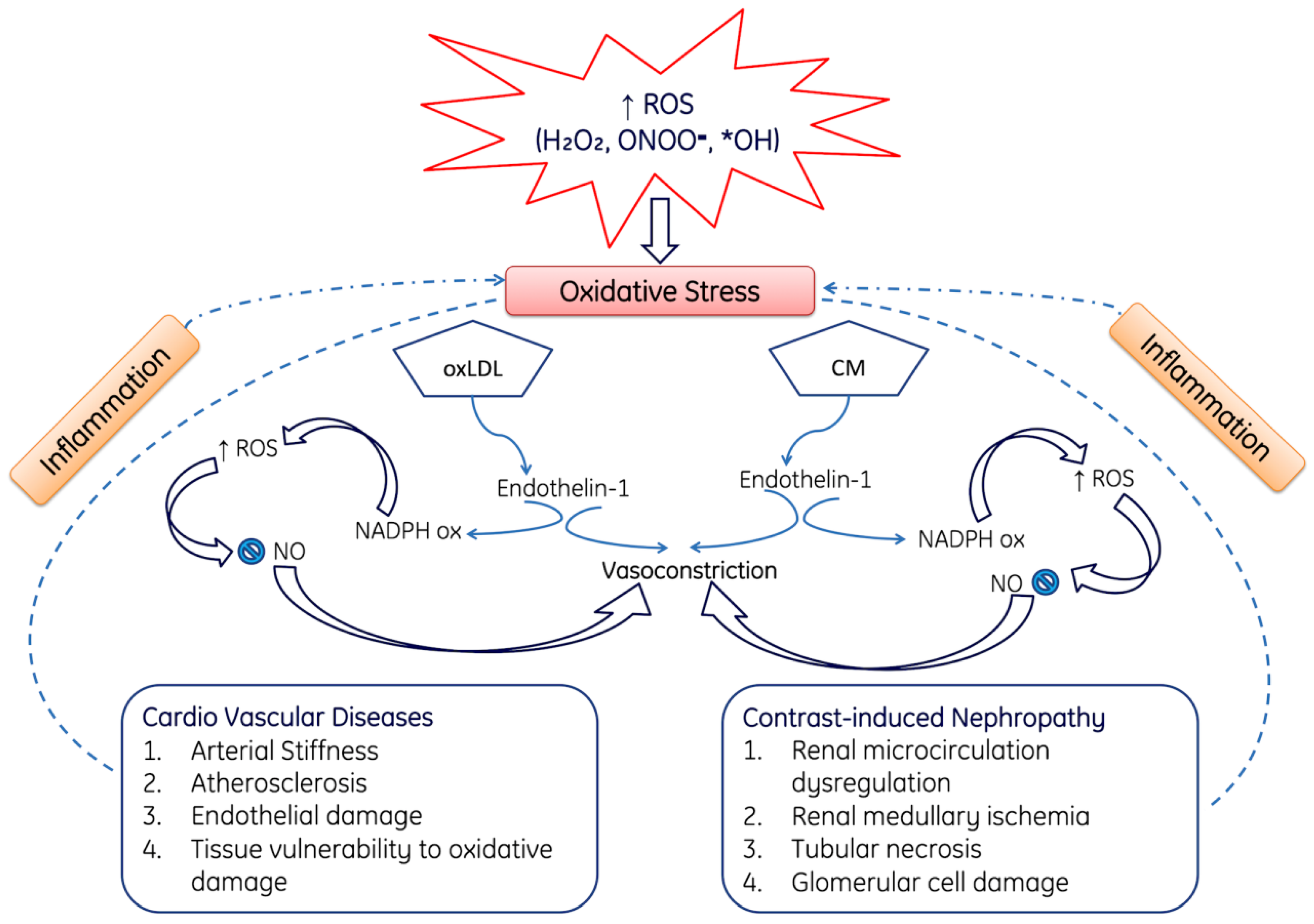
ROS-Mediated Cardio Vascular and Nephropathy
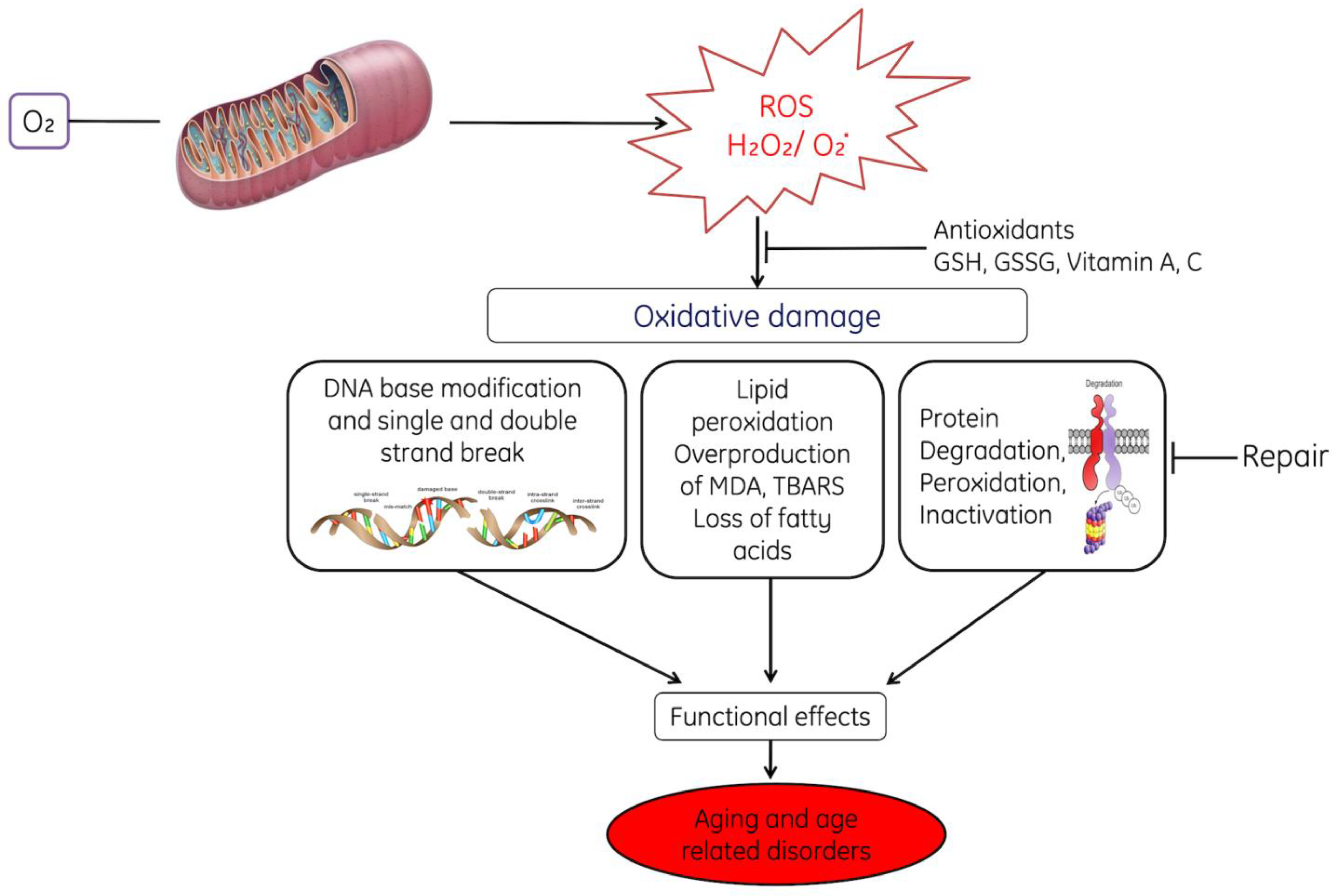
5. ROS in Aging and Age-Related Diseases
Mitochondria-Associated Membranes Involvement with Aging
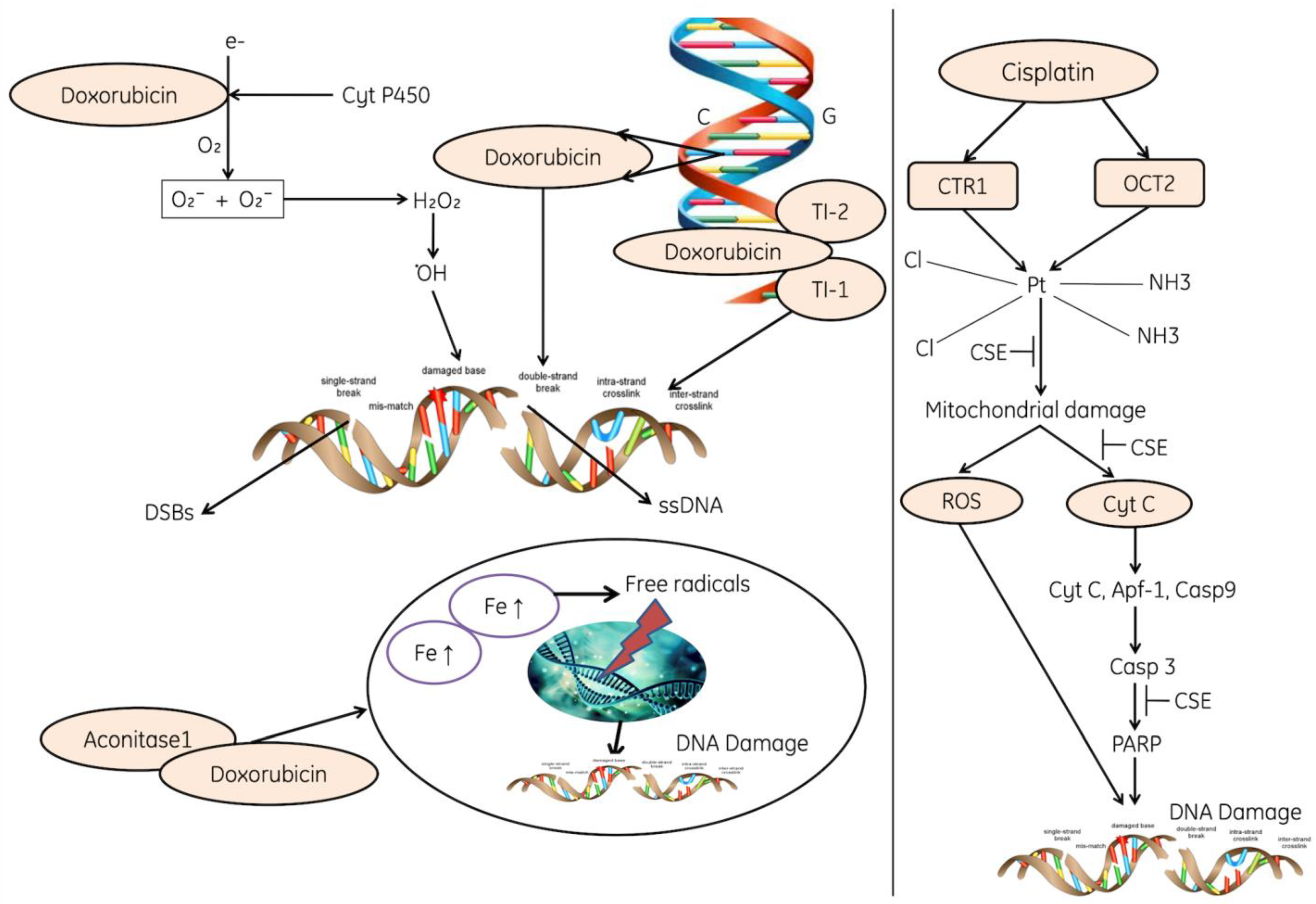
6. The Role of ROS in the Induction of DNA Damage
6.1. Role of ROS in Mediating Genotoxin-Induced Damage
6.2. Role of ROS-NO in DNA Damage by Oncogenic Replication Stress
7. Conclusions
8. Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Merry, T.L.; Ristow, M. Mitohormesis in exercise training. Free Radic. Biol. Med. 2016, 98, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, K.N.; Cruzat, V.F.; Carlessi, R.; de Bittencourt, P.I.H.; Newsholme, P. Molecular events linking oxidative stress and inflammation to insulin resistance and β-cell dysfunction. Oxid. Med. Cell. Longev. 2015, 2015, 181643. [Google Scholar] [CrossRef] [Green Version]
- Araki, E.; Nishikawa, T. Oxidative stress: A cause and therapeutic target of diabetic complications. J. Diabetes Investig. 2010, 1, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T.; Holbrook, N.J. Oxidative stress and aging: Catalase is a longevity determinant enzyme. Nature 2000, 408, 239. [Google Scholar] [CrossRef]
- Newsholme, P.; Cruzat, V.; Arfuso, F.; Keane, K. Nutrient regulation of insulin secretion and action. J. Endocrinol. 2014, 221, 105–120. [Google Scholar] [CrossRef]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and β-cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Haber, E.P.; Hirabara, S.M.; Rebelato, E.L.O.; Procópio, J.; Morgan, D.; Oliveira-Emilio, H.C.; Carpinelli, A.R.; Curi, R. Diabetes associated cell stress and dysfunction: Role of mitochondrial and non-mitochondrial ROS production and activity. J. Physiol. 2007, 583, 9–24. [Google Scholar] [CrossRef]
- Newsholme, P.; Gaudel, C.; Krause, M. Mitochondria and diabetes. An intriguing pathogenetic role. Adv. Mitochon. Med. 2012, 235–247. [Google Scholar]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; TsouhFokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, oxidative stress, and antioxidants: Back and forth in the pathophysiology of chronic diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vásquez-Vivar, J.; Kalyanaraman, B.; Kennedy, M.C. Mitochondrial aconitase is a source of hydroxyl radical: An electron spin resonance investigation. J. Biol. Chem. 2000, 275, 14064–14069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [Green Version]
- Yasui, H.; Hayashi, S.; Sakurai, H. Possible involvement of singlet oxygen species as multiple oxidants in p450 catalytic reactions. Drug Metab. Pharmacokinet. 2005, 20, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omura, T. Mitochondrial P450s. Chem. Biol. Interact. 2006, 163, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Zumárraga, M.; Andía, I.; Dávila, R.; Miller, J.C.; Friedhoff, A.J. Expression in normal and in subjects with schizophrenia of a novel gene fragment originally isolated from monozygotic twins discordant for schizophrenia. Genet. Mol. Biol. 2004, 27, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Tao, L.; Zhou, X.; Zuo, Z.; Gong, J.; Liu, X.; Zhou, Y.; Liu, C.; Sang, N.; Liu, H.; et al. DHODH and cancer: Promising prospects to be explored. Cancer Metab. 2021, 9, 1–25. [Google Scholar] [CrossRef]
- Hey-Mogensen, M.; Goncalves, R.L.; Orr, A.L.; Brand, M.D. Production of superoxide/H2O2 by dihydroorotate dehydrogenase in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 2014, 72, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Popugaeva, E.; Bezprozvanny, I. Role of endoplasmic reticulum Ca2+ signaling in the pathogenesis of Alzheimer disease. Front. Mol. Neurosci. 2013, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell. Cardiol. 2014, 73, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabilloud, T.; Heller, M.; Rigobello, M.P.; Bindoli, A.; Aebersold, R.; Lunardi, J. The mitochondrial antioxidant defence system and its response to oxidative stress. Proteomics 2001, 1, 1105–1110. [Google Scholar] [CrossRef]
- Hanukoglu, I. Antioxidant protective mechanisms against reactive oxygen species (ROS) generated by mitochondrial P450 systems in steroidogenic cells. Drug Metab. Rev. 2006, 38, 171–196. [Google Scholar] [CrossRef]
