
Valproic Acid-Induced CCN1 Promotes Osteogenic Differentiation by Increasing CCN1 Protein Stability through HDAC1 Inhibition in Tonsil-Derived Mesenchymal Stem Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. TMSC Isolation and Culture
2.2. MTT Cell-Proliferation Assay
2.3. Bromodeoxyuridine (BrdU) Incorporation
2.4. Apoptosis Analysis
2.5. Flow Cytometry Analysis
2.6. Adipogenic, Chondrogenic and Osteogenic Differentiation
2.7. Reverse-Transcription Polymerase Chain Reaction (RT-PCR) Assay
2.8. Western Blot Analysis
2.9. CCN1 Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. Transfection
2.11. Statistical Analysis
3. Results
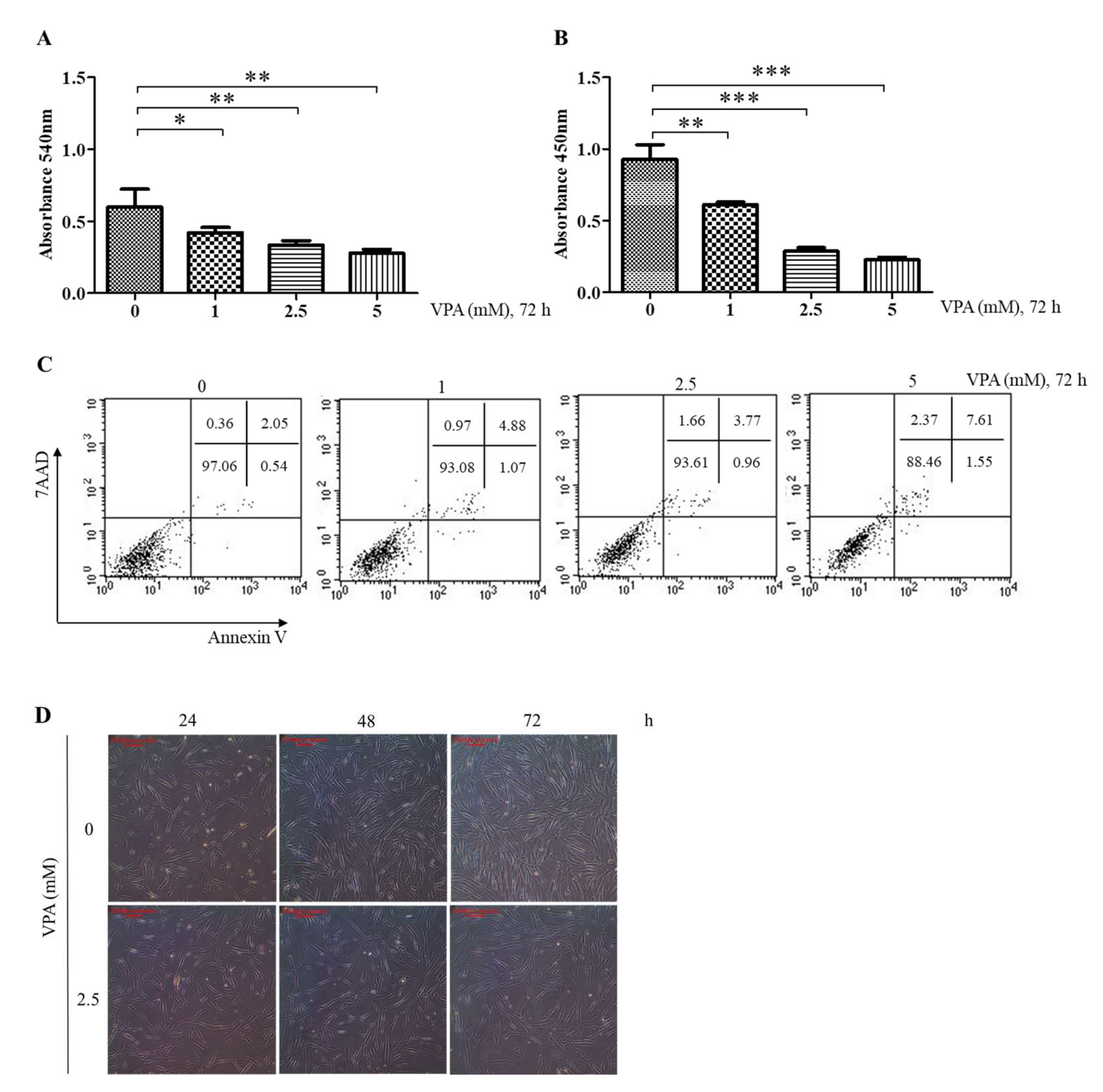
3.1. VPA Decreases the Proliferation of TMSCs by Inhibiting DNA Synthesis
3.2. Immunophenotypic Profiling of the TMSCs Exposed to VPA
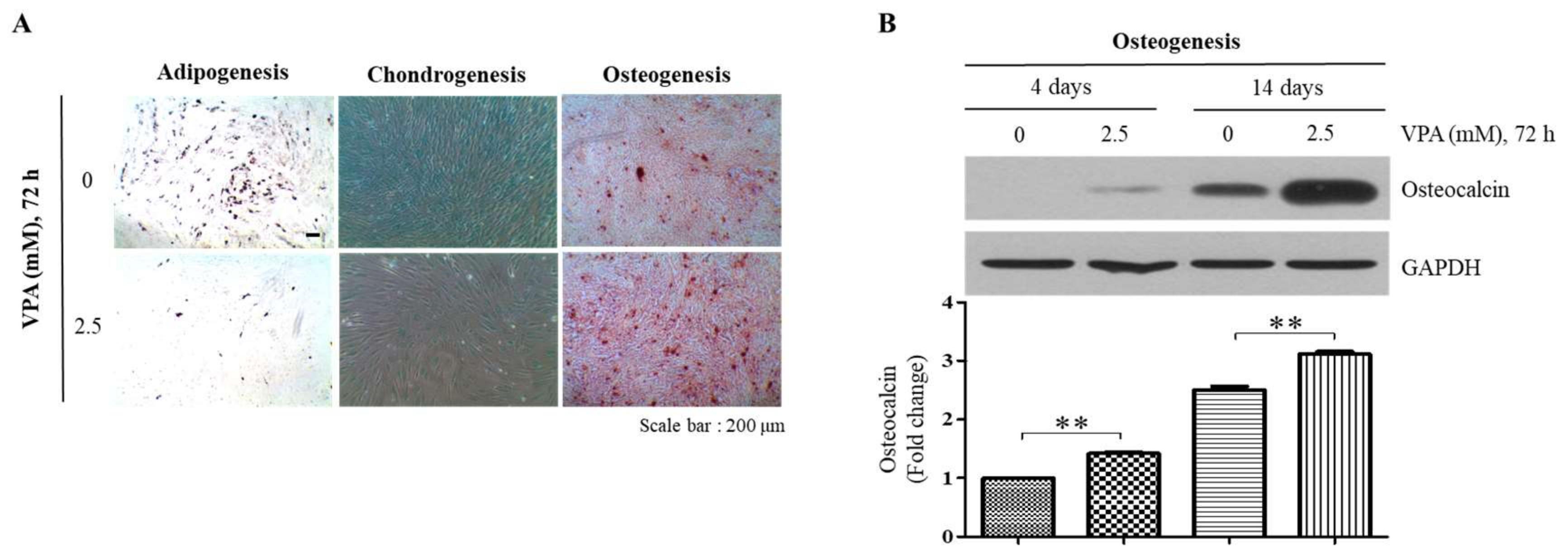
3.3. VPA Increases Osteogenic Differentiation but Decreases Adipogenic and Chondrogenic Differentiation of the TMSCs
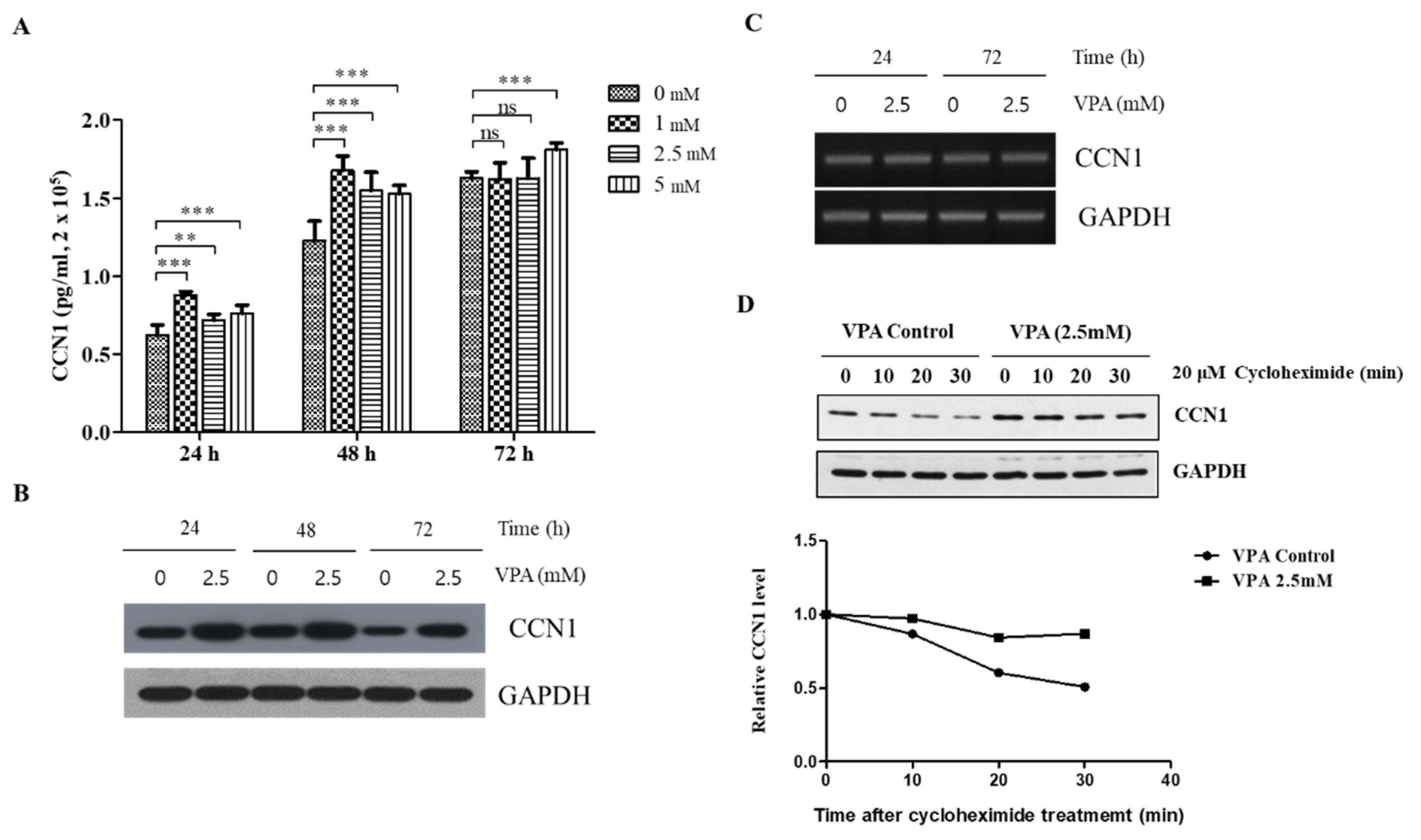
3.4. VPA Induces the Protein Level of CCN1 in a Dose-Dependent Manner but Not the Protein Level of TAZ, OPN and RUNX2
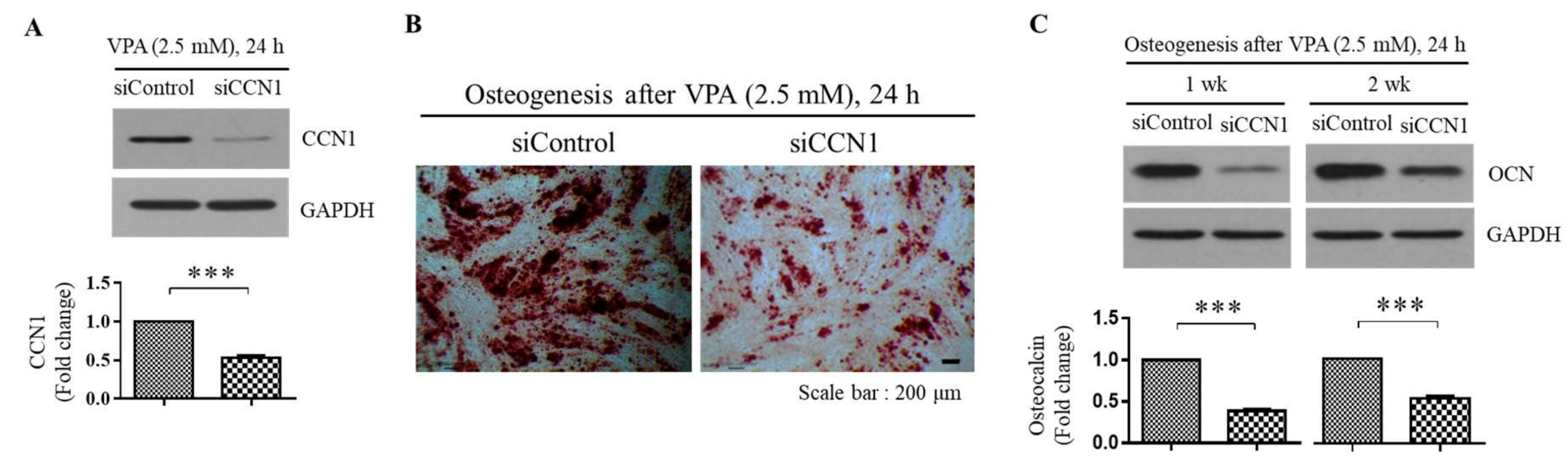
3.5. VPA Induces the Osteogenesis of the TMSCs by Inducing the CCN1 Level
3.6. VPA Increases the Protein Level of CCN1 by Inducing the Stability of the CCN1 Protein
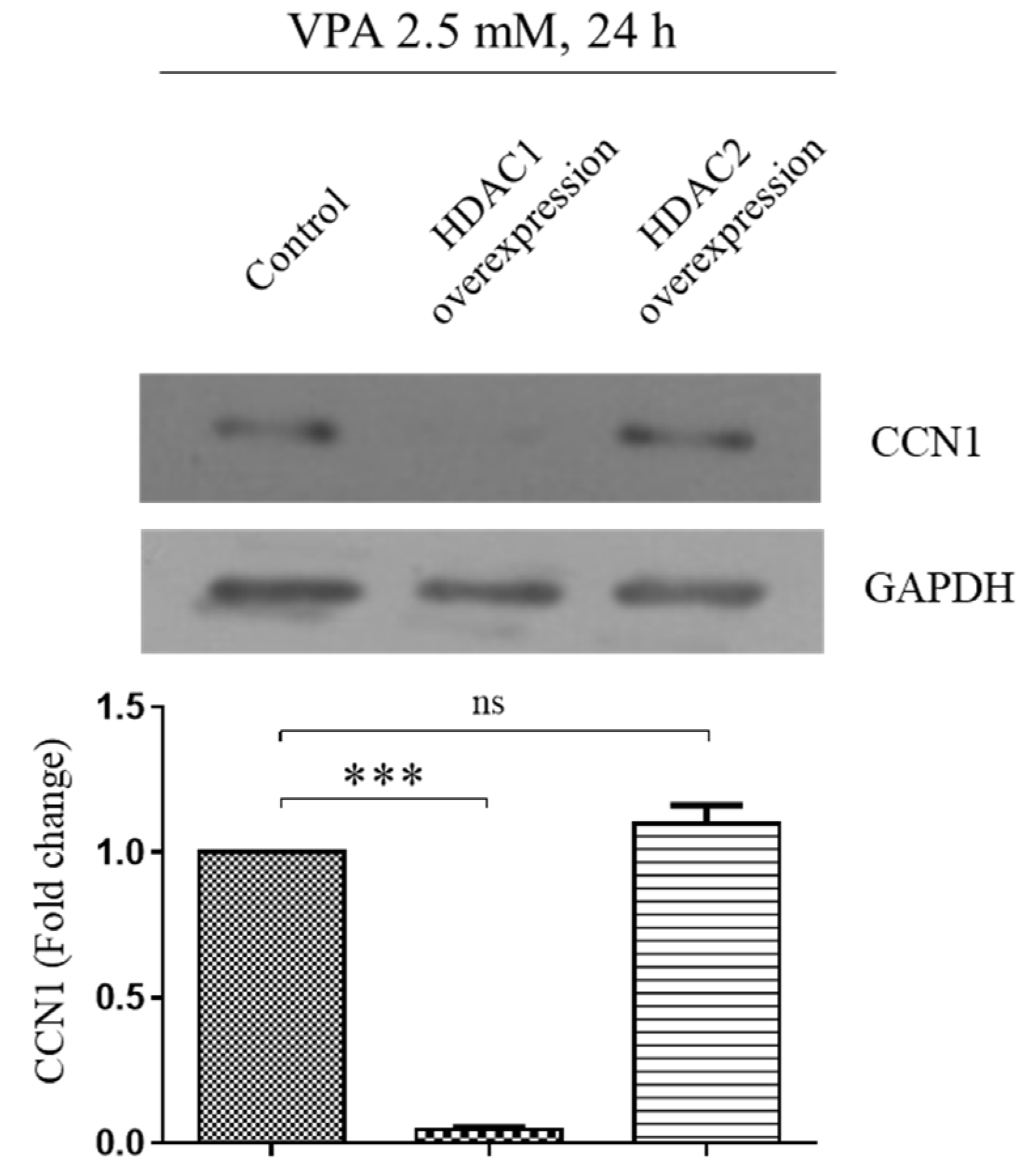
3.7. VPA Induces the CCN1 by Inhibiting Histone Deacetylase 1 (HDAC1) but Not HDAC2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oh, S.; Choi, Y.M.; Kim, H.Y.; Park, Y.S.; Jung, S.; Park, J.; Woo, S.; Ryu, K.; Kim, H.S.; Jo, I. Application of Tonsil-Derived Mesenchymal Stem Cells in Tissue Regeneration: Concise Review. Stem Cells 2019, 37, 1252–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.S.; Kim, H.S.; Jin, Y.M.; Yu, Y.; Kim, H.Y.; Park, H.S.; Jung, S.-C.; Han, K.-H.; Park, Y.J.; Ryu, K.