
CNOT6: A Novel Regulator of DNA Mismatch Repair

 , , , , , ,
, , , , , ,
Abstract
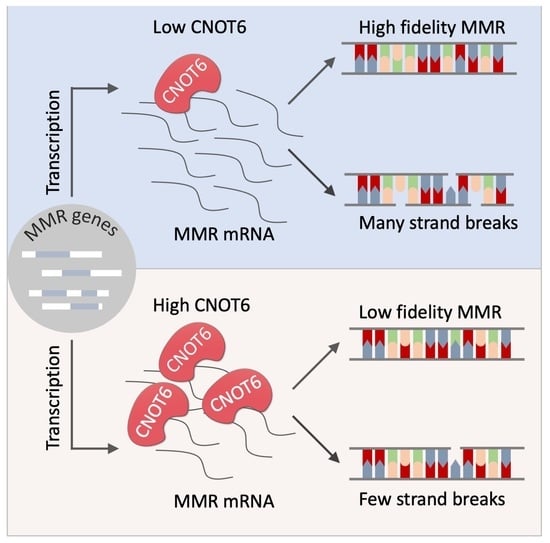
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. High-Throughput Screening
2.3. siRNA Sequences, DNA Constructs and Antibodies
2.4. Cell Transfection
2.5. Protein Immunoblotting and Immunoprecipitation
2.6. RNA Isolation and Quantitative Real-Time PCR
2.7. Cell Viability (MTS) Assay
2.8. Apoptosis Detection by Flow Cytometry
2.9. mRNA Stability Assay
2.10. HPRT Mutation Assay
2.11. In Vitro MMR Assay
3. Results
3.1. Depletion of the Deadenylase Subunit of the CCR4-NOT Complex Sensitizes U2OS Cells to MNNG
3.2. Knockdown of CNOT6 Increases the Frequency of MNNG-Induced Apoptosis
3.3. CNOT6 Regulates MMR Activity In Vitro
3.4. Depletion of CNOT6 Decreases Mutation Frequency in MMR-Proficient Cells, but Not in MMR-Deficient Cells
3.5. Absence of Physical Interaction between CNOT6 and MMR Proteins
3.6. Knockdown of CNOT6 Stabilizes mRNA Transcripts through Decreased Deadenylation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schofield, M.J.; Hsieh, P. DNA Mismatch Repair: Molecular Mechanisms and Biological Function. Annu. Rev. Microbiol. 2003, 57, 579–608. [Google Scholar] [CrossRef]
- Li, G.-M. Mechanisms and Functions of DNA Mismatch Repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Alani, E.; Lee, J.Y.; Schofield, M.J.; Kijas, A.W.; Hsieh, P.; Yang, W. Crystal Structure and Biochemical Analysis of the MutS·ADP·Beryllium Fluoride Complex Suggests a Conserved Mechanism for ATP Interactions in Mismatch Repair. J. Biol. Chem. 2003, 278, 16088–16094. [Google Scholar] [CrossRef] [Green Version]
- Qiu, R.; Sakato, M.; Sacho, E.J.; Wilkins, H.; Zhang, X.; Modrich, P.; Hingorani, M.M.; Erie, D.A.; Weninger, K.R. MutL Traps MutS at a DNA Mismatch. Proc. Natl. Acad. Sci. USA 2015, 112, 10914–10919. [Google Scholar] [CrossRef] [Green Version]
- Pluciennik, A.; Dzantiev, L.; Iyer, R.R.; Constantin, N.; Kadyrov, F.A.; Modrich, P. PCNA Function in the Activation and Strand Direction of MutL Endonuclease in Mismatch Repair. Proc. Natl. Acad. Sci. USA 2010, 107, 16066–16071. [Google Scholar] [CrossRef] [Green Version]
- Kadyrov, F.A.; Holmes, S.F.; Arana, M.E.; Lukianova, O.A.; O’Donnell, M.; Kunkel, T.A.; Modrich, P. Saccharomyces Cerevisiae MutLα Is a Mismatch Repair Endonuclease. J. Biol. Chem. 2007, 282, 37181–37190. [Google Scholar] [CrossRef] [Green Version]
- Schanz, S.; Castor, D.; Fischer, F.; Jiricny, J. Interference of Mismatch and Base Excision Repair during the Processing of Adjacent U/G Mispairs May Play a Key Role in Somatic Hypermutation. Proc. Natl. Acad. Sci. USA 2009, 106, 5593–5598. [Google Scholar] [CrossRef] [Green Version]
