
Innate Immune Pathways Promote Oligodendrocyte Progenitor Cell Recruitment to the Injury Site in Adult Zebrafish Brain

 , , , ,
, , , ,
Abstract
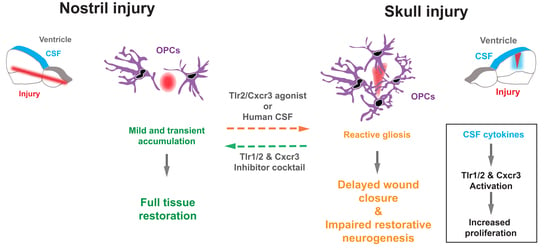
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Stab Wound Injuries
2.3. Tlr2 Agonist (Zymosan A Bioparticles, Invitrogen) Administration
2.4. Cxcr3 Agonist (VUF 11222, R & D Systems) Administration
2.5. Inhibitor Administration
2.6. BrdU Labelling Experiments
2.7. Immune Cell Depletion Assay
2.8. Human CSF Sample Collection
2.9. Human Plasma, Cerebrospinal Fluid, and Heat-Inactivated Cerebrospinal Fluid Administration
2.10. Plasmid Electroporation
2.11. Tissue Preparation and Immunohistochemistry
2.12. RNAscope
2.13. Image Acquisition and Processing
2.14. Quantitative Analysis
2.15. Statistical Analysis
2.16. Analysis of Restorative Neurogenesis
2.17. RNA Extraction, cDNA Synthesis, and RT-qPCR
2.18. Microarray Analysis
2.19. Assignment of Zebrafish Array Probes to Homologous Mouse Genes
2.20. FACS Analysis
2.21. Preparation of Libraries for Deep Sequencing
2.22. RNAseq Analysis
2.23. Primary OPC Culture and Clonal Analysis
2.24. Generation of gRNAs for CRISPR/Cas9-Mediated Deletion
2.25. DNA Extraction and PCR
2.26. Generation of the Oli-Neu Cell Line Deficient for Cxcr3 and Tlr2
2.27. Screen for Cxcr3 Ligands from the CSF
2.28. Human Cytokine Antibody Array
3. Results
3.1. Skull and Nostril Models of Zebrafish Telencephalon Injury Differ in the Kinetics of the Glial Reaction
3.2. Activation of Innate Immunity Pathways Induces Prolonged Glia Reactivity after Injury in the Zebrafish Telencephalon
3.3. Tlr1/2 and Cxcr3 Pathways Cooperatively Control Reactive Gliosis after Injury in the Zebrafish Telencephalon
3.4. Reduction in Glial Accumulation Correlates with Better Tissue Recovery
3.5. Microglia/Monocytes Depletion Does Not Alter the Innate Immunity-Regulated Accumulation of Olig2:GFP+ Cells at the Injury Site
3.6. Olig2:dsRed+ Cells Activate Both Innate Immunity Pathways and Transcription Programs Involved in Cell Proliferation in Response to an Injury
3.7. Regulation of Oligodendrocyte Progenitor Cell Proliferation by Tlr1/2 and Cxcr3 Signaling
3.8. Cerebrospinal Fluid Induces Exacerbated Glial Reactivity by Increasing OPC Proliferation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zambusi, A.; Ninkovic, J. Regeneration of the central nervous system-principles from brain regeneration in adult zebrafish. World J. Stem Cells 2020, 12, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Kyritsis, N.; Kizil, C.; Zocher, S.; Kroehne, V.; Kaslin, J.; Freudenreich, D.; Iltzsche, A.; Brand, M. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science 2012, 338, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41. [Google Scholar] [CrossRef] [PubMed]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial activation in traumatic brain injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O′Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation AIDS central nervous system axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Dias, D.O.; Kim, H.; Holl, D.; Solnestam, B.W.; Lundeberg, J.; Carlen, M.; Goritz, C.; Frisen, J. Reducing Pericyte-Derived Scarring Promotes Recovery after Spinal Cord Injury. Cell 2018, 173, 153–165.e22. [Google Scholar] [CrossRef] [Green Version]