-H.; et al. Differentiated Tonsil-Derived Mesenchymal Stem Cells Embedded in Matrigel Restore Parathyroid Cell Functions in Rats with Parathyroidectomy. Biomaterials 2015, 65, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Kim, Y.-H.; Woo, S.-Y.; Lee, H.J.; Yu, Y.; Kim, H.S.; Park, Y.S.; Jo, I.; Park, J.-W.; Jung, S.-C.; et al. Tonsil-Derived Mesenchymal Stem Cells Ameliorate CCl4-Induced Liver Fibrosis in Mice via Autophagy Activation. Sci. Rep. 2015, 5, 8616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-Y.; Lee, Y.; Yoon, H.-S.; Kim, Y.-H.; Cho, K.-A.; Woo, S.-Y.; Kim, H.S.; Park, B.-Y.; Jung, S.-C.; Jo, I.; et al. A Novel Method to Differentiate Tonsil-Derived Mesenchymal Stem Cells In Vitro into Estrogen-Secreting Cells. Tissue Eng. Regen. Med. 2021, 18, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Park, Y.S.; Kim, H.S.; Kim, H.Y.; Jin, Y.M.; Jung, S.-C.; Ryu, K.-H.; Jo, I. Characterization of Long-Term in Vitro Culture-Related Alterations of Human Tonsil-Derived Mesenchymal Stem Cells: Role for CCN1 in Replicative Senescence-Associated Increase in Osteogenic Differentiation. J. Anat. 2014, 225, 510–518. [Google Scholar] [CrossRef]
- Holbourn, K.P.; Acharya, K.R.; Perbal, B. The CCN Family of Proteins: Structure-Function Relationships. Trends Biochem. Sci. 2008, 33, 461–473. [Google Scholar] [CrossRef]
- Yu, Y.; Gao, Y.; Qin, J.; Kuang, C.-Y.; Song, M.-B.; Yu, S.-Y.; Cui, B.; Chen, J.-F.; Huang, L. CCN1 Promotes the Differentiation of Endothelial Progenitor Cells and Reendothelialization in the Early Phase after Vascular Injury. Basic Res. Cardiol. 2010, 105, 713–724. [Google Scholar] [CrossRef]
- Liu, H.; Peng, F.; Liu, Z.; Jiang, F.; Li, L.; Gao, S.; Wang, G.; Song, J.; Ruan, E.; Shao, Z.; et al. CYR61/CCN1 Stimulates Proliferation and Differentiation of Osteoblasts in Vitro and Contributes to Bone Remodelling in Vivo in Myeloma Bone Disease. Int. J. Oncol. 2017, 50, 631–639. [Google Scholar] [CrossRef] [Green Version]