- Shahi, A.; Lee, J.-H.; Kang, Y.; Lee, S.H.; Hyun, J.-W.; Chang, I.-Y.; Jun, J.-Y.; You, H.J. Mismatch-Repair Protein MSH6 Is Associated with Ku70 and Regulates DNA Double-Strand Break Repair. Nucleic Acids Res. 2011, 39, 2130–2143. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, P.; Tishkoff, D.X.; Filosi, N.; Dasgupta, R.; Kolodner, R.D. Physical Interaction between Components of DNA Mismatch Repair and Nucleotide Excision Repair. Proc. Natl. Acad. Sci. USA 1998, 95, 14278–14283. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Litman, R.; Xie, J.; Sharma, S.; Brosh, R.M.; Cantor, S.B. The FANCJ/MutLα Interaction Is Required for Correction of the Cross-Link Response in FA-J Cells. EMBO J. 2007, 26, 3238–3249. [Google Scholar] [CrossRef]
- Spies, M.; Fishel, R. Mismatch Repair during Homologous and Homeologous Recombination. Cold Spring Harb. Perspect. Biol. 2015, 7, a022657. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, N.; Goldfarb, T.; Studamire, B.; Alani, E.; Haber, J.E. Heteroduplex Rejection during Single-Strand Annealing Requires Sgs1 Helicase and Mismatch Repair Proteins Msh2 and Msh6 but Not Pms1. Proc. Natl. Acad. Sci. USA 2004, 101, 9315–9320. [Google Scholar] [CrossRef] [Green Version]
- Heyer, W.-D.; Ehmsen, K.T.; Liu, J. Regulation of Homologous Recombination in Eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [Green Version]
- Welz-Voegele, C.; Jinks-Robertson, S. Sequence Divergence Impedes Crossover More than Noncrossover Events during Mitotic Gap Repair in Yeast. Genetics 2008, 179, 1251–1262. [Google Scholar] [CrossRef] [Green Version]
- Karran, P. Mechanisms of Tolerance to DNA Damaging Therapeutic Drugs. Carcinogenesis 2001, 22, 1931–1937. [Google Scholar] [CrossRef] [Green Version]
- Bronner, C.E.; Baker, S.M.; Morrison, P.T.; Warren, G.; Smith, L.G.; Lescoe, M.K.; Kane, M.; Earabino, C.; Lipford, J.; Lindblom, A.; et al. Mutation in the DNA Mismatch Repair Gene Homologue HMLH 1 Is Associated with Hereditary Non-Polyposis Colon Cancer. Nature 1994, 368, 258–261. [Google Scholar] [CrossRef]
- Miyaki, M.; Konishi, M.; Tanaka, K.; Kikuchi-Yanoshita, R.; Muraoka, M.; Yasuno, M.; Igari, T.; Koike, M.; Chiba, M.; Mori, T. Germline Mutation of MSH6 as the Cause of Hereditary Nonpolyposis Colorectal Cancer. Nat. Genet. 1997, 17, 271–272. [Google Scholar] [CrossRef]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Li, G.M. The Role of Mismatch Repair in DNA Damage-Induced Apoptosis. Oncol. Res. 1999, 11, 393–400. [Google Scholar]
- Bellacosa, A.; Cicchillitti, L.; Schepis, F.; Riccio, A.; Yeung, A.T.; Matsumoto, Y.; Golemis, E.A.; Genuardi, M.; Neri, G. MED1, a Novel Human Methyl-CpG-Binding Endonuclease, Interacts with DNA Mismatch Repair Protein MLH1. Proc. Natl. Acad. Sci. USA 1999, 96, 3969–3974. [Google Scholar] [CrossRef] [Green Version]