- Goritz, C.; Dias, D.O.; Tomilin, N.; Barbacid, M.; Shupliakov, O.; Frisen, J. A pericyte origin of spinal cord scar tissue. Science 2011, 333, 238–242. [Google Scholar] [CrossRef]
- O′Shea, T.M.; Burda, J.E.; Sofroniew, M.V. Cell biology of spinal cord injury and repair. J. Clin. Investig. 2017, 127, 3259–3270. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef] [Green Version]
- Bardehle, S.; Kruger, M.; Buggenthin, F.; Schwausch, J.; Ninkovic, J.; Clevers, H.; Snippert, H.J.J.; Theis, F.J.J.; Meyer-Luehmann, M.; Bechmann, I.; et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat. Neurosci. 2013, 16, 580–586. [Google Scholar] [CrossRef]
- Shandra, O.; Winemiller, A.R.; Heithoff, B.P.; Munoz-Ballester, C.; George, K.K.; Benko, M.J.; Zuidhoek, I.A.; Besser, M.N.; Curley, D.E.; Edwards, G.F.; et al. Repetitive diffuse mild traumatic brain injury causes an atypical astrocyte response and spontaneous recurrent seizures. J. Neurosci. 2019, 39, 1944–1963. [Google Scholar] [CrossRef] [PubMed]
- Bush, T.G.; Puvanachandra, N.; Horner, C.H.; Polito, A.; Ostenfeld, T.; Svendsen, C.N.; Mucke, L.; Johnson, M.H.; Sofroniew, M.V. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 1999, 23, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Hol, E.M.; Pekny, M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr. Opin. Cell Biol. 2015, 32, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Willis, E.F.; MacDonald, K.P.A.; Nguyen, Q.H.; Garrido, A.L.; Gillespie, E.R.; Harley, S.B.R.; Bartlett, P.F.; Schroder, W.A.; Yates, A.G.; Anthony, D.C.; et al. Repopulating Microglia Promote Brain Repair in an IL-6-Dependent Manner. Cell 2020, 180, 833–846. [Google Scholar] [CrossRef]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.A.; Silver, J. The role of extracellular matrix in CNS regeneration. Curr. Opin. Neurobiol. 2007, 17, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Fitch, M.T.; Silver, J. CNS injury, glial scars, and inflammation: Inhibitory extracellular matrices and regeneration failure. Exp. Neurol. 2008, 209, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Frik, J.; Merl-Pham, J.; Plesnila, N.; Mattugini, N.; Kjell, J.; Kraska, J.; Gomez, R.M.; Hauck, S.M.; Sirko, S.; Gotz, M. Cross-talk between monocyte invasion and astrocyte proliferation regulates scarring in brain injury. EMBO Rep. 2018, 19, e45294. [Google Scholar] [CrossRef]
- Di Bello, I.C.; Dawson, M.R.L.; Levine, J.M.; Reynolds, R. Generation of oligodendroglial progenitors in acute inflammatory demyelinating lesions of the rat brain stem is associated with demyelination rather than inflammation. J. Neurocytol. 1999, 28, 365–381. [Google Scholar] [CrossRef]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front. Cell Dev. Biol. 2016, 4, 71. [Google Scholar] [PubMed]
- Simon, C.; Dimou, L.; Gotz, M. Progenitors in the adult cerebral cortex—Cell cycle properties and regulation by physiological stimuli and injury. Glia 2011, 59, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Li, Y.; Fukaya, M.; Lorenzini, I.; Cleveland, D.W.; Ostrow, L.W.; Rothstein, J.D.; Bergles, D.E. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 2013, 16, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, E.G.; Kang, S.H.; Fukaya, M.; Bergles, D.E. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 2013, 16, 668–676. [Google Scholar] [CrossRef] [Green Version]