- Rohan Fernando, M.; Lechner, J.M.; Löfgren, S.; Gladyshev, V.N.; Lou, M.F.; Rohan Fernando, M.; Lechner, J.M.; Löfgren, S.; Gladyshev, V.N.; Lou, M.F. Mitochondrial thioltransferase (glutaredoxin 2) has GSH-dependent and thioredoxin reductase-dependent peroxidase activities in vitro and in lens epithelial cells. FASEB J. 2006, 20, 2645–2647. [Google Scholar] [CrossRef] [Green Version]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879. [Google Scholar] [CrossRef] [Green Version]
- Kirkinezos, I.G.; Moraes, C.T. Reactive oxygen species and mitochondrial diseases. Semin. Cell Dev. Biol. 2001, 12, 449–457. [Google Scholar] [CrossRef]
- Rimessi, A.; Previati, M.; Nigro, F.; Wieckowski, M.R.; Pinton, P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 2016, 81, 281–293. [Google Scholar] [CrossRef]
- Reshi, M.S.; Yadav, D.; Uthra, C.; Shrivastava, S.; Shukla, S. Acetaminophen-induced renal toxicity: Preventive effect of silver nanoparticles. Toxicol. Res. 2020, 9, 406–412. [Google Scholar] [CrossRef]
- Księżakowska-Łakoma, K.; Żyła, M.; Wilczyński, J.R. Mitochondrial dysfunction in cancer. Prz. = Menopause Rev. 2014, 13, 136. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R.; Levine, R.L. Protein oxidation. Ann. N. Y. Acad. Sci. 2000, 899, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Fruhwirth, G.O.; Hermetter, A. Mediation of apoptosis by oxidized phospholipids. Lipids Health Dis. 2008, 49, 351–367. [Google Scholar]
- Auten, R.L.; Whorton, M.H.; Nicholas Mason, S. Blocking neutrophil influx reduces DNA damage in hyperoxia-exposed newborn rat lung. Am. J. Respir. Cell Mol. Biol. 2002, 26, 391–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parinandi, N.L.; Kleinberg, M.A.; Usatyuk, P.V.; Cummings, R.J.; Pennathur, A.; Cardounel, A.J.; Zweier, J.L.; Garcia, J.G.; Natarajan, V. Hyperoxia-induced NAD (P) H oxidase activation and regulation by MAP kinases in human lung endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, 26–38. [Google Scholar] [CrossRef] [Green Version]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the BCL2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The role of hydrogen peroxide in redox-dependent signaling: Homeostatic and pathological responses in mammalian cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef] [Green Version]
- Linley, E.; Denyer, S.P.; McDonnell, G.; Simons, C.; Maillard, J.-Y. Use of hydrogen peroxide as a biocide: New consideration of its mechanisms of biocidal action. J. Antimicrob. Chemother. 2012, 67, 1589–1596. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Lorenzen, I.; Mullen, L.; Bekeschus, S.; Hanschmann, E.-M. Redox regulation of inflammatory processes is enzymatically controlled. Oxid. Med. Cell. Longev. 2017, 2017, 8459402. [Google Scholar] [CrossRef] [Green Version]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef]
- Bienert, G.P.; Møller, A.L.B.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide- production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Miller, A.-F. Superoxide dismutases: Ancient enzymes and new insights. FEBS Lett. 2012, 586, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Sumimoto, H.; Miyano, K.; Takeya, R. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem. Biophys. Res. Commun. 2005, 338, 677–686. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.W.; Dickinson, B.C.; Chang, C.J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 15681–15686. [Google Scholar] [CrossRef] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta. Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Willcox, J.K.; Ash, S.L.; Catignani, G.L. Antioxidants and prevention of chronic disease. Crit. Rev. Food Sci. Nutr. 2004, 44, 275–295. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Genestra, M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell. Signal. 2007, 19, 1807–1819. [Google Scholar] [CrossRef]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Young, I.S.; Woodside, J.V. Antioxidants in health and disease. J. Clin. Pathol. 2001, 54, 176–186. [Google Scholar] [CrossRef] [Green Version]
- Han, R.N.; Stewart, D.J. Defective lung vascular development in endothelial nitric oxide synthase-deficient mice. Trends Cardiovasc. Med. 2006, 16, 29–34. [Google Scholar] [CrossRef]
- Frei, B. Reactive oxygen species and antioxidant vitamins. Am. J. Med. 1994, 97, 5S–13S. [Google Scholar] [CrossRef]