- Her, C.; Vo, A.T.; Wu, X. Evidence for a Direct Association of HMRE11 with the Human Mismatch Repair Protein HMLH1. DNA Repair 2002, 1, 719–729. [Google Scholar] [CrossRef]
- Traver, S.; Coulombe, P.; Peiffer, I.; Hutchins, J.R.A.; Kitzmann, M.; Latreille, D.; Méchali, M. MCM9 Is Required for Mammalian DNA Mismatch Repair. Mol. Cell 2015, 59, 831–839. [Google Scholar] [CrossRef] [Green Version]
- Collart, M.A. The Ccr4-Not Complex Is a Key Regulator of Eukaryotic Gene Expression. Wiley Interdiscip. Rev. RNA 2016, 7, 438–454. [Google Scholar] [CrossRef] [Green Version]
- Collart, M.A.; Panasenko, O.O. The Ccr4-Not Complex: Architecture and Structural Insights. Subcell Biochem. 2017, 83, 349–379. [Google Scholar] [CrossRef]
- Dlakić, M. Functionally Unrelated Signalling Proteins Contain a Fold Similar to Mg2+-Dependent Endonucleases. Trends Biochem. Sci. 2000, 25, 272–273. [Google Scholar] [CrossRef]
- Yamashita, A.; Chang, T.-C.; Yamashita, Y.; Zhu, W.; Zhong, Z.; Chen, C.-Y.A.; Shyu, A.-B. Concerted Action of Poly(A) Nucleases and Decapping Enzyme in Mammalian MRNA Turnover. Nat. Struct. Mol. Biol. 2005, 12, 1054–1063. [Google Scholar] [CrossRef]
- Tucker, M.; Valencia-Sanchez, M.A.; Staples, R.R.; Chen, J.; Denis, C.L.; Parker, R. The Transcription Factor Associated Ccr4 and Caf1 Proteins Are Components of the Major Cytoplasmic MRNA Deadenylase in Saccharomyces Cerevisiae. Cell 2001, 104, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Chiang, Y.-C.; Denis, C.L. CCR4, a 3′–5′ Poly(A) RNA and SsDNA Exonuclease, Is the Catalytic Component of the Cytoplasmic Deadenylase. EMBO J. 2002, 21, 1414–1426. [Google Scholar] [CrossRef]
- Traven, A.; Hammet, A.; Tenis, N.; Denis, C.L.; Heierhorst, J. Ccr4-Not Complex MRNA Deadenylase Activity Contributes to DNA Damage Responses in Saccharomyces Cerevisiae. Genetics 2005, 169, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Perez, I.; Manguan-Garcia, C.; Menacho-Marquez, M.; Murguía, J.R.; Perona, R. HCCR4/CNOT6 Targets DNA-Damage Response Proteins. Cancer Lett. 2009, 273, 281–291. [Google Scholar] [CrossRef]
- Zukeran, A.; Takahashi, A.; Takaoka, S.; Mohamed, H.M.A.; Suzuki, T.; Ikematsu, S.; Yamamoto, T. The CCR4-NOT Deadenylase Activity Contributes to Generation of Induced Pluripotent Stem Cells. Mol. Biol. Cell 2016, 474, 233–239. [Google Scholar] [CrossRef]
- Maragozidis, P.; Karangeli, M.; Labrou, M.; Dimoulou, G.; Papaspyrou, K.; Salataj, E.; Pournaras, S.; Matsouka, P.; Gourgoulianis, K.I.; Balatsos, N.A.A. Alterations of Deadenylase Expression in Acute Leukemias: Evidence for Poly(A)-Specific Ribonuclease as a Potential Biomarker. Acta Haematol. 2012, 128, 39–46. [Google Scholar] [CrossRef]