- Von Streitberg, A.; Jäkel, S.; Eugenin von Bernhardi, J.; Straube, C.; Buggenthin, F.; Marr, C.; Dimou, L. NG2-Glia Transiently Overcome Their Homeostatic Network and Contribute to Wound Closure After Brain Injury. Front. Cell Dev. Biol. 2021, 9, 762. [Google Scholar] [CrossRef]
- Komitova, M.; Zhu, X.; Serwanski, D.R.; Nishiyama, A. NG2 cells are distinct from neurogenic cells in the postnatal mouse subventricular zone. J. Comp. Neurol. 2009, 512, 702–716. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, A.; Komitova, M.; Suzuki, R.; Zhu, X. Polydendrocytes (NG2 cells): Multifunctional cells with lineage plasticity. Nat. Rev. Neurosci. 2009, 10, 9–22. [Google Scholar] [CrossRef]
- Buffo, A.; Vosko, M.R.; Erturk, D.; Hamann, G.F.; Jucker, M.; Rowitch, D.; Gotz, M. Expression pattern of the transcription factor Olig2 in response to brain injuries: Implications for neuronal repair. Proc. Natl. Acad. Sci. USA 2005, 102, 18183–18188. [Google Scholar] [CrossRef] [Green Version]
- Wellman, S.M.; Kozai, T.D.Y. In vivo spatiotemporal dynamics of NG2 glia activity caused by neural electrode implantation. Biomaterials 2018, 164, 121–133. [Google Scholar] [CrossRef]
- Rhodes, K.E.; Moon, L.D.F.; Fawcett, J.W. Inhibiting cell proliferation during formation of the glial scar: Effects on axon regeneration in the CNS. Neuroscience 2003, 120, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Busch, S.A.; Horn, K.P.; Cuascut, F.X.; Hawthorne, A.L.; Bai, L.; Miller, R.H.; Silver, J. Adult NG2+cells are permissive to neurite outgrowth and stabilize sensory axons during macrophage-induced axonal dieback after spinal cord injury. J. Neurosci. 2010, 30, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.J.; Ughrin, Y.; Levine, J.M. Inhibition of axon growth by oligodendrocyte precursor cells. Mol. Cell. Neurosci. 2002, 20, 125–139. [Google Scholar] [CrossRef] [Green Version]
- Kroehne, V.; Freudenreich, D.; Hans, S.; Kaslin, J.; Brand, M. Regeneration of the adult zebrafish brain from neurogenic radial glia-type progenitors. Development 2011, 138, 4831–4841. [Google Scholar] [CrossRef] [Green Version]
- Dimou, L.; Gotz, M. Glial cells as progenitors and stem cells: New roles in the healthy and diseased brain. Physiol. Rev. 2014, 94, 709–737. [Google Scholar] [CrossRef] [Green Version]
- Reimer, M.M.; Kuscha, V.; Wyatt, C.; Sorensen, I.; Frank, R.E.; Knuwer, M.; Becker, T.; Becker, C.G. Sonic hedgehog is a polarized signal for motor neuron regeneration in adult zebrafish. J. Neurosci. 2009, 29, 15073–15082. [Google Scholar] [CrossRef]
- Reimer, M.M.; Sorensen, I.; Kuscha, V.; Frank, R.E.; Liu, C.; Becker, C.G.; Becker, T. Motor neuron regeneration in adult zebrafish. J. Neurosci. 2008, 28, 8510–8516. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, E.V.V.; Barbosa, J.S.S.; Bally-Cuif, L.; Gotz, M.; Ninkovic, J.; Götz, M.; Ninkovic, J. Stab wound injury of the zebrafish telencephalon: A model for comparative analysis of reactive gliosis. Glia 2012, 60, 343–357. [Google Scholar] [CrossRef]