- Nishida, N.; Arizumi, T.; Takita, M.; Kitai, S.; Yada, N.; Hagiwara, S.; Inoue, T.; Minami, Y.; Ueshima, K.; Sakurai, T.; et al. Reactive oxygen species induce epigenetic instability through the formation of 8-hydroxydeoxyguanosine in human hepatocarcinogenesis. Dig. Dis. 2013, 31, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Yasui, M.; Kanemaru, Y.; Kamoshita, N.; Suzuki, T.; Arakawa, T.; Honma, M. Tracing the fates of site-specifically introduced DNA adducts in the human genome. DNA Repair 2014, 15, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, H.M.; Wang, W.; Sen, S.; Shields, C.D.; Lee, S.S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Van Neste, L.; Easwaran, H.; et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.F.; Ward, P.A. Role of oxidants in lung injury during sepsis. Antioxid. Redox Signal. 2007, 9, 1991–2002. [Google Scholar] [CrossRef]
- Hoshino, Y.; Mishima, M. Redox-based therapeutics for lung diseases. Antioxid. Redox Signal. 2008, 10, 701–704. [Google Scholar] [CrossRef]
- Mahajan, A.; Tandon, V.R. Antioxidants and rheumatoid arthritis. J. Indian Rheumatol. Assoc. 2004, 12, 139–142. [Google Scholar]
- Allan Butterfield, D. Amyloid β-peptide (1-42)-induced oxidative stress and neurotoxicity: Implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic. Res. 2002, 36, 1307–1313. [Google Scholar] [CrossRef]
- Rai, S.; Hajam, Y.A.; Basheer, M.; Ghosh, H. Biochemical and histopathological inflections in hepato-renal tissues of streptozotocin (STZ) induced diabetic male rats: Impact of exogenous melatonin administration. J. Clin. Res. Bioeth. 2016, 7, 2. [Google Scholar] [CrossRef]
- Hajam, Y.A.; Rai, S.; Shree, S.; Basheer, M.; Ghosh, H. Retrieval of reproductive complications by exogenous melatonin treatment in streptozotocin induced diabetic rat model. Res. Rev. J. Zool. Sci. 2017, 5, 96–104. [Google Scholar]
- Reichmann, D.; Voth, W.; Jakob, U. Maintaining a healthy proteome during oxidative stress. Mol. Cell 2018, 69, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxid. Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Yatmaz, S.; Seow, H.J.; Gualano, R.C.; Wong, Z.X.; Stambas, J.; Selemidis, S.; Crack, P.J.; Bozinovski, S.; Anderson, G.P.; Vlahos, R. Glutathione peroxidase-1 reduces influenza A virus–induced lung inflammation. Am. J. Respir. Cell Mol. Biol. 2013, 48, 17–26. [Google Scholar] [CrossRef]
- Xu, J.; Li, T.; Wu, H.; Xu, T. Role of thioredoxin in lung disease. Pulm. Pharmacol. Ther. 2012, 25, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Groitl, B.; Jakob, U. Thiol-based redox switches. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 1335–1343. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Santamarina, S.; Boronat, S.; Hidalgo, E. Reversible cysteine oxidation in hydrogen peroxide sensing and signal transduction. Biochemistry 2014, 53, 2560–2580. [Google Scholar] [CrossRef] [Green Version]
- Go, Y.M.; Jones, D.P. Redox control systems in the nucleus: Mechanisms and functions. Antioxid. Redox Signal. 2010, 13, 489–509. [Google Scholar] [CrossRef] [Green Version]
- Roos, G.; Messens, J. Protein sulfenic acid formation: From cellular damage to redox regulation. Free Radic. Biol. Med. 2011, 51, 314–326. [Google Scholar] [CrossRef]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef] [PubMed]
- Östman, A.; Frijhoff, J.; Sandin, Å.; Böhmer, F.D. Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem. 2011, 150, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006, 27, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Bao, S.; Ramanadham, S.; Turk, J. Effects of biological oxidants on the catalytic activity and structure of group VIA phospholipase A2. Biochem. 2006, 45, 6392–6406. [Google Scholar] [CrossRef] [Green Version]
- Hajam, Y.A.; Rai, S.; Ghosh, H.; Basheer, M. Combined administration of exogenous melatonin and insulin ameliorates streptozotocin induced toxic alteration on hematological parameters in diabetic male Wistar rats. Toxicol. Rep. 2020, 7, 353–359. [Google Scholar] [CrossRef]
- Uthra, C.; Shrivastava, S.; Jaswal, A.; Sinha, N.; Reshi, M.S.; Shukla, S. Therapeutic potential of quercetin against acrylamide induced toxicity in rats. Biomed. Pharmacother. 2017, 86, 705–714. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118. [Google Scholar] [CrossRef] [Green Version]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. 2008, 4, 89. [Google Scholar]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K.; Loridas, S. Pulmonary oxidative stress, inflammation and cancer: Respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int. J. Environ. Res. Public Health 2013, 10, 3886–3907. [Google Scholar] [CrossRef]