- Gutierrez-Camino, A.; Lopez-Lopez, E.; Martin-Guerrero, I.; Piñan, M.A.; Garcia-Miguel, P.; Sanchez-Toledo, J.; Carbone Bañeres, A.; Uriz, J.; Navajas, A.; Garcia-Orad, A. Noncoding RNA–Related Polymorphisms in Pediatric Acute Lymphoblastic Leukemia Susceptibility. Pediatr. Res. 2014, 75, 767–773. [Google Scholar] [CrossRef]
- Jiricny, J. The Multifaceted Mismatch-Repair System. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Aslam, A.; Mittal, S.; Koch, F.; Andrau, J.-C.; Winkler, G.S. The Ccr4–Not Deadenylase Subunits CNOT7 and CNOT8 Have Overlapping Roles and Modulate Cell Proliferation. Mol. Biol. Cell 2009, 20, 3840–3850. [Google Scholar] [CrossRef] [Green Version]
- Braun, J.E.; Huntzinger, E.; Fauser, M.; Izaurralde, E. GW182 Proteins Directly Recruit Cytoplasmic Deadenylase Complexes to MiRNA Targets. Mol. Cell 2011, 44, 120–133. [Google Scholar] [CrossRef]
- Drost, M.; Zonneveld, J.É.B.M.; van Dijk, L.; Morreau, H.; Tops, C.M.; Vasen, H.F.A.; Wijnen, J.T.; de Wind, N. A Cell-Free Assay for the Functional Analysis of Variants of the Mismatch Repair Protein MLH1. Hum. Mutat. 2010, 31, 247–253. [Google Scholar] [CrossRef]
- Duan, S.; Han, X.; Akbari, M.; Croteau, D.L.; Rasmussen, L.J.; Bohr, V.A. Interaction between RECQL4 and OGG1 Promotes Repair of Oxidative Base Lesion 8-OxoG and Is Regulated by SIRT1 Deacetylase. Nucleic Acids Res. 2020, 48, 6530–6546. [Google Scholar] [CrossRef]
- Mittal, S.; Aslam, A.; Doidge, R.; Medica, R.; Winkler, G.S. The Ccr4a (CNOT6) and Ccr4b (CNOT6L) Deadenylase Subunits of the Human Ccr4–Not Complex Contribute to the Prevention of Cell Death and Senescence. Mol. Biol. Cell 2011, 22, 748–758. [Google Scholar] [CrossRef]
- Johnson, G.E. Mammalian Cell HPRT Gene Mutation Assay: Test Methods. Methods Mol. Biol. 2012, 817, 55–67. [Google Scholar] [CrossRef]
- Wang, H.; Hays, J.B. Preparation of DNA Substrates for In Vitro Mismatch Repair. Mol. Biotechnol. 2000, 15, 97–104. [Google Scholar] [CrossRef]
- Sakamoto, M.; Iwama, K.; Sekiguchi, F.; Mashimo, H.; Kumada, S.; Ishigaki, K.; Okamoto, N.; Behnam, M.; Ghadami, M.; Koshimizu, E.; et al. Novel EXOSC9 Variants Cause Pontocerebellar Hypoplasia Type 1D with Spinal Motor Neuronopathy and Cerebellar Atrophy. J. Hum. Genet. 2020, 66, 401–407. [Google Scholar] [CrossRef]
- Loveless, A. Possible Relevance of O–6 Alkylation of Deoxyguanosine to the Mutagenicity and Carcinogenicity of Nitrosamines and Nitrosamides. Nature 1969, 223, 206–207. [Google Scholar] [CrossRef]
- Nowosielska, A.; Marinus, M.G. DNA Mismatch Repair-Induced Double-Strand Breaks. DNA Repair 2008, 7, 48–56. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, V.; Brown, R. Signalling Cell Cycle Arrest and Cell Death through the MMR System. Carcinogenesis 2006, 27, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Webster, M.W.; Chen, Y.-H.; Stowell, J.A.W.; Alhusaini, N.; Sweet, T.; Graveley, B.R.; Coller, J.; Passmore, L.A. MRNA Deadenylation Is Coupled to Translation Rates by the Differential Activities of Ccr4-Not Nucleases. Mol. Cell 2018, 70, 1089–1100.e8. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, A.L.; Pasquinelli, A.E. Tales of Detailed Poly(A) Tails. Trends Cell Biol. 2019, 29, 191–200. [Google Scholar] [CrossRef]