- Ayari, B.; Elhachimi, K.H.; Yanicostas, C.; Landoulsi, A.; Soussi-Yanicostas, N. Prokineticin 2 expression is associated with neural repair of injured adult zebrafish telencephalon. J. Neurotrauma 2010, 27, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Marz, M.; Schmidt, R.; Rastegar, S.; Strahle, U. Regenerative response following stab injury in the adult zebrafish telencephalon. Dev. Dyn. 2011, 240, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Park, H.C.; Topczewska, J.M.; Mawdsley, D.J.; Appel, B. Neural cell fate analysis in zebrafish using olig2 BAC transgenics. Methods Cell Sci 2003, 25, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Lawson, N.D.; Weinstein, B.M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol. 2002, 248, 307–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardos, R.L.; Raymond, P.A. GFAP transgenic zebrafish. Gene Expr. Patterns 2006, 6, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Kim, S.; Chung, A.Y.; Kim, H.T.; So, J.H.; Ryu, J.; Park, H.C.; Kim, C.H. Visualization of myelination in GFP-transgenic zebrafish. Dev. Dyn. 2010, 239, 592–597. [Google Scholar] [CrossRef]
- Bernut, A.; Herrmann, J.L.; Kissa, K.; Dubremetz, J.F.; Gaillard, J.L.; Lutfalla, G.; Kremer, L. Mycobacterium abscessus cording prevents phagocytosis and promotes abscess formation. Proc. Natl. Acad. Sci. USA 2014, 111, E943–E952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerfield, M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio Rerio), 4th ed.; Univ. of Oregon Press: Eugene, OR, USA, 2000. [Google Scholar]
- Durovic, T.; Ninkovic, J. Electroporation method for in vivo delivery of plasmid dna in the adult zebrafish telencephalon. J. Vis. Exp. 2019, 13, e60066. [Google Scholar] [CrossRef]
- Barbosa, J.S.S.; Sanchez-Gonzalez, R.; Di Giaimo, R.; Baumgart, E.V.V.; Theis, F.J.J.; Götz, M.; Ninkovic, J.; Gotz, M.; Ninkovic, J.; Gonzalez-Sanchez, R.; et al. Live imaging of adult neural stem cell behavior in the intact and injured zebrafish brain. Science 2015, 348, 789–793. [Google Scholar] [CrossRef]
- Barbosa, J.S.S.; Di Giaimo, R.; Götz, M.; Ninkovic, J.; Gotz, M.; Ninkovic, J. Single-cell in vivo imaging of adult neural stem cells in the zebrafish telencephalon. Nat. Protoc. 2016, 11, 1360–1370. [Google Scholar] [CrossRef] [Green Version]
- Di Giaimo, R.; Durovic, T.; Barquin, P.; Kociaj, A.; Lepko, T.; Aschenbroich, S.; Breunig, C.T.T.; Irmler, M.; Cernilogar, F.M.M.; Schotta, G.; et al. The Aryl Hydrocarbon Receptor Pathway Defines the Time Frame for Restorative Neurogenesis. Cell Rep. 2018, 25, 3241–3251. [Google Scholar] [CrossRef] [Green Version]
- Rainer, J.; Sanchez-Cabo, F.; Stocker, G.; Sturn, A.; Trajanoski, Z. CARMAweb: Comprehensive R- and bioconductor-based web service for microarray data analysis. Nucleic Acids Res. 2006, 34, W498–W503. [Google Scholar] [CrossRef] [Green Version]
- Herrero, J.; Muffato, M.; Beal, K.; Fitzgerald, S.; Gordon, L.; Pignatelli, M.; Vilella, A.J.; Searle, S.M.J.; Amode, R.; Brent, S.; et al. Ensembl comparative genomics resources. Database 2016, 2016, baw053. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, J.; Beckervordersandforth, R.; Tripathi, P.; Steiner-Mezzadri, A.; Ninkovic, J.; Gotz, M.; Götz, M. Prospective isolation of adult neural stem cells from the mouse subependymal zone. Nat. Protoc 2011, 6, 1981–1989. [Google Scholar] [CrossRef] [PubMed]
- Di Giaimo, R.; Aschenbroich, S.; Ninkovic, J. Fluorescence-Activated Cell Sorting-Based Isolation and Characterization of Neural Stem Cells from the Adult Zebrafish Telencephalon. Adv. Struct. Saf. Stud. 2019, 1938, 49–66. [Google Scholar]