- Maechler, P.; Wollheim, C.B. Mitochondrial function in normal and diabetic β-cells. Nature 2001, 414, 807–812. [Google Scholar] [CrossRef]
- Hastoy, B.; Godazgar, M.; Clark, A.; Nylander, V.; Spiliotis, I.; van de Bunt, M.; Chibalina, M.V.; Barrett, A.; Burrows, C.; Tarasov, A.I.; et al. Electrophysiological properties of human beta-cell lines EndoC-βH1 and-βH2 conform with human beta-cells. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rustenbeck, I.; Schulze, T.; Morsi, M.; Alshafei, M.; Panten, U. What is the metabolic amplification of insulin secretion and is it (still) relevant? Metabolites 2021, 11, 355. [Google Scholar] [CrossRef]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.P.; Köhler, M.; Graff, C.; Oldfors, A.; Magnuson, M.A.; Berggren, P.O.; Larsson, N.G. Impaired insulin secretion and β-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat. Genet. 2000, 26, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F. Mitochondrial disease in adults: What’s old and what’s new? EMBO Mol. Med. 2015, 7, 1503–1512. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys) function and insulin resistance: From pathophysiological molecular mechanisms to the impact of diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Alfadda, A.A.; Sallam, R.M. Reactive oxygen species in health and disease. J. Biomed. Biotechnol. 2012, 2012, 936486. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed]
- Acharya, J.D.; Ghaskadbi, S.S. Islets and their antioxidant defense. Islets 2010, 2, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.B.; De Leo, D.; Joseph, J.W.; McQuaid, T.S.; Ha, X.F.; Xu, F.; Tsushima, R.G.; Pennefather, P.S.; Salapatek, A.M.F.; Wheeler, M.B. Increased uncoupling protein-2 levels in β-cells are associated with impaired glucose-stimulated insulin secretion: Mechanism of action. Diabetes 2001, 50, 1302–1310. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.F.; Zou, H.D. PEDF in diabetic retinopathy: A protective effect of oxidative stress. J. Biomed. Biotechnol. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.X.; Stinnett, A. Critical role of the NADPH oxidase subunit p47 phox on vascular TLR expression and neointimal lesion formation in high-fat diet-induced obesity. Lab. Investig. 2008, 88, 1316–1328. [Google Scholar] [CrossRef] [Green Version]
- El-Haschimi, K.; Pierroz, D.D.; Hileman, S.M.; Bjorbaek, C.; Flier, J.S. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J. Clin. Investig. 2000, 105, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Horvath, T.L.; Andrews, Z.B.; Diano, S. Fuel utilization by hypothalamic neurons: Roles for ROS. Trends Endocrinol. Metab. 2009, 20, 78–87. [Google Scholar] [CrossRef]
- Shih, N.L.; Cheng, T.H.; Loh, S.H.; Cheng, P.Y.; Wang, D.L.; Chen, Y.S.; Liu, S.H.; Liew, C.C.; Chen, J.J. Reactive oxygen species modulate angiotensin II-induced β-myosin heavy chain gene expression via ras/raf/extracellular signal-regulated kinase pathway in neonatal rat cardiomyocytes. Biochem. Biophys. Res. Commun. 2001, 283, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Gall, N.P.; Grieve, D.J.; Chen, M.; Shah, A.M. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 2002, 40, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Satoh, M.; Ogita, H.; Takeshita, K.; Mukai, Y.; Kwiatkowski, D.J.; Liao, J.K. Requirement of Rac1 in the development of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 7432–7437. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, Y.; Otsu, K.; Nishida, K.; Hirotani, S.; Nakayama, H.; Yamaguchi, O.; Hikoso, S.; Kashiwase, K.; Takeda, T.; Watanabe, T.; et al. The small GTP-binding protein Rac1 induces cardiac myocyte hypertrophy through the activation of apoptosis signal-regulating kinase 1 and nuclear factor-κB. J. Biol. Chem. 2003, 278, 20770–20777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumiya, Y.; Kim, S.; Izumi, Y.; Yoshida, K.; Yoshiyama, M.; Matsuzawa, A.; Ichijo, H.; Iwao, H. Apoptosis signal-regulating kinase 1 plays a pivotal role in angiotensin II–induced cardiac hypertrophy and remodeling. Circ. Res. 2003, 93, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Sargent, M.A.; York, A.J.; Molkentin, J.D. ASK1 regulates cardiomyocyte death but not hypertrophy in transgenic mice. Circ. Res. 2009, 105, 1110–1117. [Google Scholar] [CrossRef] [Green Version]