- You, J.-J.; Yang, C.-M.; Chen, M.-S.; Yang, C.-H. Regulation of Cyr61/CCN1 Expression by Hypoxia through Cooperation of c-Jun/AP-1 and HIF-1α in Retinal Vascular Endothelial Cells. Exp. Eye Res. 2010, 91, 825–836. [Google Scholar] [CrossRef]
- Quan, T.; Qin, Z.; Xu, Y.; He, T.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Ultraviolet Irradiation Induces CYR61/CCN1, a Mediator of Collagen Homeostasis, through Activation of Transcription Factor AP-1 in Human Skin Fibroblasts. J. Invest. Dermatol. 2010, 130, 1697–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, M.; Liu, H.; Amir, J.; Sun, Y.; Morris, S.W.; Siddiqui, M.a.Q.; Lau, L.F.; Chaqour, B. Mechanical Regulation of the Proangiogenic Factor CCN1/CYR61 Gene Requires the Combined Activities of MRTF-A and CREB-Binding Protein Histone Acetyltransferase. J. Biol. Chem. 2009, 284, 23125–23136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, J.-I.; Lau, L.F. The Matricellular Protein CCN1 Induces Fibroblast Senescence and Restricts Fibrosis in Cutaneous Wound Healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.W.; Shih, D.Q.; Hing, T.C.; Chen, J.; Ho, S.; Zhao, D.; Targan, S.R.; Pothoulakis, C. Substance P Induces CCN1 Expression via Histone Deacetylase Activity in Human Colonic Epithelial Cells. Am. J. Pathol. 2011, 179, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Bosak, M.; Słowik, A.; Turaj, W. Why Do Some Women with Epilepsy Use Valproic Acid despite Current Guidelines? A Single-Center Cohort Study. Epilepsy Behav. 2019, 98, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Cheema, M.; Parker, D.; Wiegand, A.; Bosch, R.J.; Coffin, J.M.; Eron, J.; Cohen, M.; Margolis, D.M. Antiretroviral Intensification and Valproic Acid Lack Sustained Effect on Residual HIV-1 Viremia or Resting CD4+ Cell Infection. PLoS ONE 2010, 5, e9390. [Google Scholar] [CrossRef]