- He, D.; Meyer, B.; Lu, R. Isolation and Culture of Oligodendrocyte Precursor Cells from Prenatal and Postnatal Rodent Brain. In Stem Cell Technologies in Neuroscience; Humana Press: New York, NY, USA, 2017. [Google Scholar]
- Walcher, T.; Xie, Q.; Sun, J.; Irmler, M.; Beckers, J.; Öztürk, T.; Niessing, D.; Stoykova, A.; Cvekl, A.; Ninkovic, J.; et al. Functional dissection of the paired domain of Pax6 reveals molecular mechanisms of coordinating neurogenesis and proliferation. Development 2013, 140, 1123–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breunig, C.T.; Durovic, T.; Neuner, A.M.; Baumann, V.; Wiesbeck, M.F.; Köferle, A.; Götz, M.; Ninkovic, J.; Stricker, S.H. One step generation of customizable gRNA vectors for multiplex CRISPR approaches through string assembly gRNA cloning (STAgR). PLoS ONE 2018, 13, e0196015. [Google Scholar] [CrossRef]
- Koferle, A.; Worf, K.; Breunig, C.; Baumann, V.; Herrero, J.; Wiesbeck, M.; Hutter, L.H.; Gotz, M.; Fuchs, C.; Beck, S.; et al. CORALINA: A universal method for the generation of gRNA libraries for CRISPR-based screening. BMC Genomics 2016, 17, 917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef]
- Adolf, B.; Chapouton, P.; Lam, C.S.; Topp, S.; Tannhauser, B.; Strahle, U.; Gotz, M.; Bally-Cuif, L. Conserved and acquired features of adult neurogenesis in the zebrafish telencephalon. Dev. Biol. 2006, 295, 278–293. [Google Scholar] [CrossRef] [Green Version]
- Horvat, A.; Schwaiger, F.W.; Hager, G.; Bröcker, F.; Streif, R.; Knyazev, P.G.; Ullrich, A.; Kreutzberg, G.W. A novel role for protein tyrosine phosphatase SHP1 in controlling glial activation in the normal and injured nervous system. J. Neurosci. 2001, 21, 865–874. [Google Scholar] [CrossRef]
- Zhu, Y.M.; Gao, X.; Ni, Y.; Li, W.; Kent, T.A.; Qiao, S.G.; Wang, C.; Xu, X.X.; Zhang, H.L. Sevoflurane postconditioning attenuates reactive astrogliosis and glial scar formation after ischemia–reperfusion brain injury. Neuroscience 2017, 356, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.Y.C.; Bourguignon, L.Y.W.; Adams, C.M.; Peyrollier, K.; Zhang, H.; Fandel, T.; Cun, C.L.; Werb, Z.; Noble-Haeusslein, L.J. Matrix metalloproteinase-9 facilitates glial scar formation in the injured spinal cord. J. Neurosci. 2008, 28, 13467–13477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schachtrup, C.; Ryu, J.K.; Helmrick, M.J.; Vagena, E.; Galanakis, D.K.; Degen, J.L.; Margolis, R.U.; Akassoglou, K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-β after vascular damage. J. Neurosci. 2010, 30, 5843–5854. [Google Scholar] [CrossRef] [PubMed]
- Rünger, T.M.; Quintanilla-Dieck, M.J.; Bhawan, J. Role of cathepsin K in the turnover of the dermal extracellular matrix during scar formation. J. Investig. Dermatol. 2007, 127, 293–297. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wu, Z.; Ni, J.; Liu, Y.; Meng, J.; Yu, W.; Nakanishi, H.; Zhou, Y. Cathepsin B Regulates Collagen Expression by Fibroblasts via Prolonging TLR2/NF-κB Activation. Oxid. Med. Cell. Longev. 2016, 2016, 7894247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalmanach, G.; Saidi, A.; Marchand-Adam, S.; Lecaille, F.; Kasabova, M. Cysteine cathepsins and cystatins: From ancillary tasks to prominent status in lung diseases. Biol. Chem. 2015, 396, 111–130. [Google Scholar] [CrossRef]
- Pan, Z.; Yang, K.; Wang, H.; Xiao, Y.; Zhang, M.; Yu, X.; Xu, T.; Bai, T.; Zhu, H. MFAP4 deficiency alleviates renal fibrosis through inhibition of NF-κB and TGF-β/Smad signaling pathways. FASEB J. 2020, 34, 14250–14263. [Google Scholar] [CrossRef]