- Shanmugam, P.; Valente, A.J.; Prabhu, S.D.; Venkatesan, B.; Yoshida, T.; Delafontaine, P.; Chandrasekar, B. Angiotensin-II type 1 receptor and NOX2 mediate TCF/LEF and CREB dependent WISP1 induction and cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 2011, 50, 928–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochem. 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimer’s Dis. 2014, 42, 125–152. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, Y.; Picciano, M.; La Francois, J.; Duff, K. Fibrillar β-amyloid evokes oxidative damage in a transgenic mouse model of Alzheimer’s disease. Neuroscience 2001, 104, 609–613. [Google Scholar] [CrossRef]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K.; et al. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid β protein oligomerization and memory loss in mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 44557–44568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Na, R.; Gu, M.; Richardson, A.; Ran, Q. Lipid peroxidation up-regulates BACE1 expression in vivo: A possible early event of amyloidogenesis in Alzheimer’s disease. J. Neurochem. 2008, 107, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- David, D.C.; Hauptmann, S.; Scherping, I.; Schuessel, K.; Keil, U.; Rizzu, P.; Ravid, R.; Dröse, S.; Brandt, U.; Müller, W.E.; et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 2005, 280, 23802–23814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halverson, R.A.; Lewis, J.; Frausto, S.; Hutton, M.; Muma, N.A. Tau protein is cross-linked by transglutaminase in P301L tau transgenic mice. J. Neurosci. 2005, 25, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Castellani, R.; Hirai, K.; Aliev, G.; Drew, K.L.; Nunomura, A.; Takeda, A.; Cash, A.D.; Obrenovich, M.E.; Perry, G.; Smith, M.A. Role of mitochondrial dysfunction in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 357–360. [Google Scholar] [CrossRef]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta. Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.E.; Paek, S.H. Mitochondrial dysfunction in Parkinson’s disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. The Parkinson disease mitochondrial hypothesis: Where are we at? Neuroscientist 2016, 22, 266–277. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Hauser, D.N.; Hastings, T.G. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic parkinsonism. Neurobiol. Dis. 2013, 51, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buneeva, O.; Fedchenko, V.; Kopylov, A.; Medvedev, A. Mitochondrial dysfunction in Parkinson’s disease: Focus on Mitochondrial DNA. Biomedicines 2020, 8, 591. [Google Scholar] [CrossRef]
- Simon, D.K.; Pankratz, N.; Kissell, D.K.; Pauciulo, M.W.; Halter, C.A.; Rudolph, A.; Pfeiffer, R.F.; Nichols, W.C.; Foroud, T. Maternal inheritance and mitochondrial DNA variants in familial Parkinson’s disease. BMC Med. Genet. 2010, 11, 1–9. [Google Scholar] [CrossRef]
- Ellmore, T.M.; Suescun, J.; Castriotta, R.J.; Schiess, M.C. A study of the relationship between uric acid and substantia nigra brain connectivity in patients with REM sleep behavior disorder and Parkinson’s disease. Front. Neurol. 2020, 11, 815. [Google Scholar] [CrossRef]
- Ma, L.; Azad, M.G.; Dharmasivam, M.; Richardson, V.; Quinn, R.J.; Feng, Y.; Pountney, D.L.; Tonissen, K.F.; Mellick, G.D.; Yanatori, I.; et al. Parkinson’s disease: Alterations in iron and redox biology as a key to unlock therapeutic strategies. Redox Biol. 2021, 41, 101896. [Google Scholar] [CrossRef]
- Oakley, A.E.; Collingwood, J.F.; Dobson, J.; Love, G.; Perrott, H.R.; Edwardson, J.A.; Elstner, M.; Morris, C.M. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology 2007, 68, 1820–1825. [Google Scholar] [CrossRef]
- Locatelli, F.; Canaud, B.; Eckardt, K.U.; Stenvinkel, P.; Wanner, C.; Zoccali, C. Oxidative stress in end-stage renal disease: An emerging threat to patient outcome. Nephrol. Dial. Transplant. 2003, 18, 1272–1280. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [Green Version]
- Annuk, M.; Zilmer, M.; Lind, L.; Linde, T.; Fellström, B. Oxidative stress and endothelial function in chronic renal failure. J. Am. Soc. Nephrol. 2001, 12, 2747–2752. [Google Scholar] [CrossRef]