- Divsalar, D.N.; Simoben, C.V.; Schonhofer, C.; Richard, K.; Sippl, W.; Ntie-Kang, F.; Tietjen, I. Novel Histone Deacetylase Inhibitors and HIV-1 Latency-Reversing Agents Identified by Large-Scale Virtual Screening. Front. Pharmacol. 2020, 11, 905. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, P.; Ge, J.; Cheng, J.; Dong, W.; Yuan, H.; Du, Y.; Yang, M.; Sun, R.; Jiang, H. Histone Deacetylase 8 Suppresses Osteogenic Differentiation of Bone Marrow Stromal Cells by Inhibiting Histone H3K9 Acetylation and RUNX2 Activity. Int. J. Biochem. Cell Biol. 2014, 54, 68–77. [Google Scholar] [CrossRef]
- Lee, S.; Park, J.-R.; Seo, M.-S.; Roh, K.-H.; Park, S.-B.; Hwang, J.-W.; Sun, B.; Seo, K.; Lee, Y.-S.; Kang, S.-K.; et al. Histone Deacetylase Inhibitors Decrease Proliferation Potential and Multilineage Differentiation Capability of Human Mesenchymal Stem Cells. Cell Prolif. 2009, 42, 711–720. [Google Scholar] [CrossRef]
- Dong, X.; Pan, R.; Zhang, H.; Yang, C.; Shao, J.; Xiang, L. Modification of Histone Acetylation Facilitates Hepatic Differentiation of Human Bone Marrow Mesenchymal Stem Cells. PloS One 2013, 8, e63405. [Google Scholar] [CrossRef] [Green Version]
- Janjanin, S.; Djouad, F.; Shanti, R.M.; Baksh, D.; Gollapudi, K.; Prgomet, D.; Rackwitz, L.; Joshi, A.S.; Tuan, R.S. Human Palatine Tonsil: A New Potential Tissue Source of Multipotent Mesenchymal Progenitor Cells. Arthritis Res. Ther. 2008, 10, R83. [Google Scholar] [CrossRef] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells. The International Society for Cellular Therapy Position Statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Virdi, J.K.; Pethe, P. Biomaterials Regulate Mechanosensors YAP/TAZ in Stem Cell Growth and Differentiation. Tissue Eng. Regen. Med. 2021, 18, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Standal, T.; Borset, M.; Sundan, A. Role of Osteopontin in Adhesion, Migration, Cell Survival and Bone Remodeling. Exp. Oncol. 2004, 26, 179–184. [Google Scholar]