- Sulimai, N.; Lominadze, D. Fibrinogen and Neuroinflammation During Traumatic Brain Injury. Mol. Neurobiol. 2020, 57, 4692–4703. [Google Scholar] [CrossRef]
- Dietrich, N.; Lienenklaus, S.; Weiss, S.; Gekara, N.O. Murine Toll-Like Receptor 2 Activation Induces Type I Interferon Responses from Endolysosomal Compartments. PLoS ONE 2010, 5, e10250. [Google Scholar] [CrossRef] [Green Version]
- Owens, B.M.J.; Moore, J.W.J.; Kaye, P.M. IRF7 regulates TLR2-mediated activation of splenic CD11chi dendritic cells. PLoS ONE 2012, 7, e41050. [Google Scholar] [CrossRef] [Green Version]
- Liljeroos, M.; Vuolteenaho, R.; Rounioja, S.; Henriques-Normark, B.; Hallman, M.; Ojaniemi, M. Bacterial ligand of TLR2 signals Stat activation via induction of IRF1/2 and interferon-α production. Cell. Signal. 2008, 20, 1873–1881. [Google Scholar] [CrossRef]
- Nomiyama, H.; Osada, N.; Yoshie, O. The evolution of mammalian chemokine genes. Cytokine Growth Factor Rev. 2010, 21, 253–262. [Google Scholar] [CrossRef]
- Portou, M.J.; Baker, D.; Abraham, D.; Tsui, J. The innate immune system, toll-like receptors and dermal wound healing: A review. Vasc. Pharmacol. 2015, 71, 31–36. [Google Scholar] [CrossRef]
- Mann, D.L.; Topkara, V.K.; Evans, S.; Barger, P.M. Innate immunity in the adult mammalian heart: For whom the cell tolls. Trans. Am. Clin. Clim. Assoc. 2010, 121, 31–34. [Google Scholar]
- Griffiths, M.R.; Gasque, P.; Neal, J.W. The regulation of the CNS innate immune response is vital for the restoration of tissue homeostasis (repair) after acute brain injury: A brief review. Int. J. Inflam 2010, 2010, 151097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Sano, H.; Iwaki, D.; Kudo, K.; Konishi, M.; Takahashi, H.; Takahashi, T.; Imaizumi, H.; Asai, Y.; Kuroki, Y. Direct Binding of Toll-Like Receptor 2 to Zymosan, and Zymosan-Induced NF-κB Activation and TNF-α Secretion Are Down-Regulated by Lung Collectin Surfactant Protein A. J. Immunol. 2003, 171, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcel, L.; Pius, L.; Nicole, B.; Eckart, M.; Bernhard, M. Lymphocyte-specific chemokine receptor CXCR3: Regulation, chemokine binding and gene localization. Eur. J. Immunol. 1998, 28, 3696–3705. [Google Scholar]
- Wijtmans, M.; Scholten, D.J.; Roumen, L.; Canals, M.; Custers, H.; Glas, M.; Vreeker, M.C.A.; de Kanter, F.J.J.; de Graaf, C.; Smit, M.J.; et al. Chemical subtleties in small-molecule modulation of peptide receptor function: The case of CXCR3 biaryl-type ligands. J. Med. Chem. 2012, 55, 10572–10583. [Google Scholar] [CrossRef]
- Zambusi, A.; Pelin Burhan, Ö.; Di Giaimo, R.; Schmid, B.; Ninkovic, J. Granulins Regulate Aging Kinetics in the Adult Zebrafish Telencephalon. Cells 2020, 9, 350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, M.; Muramatsu, R.; Maedera, N.; Koyama, Y.; Hamaguchi, M.; Fujimura, H.; Yoshida, M.; Konishi, M.; Itoh, N.; Mochizuki, H.; et al. Peripherally derived FGF21 promotes remyelination in the central nervous system. J. Clin. Investig. 2017, 127, 3496–3509. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Cheng, X.; Qi, J.; Xie, B.; Zhao, X.; Zheng, K.; Zhang, Z.; Qiu, M. EGF enhances oligodendrogenesis from glial progenitor cells. Front. Mol. Neurosci. 