- Dounousi, E.; Papavasiliou, E.; Makedou, A.; Ioannou, K.; Katopodis, K.P.; Tselepis, A.; Siamopoulos, K.C.; Tsakiris, D. Oxidative stress is progressively enhanced with advancing stages of CKD. Am. J. Kidney Dis. 2006, 48, 752–760. [Google Scholar] [CrossRef] [Green Version]
- Ferraro, B.; Galli, F.; Frei, B.; Kingdon, E.; Canestrari, F.; Rice-Evans, C.; Buoncristiani, U.; Davenport, A.; Moore, K.P. Peroxynitrite-induced oxidation of plasma lipids is enhanced in stable hemodialysis patients. Kidney Inter. 2003, 63, 2207–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liakopoulos, V.; Roumeliotis, S.; Gorny, X.; Dounousi, E.; Mertens, P.R. Oxidative stress in hemodialysis patients: A review of the literature. Oxid. Med. Cell. Longev. 2017, 2014, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Krata, N.; Zagożdżon, R.; Foroncewicz, B.; Mucha, K. Oxidative stress in kidney diseases: The cause or the consequence? Arch. Immunol. Ther. Exp. 2018, 66, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Wu, F.R.; Wang, J.N.; Gao, L.; Jiang, L.; Li, H.D.; Ma, Q.; Liu, X.Q.; Wei, B.; Zhou, L.; et al. Nox4 in renal diseases: An update. Free Radic. Biol. Med. 2018, 124, 466–472. [Google Scholar] [CrossRef]
- Holterman, C.E.; Thibodeau, J.F.; Kennedy, C.R. NADPH oxidase 5 and renal disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 81–87. [Google Scholar] [CrossRef]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [Green Version]
- Monari, A.; Dumont, E. Understanding DNA under oxidative stress and sensitization: The role of molecular modeling. Front. Chem. 2015, 3, 43. [Google Scholar]
- Descamps-Latscha, B.; Drüeke, T.; Witko-Sarsat, V. Dialysis-induced oxidative stress: Biological aspects, clinical consequences, and therapy. Semin. Dial. 2001, 14, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Hajam, Y.A.; Rai, S.; Pandi-Perumal, S.R.; Brown, G.M.; Reiter, R.J.; Cardinali, D.P. Coadministration of melatonin and insulin improves diabetic-induced impairment of rat kidney function. Neuroendocrinology 2021. [Google Scholar] [CrossRef] [PubMed]
- Zill, H.; Günther, R.; Erbersdobler, H.F.; Fölsch, U.R.; Faist, V. RAGE expression and AGE-induced MAP kinase activation in Caco-2 cells. Biochem. Biophys. Res. Commun. 2001, 288, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, E.; Wautier, M.P.; Wautier, J.L.; Boval, B.; Panis, Y.; Wernert, N.; Danze, P.M.; Dequiedt, P. AGEs bind to mesothelial cells via RAGE and stimulate VCAM-1 expression. Kidney Inter. 2002, 61, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Inter. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Reshi, M.S.; Shrivastava, S.; Jaswal, A.; Sinha, N.; Uthra, C.; Shukla, S. Gold nanoparticles ameliorate acetaminophen induced hepato-renal injury in rats. Exp. Toxicol. Pathol. 2017, 69, 231–240. [Google Scholar] [CrossRef]
- Modaresi, A.; Nafar, M.; Sahraei, Z. Oxidative stress in chronic kidney disease. Iran. J. Kidney. Dis. 2015, 9, 165. [Google Scholar]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [Green Version]
- Adesso, S.; Popolo, A.; Bianco, G.; Sorrentino, R.; Pinto, A.; Autore, G.; Marzocco, S. The uremic toxin indoxyl sulphate enhances macrophage response to LPS. PLoS ONE 2013, 8, 76778. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Otln, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Benz, C.C.; Yau, C. Ageing, oxidative stress and cancer: Paradigms in parallax. Nat. Rev. Cancer 2008, 8, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Bonomini, F.; Rodella, L.F.; Rezzani, R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015, 6, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, R.; Ghaffari, S. Stem cells, redox signaling, and stem cell aging. Antioxid. Redox Signal. 2014, 20, 1902–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.E.; Sheaff, M.T. The pathology of ageing: Concepts and mechanisms. J. Pathol. 2007, 211, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Biala, A.K.; Dhingra, R.; Kirshenbaum, L.A. Mitochondrial dynamics: Orchestrating the journey to advanced age. J. Mol. Cell. Cardiol. 2015, 83, 37–43. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Indo, H.P.; Yen, H.C.; Nakanishi, I.; Matsumoto, K.I.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Genova, M.L.; Lenaz, G. The interplay between respiratory supercomplexes and ROS in aging. Antioxid. Redox Signal. 2015, 23, 208–238. [Google Scholar] [CrossRef]