- Yang, G.; Kim, Y.-N.; Kim, H.; Lee, B.-K. Effect of Human Umbilical Cord Matrix-Derived Mesenchymal Stem Cells on Bisphosphonate-Related Osteonecrosis of the Jaw. Tissue Eng. Regen. Med. 2021, 18, 975–988. [Google Scholar] [CrossRef]
- Paino, F.; La Noce, M.; Tirino, V.; Naddeo, P.; Desiderio, V.; Pirozzi, G.; De Rosa, A.; Laino, L.; Altucci, L.; Papaccio, G. Histone Deacetylase Inhibition with Valproic Acid Downregulates Osteocalcin Gene Expression in Human Dental Pulp Stem Cells and Osteoblasts: Evidence for HDAC2 Involvement. Stem Cells 2014, 32, 279–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshmaiah, K.C.; Jacob, L.A.; Aparna, S.; Lokanatha, D.; Saldanha, S.C. Epigenetic Therapy of Cancer with Histone Deacetylase Inhibitors. J. Cancer Res. Ther. 2014, 10, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Tandon, N.; Ramakrishnan, V.; Kumar, S.K. Clinical Use and Applications of Histone Deacetylase Inhibitors in Multiple Myeloma. Clin. Pharmacol. Adv. Appl. 2016, 8, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Mohseni, J.; Zabidi-Hussin, Z.A.M.H.; Sasongko, T.H. Histone Deacetylase Inhibitors as Potential Treatment for Spinal Muscular Atrophy. Genet. Mol. Biol. 2013, 36, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mawatari, T.; Ninomiya, I.; Inokuchi, M.; Harada, S.; Hayashi, H.; Oyama, K.; Makino, I.; Nakagawara, H.; Miyashita, T.; Tajima, H.; et al. Valproic Acid Inhibits Proliferation of HER2-Expressing Breast Cancer Cells by Inducing Cell Cycle Arrest and Apoptosis through Hsp70 Acetylation. Int. J. Oncol. 2015, 47, 2073–2081. [Google Scholar] [CrossRef] [Green Version]
- Venkataramani, V.; Rossner, C.; Iffland, L.; Schweyer, S.; Tamboli, I.Y.; Walter, J.; Wirths, O.; Bayer, T.A. Histone Deacetylase Inhibitor Valproic Acid Inhibits Cancer Cell Proliferation via Down-Regulation of the Alzheimer Amyloid Precursor Protein. J. Biol. Chem. 2010, 285, 10678–10689. [Google Scholar] [CrossRef] [Green Version]