2017, 10, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinukonda, G.; Hu, F.; Mehdizadeh, R.; Dohare, P.; Kidwai, A.; Juneja, A.; Naran, V.; Kierstead, M.; Chawla, R.; Kayton, R.; et al. Epidermal growth factor preserves myelin and promotes astrogliosis after intraventricular hemorrhage. Glia 2016, 64, 1987–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, D.; Liu, N.; Li, D.; Yan, H.; Wang, Q.B.; Fang, Y.; Xie, L.; Li, H.P. Inhibition of platelet-derived growth factor receptor β reduces reactive glia and scar formation after traumatic brain injury in mice. Brain Res. Bull. 2017, 134, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Robel, S.; Berninger, B.; Götz, M.; Gotz, M. The stem cell potential of glia: Lessons from reactive gliosis. Nat. Rev. Neurosci. 2011, 12, 88–104. [Google Scholar] [CrossRef]
- Eugenín-von Bernhardi, J.; Dimou, L. NG2-glia, more than progenitor cells. In Glial Cells in Health and Disease of the CNS; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2016; pp. 27–45. [Google Scholar] [CrossRef]
- Zupanc, G.K. Adult neurogenesis and neuronal regeneration in the brain of teleost fish. J. Physiol. 2008, 102, 357–373. [Google Scholar] [CrossRef]
- Zupanc, G.K. Towards brain repair: Insights from teleost fish. Semin Cell Dev. Biol. 2009, 20, 683–690. [Google Scholar] [CrossRef]
- Zupanc, G.K. Neurogenesis and neuronal regeneration in the adult fish brain. J. Comp. Physiol. A Neuroethol. Sens Neural Behav. Physiol. 2006, 192, 649–670. [Google Scholar] [CrossRef] [PubMed]
- Alunni, A.; Bally-Cuif, L. A comparative view of regenerative neurogenesis in vertebrates. Development 2016, 143, 741–753. [Google Scholar] [CrossRef] [Green Version]
- Kizil, C.; Dudczig, S.; Kyritsis, N.; Machate, A.; Blaesche, J.; Kroehne, V.; Brand, M. The chemokine receptor cxcr5 regulates the regenerative neurogenesis response in the adult zebrafish brain. Neural Dev. 2012, 7, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbe, R.M.; Irimia, M.; Currie, K.W.; Lin, A.; Zhu, S.J.; Brown, D.D.; Ross, E.J.; Voisin, V.; Bader, G.D.; Blencowe, B.J.; et al. A comparative transcriptomic analysis reveals conserved features of stem cell pluripotency in planarians and mammals. Stem Cells 2012, 30, 1734–1745. [Google Scholar] [CrossRef] [Green Version]
- Simkin, J.; Gawriluk, T.R.; Gensel, J.C.; Seifert, A.W. Macrophages are necessary for epimorphic regeneration in African spiny mice. eLife 2017, 6, e24623. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.W.; Kiama, S.G.; Seifert, M.G.; Goheen, J.R.; Palmer, T.M.; Maden, M. Skin shedding and tissue regeneration in African spiny mice (Acomys). Nature 2012, 489, 561–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brant, J.O.; Yoon, J.H.; Polvadore, T.; Barbazuk, W.B.; Maden, M. Cellular events during scar-free skin regeneration in the spiny mouse, Acomys. Wound Repair Regen. 2016, 24, 75–88. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekny, M.; Nilsson, M. Astrocyte activation and reactive gliosis. Glia 2005, 50, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.N.; Shi, S.X.Y.; Li, Z.; Li, M.; Wood, K.; Gonzales, R.J.; Liu, Q. Depletion of microglia exacerbates postischemic inflammation and brain injury. J. Cereb. Blood Flow Metab. 2017, 37, 2224–2236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Zhao, Y.; Hu, D.; Wang, S.; Yu, T.; Zhang, L. Depletion of microglia exacerbates injury and impairs function recovery after spinal cord injury in mice. Cell Death Dis. 2020, 11, 528. [Google Scholar] [CrossRef]