- Barja, G. The mitochondrial free radical theory of aging. Prog. Mol. Biol. Transl. Sci. 2014, 127, 1–27. [Google Scholar]
- López-Lluch, G.; Santos-Ocaña, C.; Sánchez-Alcázar, J.A.; Fernández-Ayala, D.J.M.; Asencio-Salcedo, C.; Rodríguez-Aguilera, J.C.; Navas, P. Mitochondrial responsibility in ageing process: Innocent, suspect or guilty. Biogerontology 2015, 16, 599–620. [Google Scholar] [CrossRef]
- Bouzid, M.A.; Filaire, E.; McCall, A.; Fabre, C. Radical oxygen species, exercise and aging: An update. Sports Med. 2015, 45, 1245–1261. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; Lee, K.Y.; Lee, H.W.; Kim, J.H.; Kim, T.Y. SOD3 variant, R213G, altered SOD3 function, leading to ROS-mediated inflammation and damage in multiple organs of premature aging mice. Antioxid. Redox Signal. 2015, 23, 985–999. [Google Scholar] [CrossRef]
- Edrey, Y.H.; Salmon, A.B. Revisiting an age-old question regarding oxidative stress. Free Radic. Biol. Med. 2014, 71, 368–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjelakovic, G.; Nikolova, D.; Gluud, C. Antioxidant supplements and mortality. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 40–44. [Google Scholar] [CrossRef]
- Cunningham, G.M.; Roman, M.G.; Flores, L.C.; Hubbard, G.B.; Salmon, A.B.; Zhang, Y.; Gelfond, J.; Ikeno, Y. The paradoxical role of thioredoxin on oxidative stress and aging. Arch. Biochem. Biophys. 2015, 576, 32–38. [Google Scholar] [CrossRef]
- Breitenbach, M.; Rinnerthaler, M.; Hartl, J.; Stincone, A.; Vowinckel, J.; Breitenbach-Koller, H.; Ralser, M. Mitochondria in ageing: There is metabolism beyond the ROS. FEMS Yeast Res. 2014, 14, 198–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagouge, M.; Larsson, N.G. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J. Intern. Med. 2013, 273, 529–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertram, C.; Hass, R. Cellular responses to reactive oxygen species-induced DNA damage and aging. Biol. Chem. 2008, 389, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C. Role of oxidative RNA damage in chronic-degenerative diseases. Oxid. Med. Cell. Longev. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Goldman, R.D. Nuclear lamins and oxidative stress in cell proliferation and longevity. Can. Biol. Nucl. Envel. 2014, 1, 415–430. [Google Scholar]
- Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Trost, A.; Richter, K. Oxidative stress in aging human skin. Biomolecules 2015, 5, 545–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.J. Positive oxidative stress in aging and aging-related disease tolerance. Redox Biol. 2014, 2, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Guillaumet-Adkins, A.; Yañez, Y.; Peris-Diaz, M.D.; Calabria, I.; Palanca-Ballester, C.; Sandoval, J. Epigenetics and oxidative stress in aging. Oxid. Med. Cell. Longev. 2017, 2017, 9175806. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Schon, E.A. Mitochondria-associated ER membranes and Alzheimer disease. Curr. Opin. Genet. Dev. 2016, 38, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; Schon, E.A. On the pathogenesis of Alzheimer’s Disease: The MAM hypothesis. FASEB J. 2017, 31, 864–867. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef]
- Stoica, R.; De Vos, K.J.; Paillusion, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPBPTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [Green Version]
- Stoica, R.; Paillusion, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; De Vos, K.J.; Shaw, C.E.; et al. ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef]
- Tubbs, E.; Rieusset, J. Metabolic signaling functions of ER-mitochondria contact sites: Role in metabolic diseases. Soc. Endocrinol. 2016, 1, 1–55. [Google Scholar] [CrossRef] [Green Version]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.-A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arruda, A.P.; Pers, B.M.; Parlakgül, G.; Güney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulummitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Annunziata, I.; Patterson, A.; Moshiach, S.; Gomero, E.; Opferman, J.; Forte, M.; d’Azzo, A. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol. Cell 2009, 36, 500–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, C.D.; Colberg-Poley, A.M. Access of viral proteins to mitochondria via mitochondria-associated membranes. Rev. Med. Virol. 2009, 19, 147–164. [Google Scholar] [CrossRef] [Green Version]