- Witt, D.; Burfeind, P.; von Hardenberg, S.; Opitz, L.; Salinas-Riester, G.; Bremmer, F.; Schweyer, S.; Thelen, P.; Neesen, J.; Kaulfuss, S. Valproic Acid Inhibits the Proliferation of Cancer Cells by Re-Expressing Cyclin D2. Carcinogenesis 2013, 34, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- McGee-Lawrence, M.E.; McCleary-Wheeler, A.L.; Secreto, F.J.; Razidlo, D.F.; Zhang, M.; Stensgard, B.A.; Li, X.; Stein, G.S.; Lian, J.B.; Westendorf, J.J. Suberoylanilide Hydroxamic Acid (SAHA; Vorinostat) Causes Bone Loss by Inhibiting Immature Osteoblasts. Bone 2011, 48, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; De Veirman, K.; Evans, H.; Santini, G.C.; Vande Broek, I.; Leleu, X.; De Becker, A.; Van Camp, B.; Croucher, P.; Vanderkerken, K.; et al. Effect of the HDAC Inhibitor Vorinostat on the Osteogenic Differentiation of Mesenchymal Stem Cells in Vitro and Bone Formation in Vivo. Acta Pharmacol. Sin. 2013, 34, 699–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliani, N.; Morandi, F.; Tagliaferri, S.; Lazzaretti, M.; Bonomini, S.; Crugnola, M.; Mancini, C.; Martella, E.; Ferrari, L.; Tabilio, A.; et al. The Proteasome Inhibitor Bortezomib Affects Osteoblast Differentiation in Vitro and in Vivo in Multiple Myeloma Patients. Blood 2007, 110, 334–338. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Yang, S.; Shao, J.; Li, Y.-P. Signaling and Transcriptional Regulation in Osteoblast Commitment and Differentiation. Front. Biosci. J. Virtual Libr. 2007, 12, 3068–3092. [Google Scholar] [CrossRef] [Green Version]
- Safadi, F.F.; Barbe, M.F.; Abdelmagid, S.M.; Rico, M.C.; Aswad, R.A.; Litvin, J.; Popoff, S.N. Bone Structure, Development and Bone Biology. In Bone Pathology; Khurana, J.S., Ed.; Humana Press: Totowa, NJ, 2009; pp. 1–50. ISBN 978-1-58829-766-2. [Google Scholar]