- Lin, D.P.; Wang, Y.; Scherer, S.J.; Clark, A.B.; Yang, K.; Avdievich, E.; Jin, B.; Werling, U.; Parris, T.; Kurihara, N.; et al. An Msh2 Point Mutation Uncouples DNA Mismatch Repair and Apoptosis. Cancer Res. 2004, 64, 517–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Richards, B.; Wilson, T.; Lloyd, M.; Cranston, A.; Thorburn, A.; Fishel, R.; Meuth, M. Apoptosis Induced by Overexpression of HMSH2 or HMLH1. Cancer Res. 1999, 59, 3021–3027. [Google Scholar]
- Scherer, S.J.; Maier, S.M.; Seifert, M.; Hanselmann, R.G.; Zang, K.D.; Müller-Hermelink, H.K.; Angel, P.; Welter, C.; Schartl, M. P53 and C-Jun Functionally Synergize in the Regulation of the DNA Repair Gene HMSH2 in Response to UV. J. Biol. Chem. 2000, 275, 37469–37473. [Google Scholar] [CrossRef] [Green Version]
- Gazzoli, I.; Kolodner, R.D. Regulation of the Human MSH6 Gene by the Sp1 Transcription Factor and Alteration of Promoter Activity and Expression by Polymorphisms. Mol. Cell. Biol. 2003, 23, 7992–8007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maragozidis, P.; Papanastasi, E.; Scutelnic, D.; Totomi, A.; Kokkori, I.; Zarogiannis, S.G.; Kerenidi, T.; Gourgoulianis, K.I.; Balatsos, N.A.A. Poly(A)-Specific Ribonuclease and Nocturnin in Squamous Cell Lung Cancer: Prognostic Value and Impact on Gene Expression. Mol. Cancer 2015, 14, 187. [Google Scholar] [CrossRef] [PubMed]
- Nalla, A.K.; Williams, T.F.; Collins, C.P.; Rae, D.T.; Trobridge, G.D. Lentiviral Vector-Mediated Insertional Mutagenesis Screen Identifies Genes That Influence Androgen Independent Prostate Cancer Progression and Predict Clinical Outcome. Mol. Carcinog. 2016, 55, 1761–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, P.; Liu, S.; Liu, D.; Keijzers, G.; Bakula, D.; Duan, S.; de Wind, N.; Ye, Z.; Vakhrushev, S.Y.; Scheibye-Knudsen, M.; et al. CNOT6: A Novel Regulator of DNA Mismatch Repair. Cells 2022, 11, 521. https://doi.org/10.3390/cells11030521
Song P, Liu S, Liu D, Keijzers G, Bakula D, Duan S, de Wind N, Ye Z, Vakhrushev SY, Scheibye-Knudsen M, et al. CNOT6: A Novel Regulator of DNA Mismatch Repair. Cells. 2022; 11(3):521. https://doi.org/10.3390/cells11030521
Chicago/Turabian StyleSong, Peng, Shaojun Liu, Dekang Liu, Guido Keijzers, Daniela Bakula, Shunlei Duan, Niels de Wind, Zilu Ye, Sergey Y. Vakhrushev, Morten Scheibye-Knudsen, and et al. 2022. "CNOT6: A Novel Regulator of DNA Mismatch Repair" Cells 11, no. 3: 521. https://doi.org/10.3390/cells11030521