- English, A.M.; Schuler, M.H.; Xiao, T.; Kornmann, B.; Shaw, J.M.; Hughes, A.L. ERmitochondria contacts promote mitochondrial-derived compartment biogenesis. J. Cell Biol. 2020, 219, 202002144. [Google Scholar] [CrossRef]
- Goodrum, J.M.; Lever, A.R.; Coody, T.K.; Gottschling, D.E.; Hughes, A.L. Rsp5 and Mdm30 reshape the mitochondrial network in response to age-induced vacuole stress. Mol. Biol. Cell. 2019, 30, 2141–2154. [Google Scholar] [CrossRef]
- Schuler, M.H.; English, A.M.; Campbell, T.J.; Shaw, J.M.; Hughes, A.L. Mitochondrialderived compartments facilitate cellular adaptation to amino acid stress. Mol. Cell 2020, 81, 3786–3802. [Google Scholar] [CrossRef]
- Chen, Q.; Samidurai, A.; Thompson, J.; Hu, Y.; Das, A.; Willard, B.; Lesnefsky, E.J. Endoplasmic reticulum stress-mediated mitochondrial dysfunction in aged hearts. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165899. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Lavandero, S. Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef] [Green Version]
- Honrath, B.; Metz, I.; Bendridi, N.; Rieusset, J.; Culmsee, C.; Dolga, A.M. Glucoseregulated protein 75 determines ER-mitochondrial coupling and sensitivity to oxidative stress in neuronal cells. Cell Death Discov. 2017, 3, 17076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Hajnoczky, G. Ca2+-dependent regulation of mitochondrial dynamics by the Miro-Milton complex. Int. J. Biochem. Cell Biol. 2009, 41, 1972–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knupp, J.; Arvan, P.; Chang, A. Increased mitochondrial respiration promotes survival from endoplasmic reticulum stress. Cell Death Differ. 2019, 26, 487–501. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhou, R.; Zhang, C.; He, S.; Su, Z. Mitochondria-associated endoplasmic reticulum membranes in the pathogenesis of type 2 diabetes mellitus. Front. Cell Dev. Biol. 2020, 8, 935. [Google Scholar] [CrossRef] [PubMed]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, 81162. [Google Scholar] [CrossRef] [PubMed]
- Salehi, F.; Behboudi, H.; Kavoosi, G.; Ardestani, S.K. Oxidative DNA damage induced by ROS-modulating agents with the ability to target DNA: A comparison of the biological characteristics of citrus pectin and apple pectin. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cannan, W.J.; Tsang, B.P.; Wallace, S.S.; Pederson, D.S. Nucleosomes suppress the formation of double-strand DNA breaks during attempted base excision repair of clustered oxidative damages. J. Biochem. 2014, 289, 19881–19893. [Google Scholar] [CrossRef] [Green Version]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer. 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell. 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNally, J.P.; Millen, S.H.; Chaturvedi, V.; Lakes, N.; Terrell, C.E.; Elfers, E.E.; Carroll, K.R.; Hogan, S.P.; Andreassen, P.R.; Kanter, J. Manipulating DNA damage-response signaling for the treatment of immune-mediated diseases. Proc. Natl. Acad. Sci. USA 2017, 114, 4782–4791. [Google Scholar] [CrossRef] [Green Version]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 2013, 5, 012716. [Google Scholar] [CrossRef]
- Altieri, F.; Grillo, C.; Maceroni, M.; Chichiarelli, S. DNA damage and repair: From molecular mechanisms to health implications. Antioxid Redox Signal. 2008, 10, 891–938. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair, the long and short of it. Cell Mol Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajam, Y.A.; Rani, R.; Ganie, S.Y.; Sheikh, T.A.; Javaid, D.; Qadri, S.S.; Pramodh, S.; Alsulimani, A.; Alkhanani, M.F.; Harakeh, S.; et al. Oxidative Stress in Human Pathology and Aging: Molecular Mechanisms and Perspectives. Cells 2022, 11, 552. https://doi.org/10.3390/cells11030552
Hajam YA, Rani R, Ganie SY, Sheikh TA, Javaid D, Qadri SS, Pramodh S, Alsulimani A, Alkhanani MF, Harakeh S, et al. Oxidative Stress in Human Pathology and Aging: Molecular Mechanisms and Perspectives. Cells. 2022; 11(3):552. https://doi.org/10.3390/cells11030552
Chicago/Turabian StyleHajam, Younis Ahmad, Raksha Rani, Shahid Yousuf Ganie, Tariq Ahmad Sheikh, Darakhshan Javaid, Syed Sanober Qadri, Sreepoorna Pramodh, Ahmad Alsulimani, Mustfa F. Alkhanani, Steve Harakeh, and et al. 2022. "Oxidative Stress in Human Pathology and Aging: Molecular Mechanisms and Perspectives" Cells 11, no. 3: 552. https://doi.org/10.3390/cells11030552