- Matsubara, T.; Kida, K.; Yamaguchi, A.; Hata, K.; Ichida, F.; Meguro, H.; Aburatani, H.; Nishimura, R.; Yoneda, T. BMP2 Regulates Osterix through Msx2 and Runx2 during Osteoblast Differentiation. J. Biol. Chem. 2008, 283, 29119–29125. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Li, M.; Wang, S.; Xiao, Z.; Xiong, Y.; Wang, G. Recent Advances of Osterix Transcription Factor in Osteoblast Differentiation and Bone Formation. Front. Cell Dev. Biol. 2020, 8, 601224. [Google Scholar] [CrossRef]
- Koon, H.-W.; Zhao, D.; Na, X.; Moyer, M.P.; Pothoulakis, C. Metalloproteinases and Transforming Growth Factor-α Mediate Substance P-Induced Mitogen-Activated Protein Kinase Activation and Proliferation in Human Colonocytes. J. Biol. Chem. 2004, 279, 45519–45527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Zhou, Y.; Xu, W.; Wang, X.; Jin, H.; Bao, X.; Lu, C. Induction of Sestrin2 by Pterostilbene Suppresses Ethanol-Triggered Hepatocyte Senescence by Degrading CCN1 via P62-Dependent Selective Autophagy. Cell Biol. Toxicol. 2021. [Google Scholar] [CrossRef]
- Wu, W.; Luo, M.; Li, K.; Dai, Y.; Yi, H.; Zhong, Y.; Cao, Y.; Tettamanti, G.; Tian, L. Cholesterol Derivatives Induce Dephosphorylation of the Histone Deacetylases Rpd3/HDAC1 to Upregulate Autophagy. Autophagy 2021, 17, 512–528. [Google Scholar] [CrossRef]
- Wu, W.; Li, K.; Guo, S.; Xu, J.; Ma, Q.; Li, S.; Xu, X.; Huang, Z.; Zhong, Y.; Tettamanti, G.; et al. P300/HDAC1 Regulates the Acetylation/Deacetylation and Autophagic Activities of LC3/Atg8–PE Ubiquitin-like System. Cell Death Discov. 2021, 7, 128. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Huang, B.-L.; Rigueur, D.; Wang, W.; Bhoot, C.; Charles, K.R.; Baek, J.; Mohan, S.; Jiang, J.; Lyons, K.M. CYR61/CCN1 Regulates Sclerostin Levels and Bone Maintenance. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2018, 33, 1076–1089. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Zhang, L.; Zhu, Q.; Bai, D.; Zhang, C.; Wang, X. CD146 Promotes Metastasis and Predicts Poor Prognosis of Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harkness, L.; Zaher, W.; Ditzel, N.; Isa, A.; Kassem, M. CD146/MCAM Defines Functionality of Human Bone Marrow Stromal Stem Cell Populations. Stem Cell Res. Ther. 2016, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Zondler, L.; Herich, S.; Kotte, P.; Körner, K.; Schneider-Hohendorf, T.; Wiendl, H.; Schwab, N.; Zarbock, A. MCAM/CD146 Signaling via PLCγ1 Leads to Activation of Β1-Integrins in Memory T-Cells Resulting in Increased Brain Infiltration. Front. Immunol. 2020, 11, 599936. [Google Scholar] [CrossRef]
- Rahman, M.; Nguyen, H. Valproic Acid. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Park, Y.S.; Hwang, S.; Jin, Y.M.; Yu, Y.; Jung, S.-A.; Jung, S.-C.; Ryu, K.-H.; Kim, H.S.; Jo, I. CCN1 Secreted by Tonsil-Derived Mesenchymal Stem Cells Promotes Endothelial Cell Angiogenesis via Integrin αv β3 and AMPK. J. Cell. Physiol. 2015, 230, 140–149. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; Oh, S.-Y.; Kim, H.Y.; Choi, J.-Y.; Jo, S.A.; Jo, I. Valproic Acid-Induced CCN1 Promotes Osteogenic Differentiation by Increasing CCN1 Protein Stability through HDAC1 Inhibition in Tonsil-Derived Mesenchymal Stem Cells. Cells 2022, 11, 534. https://doi.org/10.3390/cells11030534
Yu Y, Oh S-Y, Kim HY, Choi J-Y, Jo SA, Jo I. Valproic Acid-Induced CCN1 Promotes Osteogenic Differentiation by Increasing CCN1 Protein Stability through HDAC1 Inhibition in Tonsil-Derived Mesenchymal Stem Cells. Cells. 2022; 11(3):534. https://doi.org/10.3390/cells11030534
Chicago/Turabian StyleYu, Yeonsil, Se-Young Oh, Ha Yeong Kim, Ji-Young Choi, Sangmee Ahn Jo, and Inho Jo. 2022. "Valproic Acid-Induced CCN1 Promotes Osteogenic Differentiation by Increasing CCN1 Protein Stability through HDAC1 Inhibition in Tonsil-Derived Mesenchymal Stem Cells" Cells 11, no. 3: 534. https://doi.org/10.3390/cells11030534