- Grade, S.; Götz, M. Neuronal replacement therapy: Previous achievements and challenges ahead. npj Regen. Med. 2017, 2, 29. [Google Scholar] [CrossRef] [Green Version]
- Grandel, H.; Brand, M. Comparative aspects of adult neural stem cell activity in vertebrates. Dev. Genes Evol. 2013, 223, 131–147. [Google Scholar] [CrossRef]
- Kizil, C.; Kaslin, J.; Kroehne, V.; Brand, M. Adult neurogenesis and brain regeneration in zebrafish. Dev. Neurobiol. 2012, 72, 429–461. [Google Scholar] [CrossRef] [PubMed]
- Dunkin, C.S.; Pleat, J.M.; Gillespie, P.H.; Tyler, M.P.; Roberts, A.H.; McGrouther, D.A. Scarring occurs at a critical depth of skin injury: Precise measurement in a graduated dermal scratch in human volunteers. Plast. Reconstr. Surg. 2007, 119, 1722–1724. [Google Scholar] [CrossRef] [PubMed]
- Bouabe, H.; Liu, Y.; Moser, M.; Bosl, M.R.; Heesemann, J. Novel highly sensitive IL-10-beta-lactamase reporter mouse reveals cells of the innate immune system as a substantial source of IL-10 in vivo. J. Immunol. 2011, 187, 3165–3176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.L.; Niemi, E.C.; Wang, S.H.; Lee, C.C.; Bingham, D.; Zhang, J.; Cozen, M.L.; Charo, I.; Huang, E.J.; Liu, J.; et al. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J. Neurotrauma 2014, 31, 1677–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wattananit, S.; Tornero, D.; Graubardt, N.; Memanishvili, T.; Monni, E.; Tatarishvili, J.; Miskinyte, G.; Ge, R.; Ahlenius, H.; Lindvall, O.; et al. Monocyte-Derived Macrophages Contribute to Spontaneous Long-Term Functional Recovery after Stroke in Mice. J. Neurosci. 2016, 36, 4182–4195. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutierrez-Vazquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef]
- Kielian, T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J. Neurosci. Res. 2006, 83, 711–730. [Google Scholar] [CrossRef] [Green Version]
- Krauthausen, M.; Saxe, S.; Zimmermann, J.; Emrich, M.; Heneka, M.T.; Muller, M. CXCR3 modulates glial accumulation and activation in cuprizone-induced demyelination of the central nervous system. J. Neuroinflammation 2014, 11, 109. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, S.H.; van der Meer, P.; Hesselgesser, J.; Jaffer, S.; Kolson, D.L.; Albright, A.V.; Gonzalez-Scarano, F.; Lavi, E. CXCR3 expression in human central nervous system diseases. Neuropathol. Appl. Neurobiol. 2001, 27, 127–138. [Google Scholar] [CrossRef]
- Bsibsi, M.; Ravid, R.; Gveric, D.; van Noort, J.M. Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Dzamko, N.; Gysbers, A.; Perera, G.; Bahar, A.; Shankar, A.; Gao, J.; Fu, Y.; Halliday, G.M. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 2017, 133, 303–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestre, H.; Du, T.; Sweeney, A.M.; Liu, G.; Samson, A.J.; Peng, W.; Mortensen, K.N.; Stæger, F.F.; Bork, P.A.R.; Bashford, L.; et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science 2020, 367, eaax7171. [Google Scholar] [CrossRef] [PubMed]
- Lepko, T.; Pusch, M.; Müller, T.; Schulte, D.; Ehses, J.; Kiebler, M.; Hasler, J.; Huttner, H.B.; Vandenbroucke, R.E.; Vandendriessche, C.; et al. Choroid plexus-derived miR-204 regulates the number of quiescent neural stem cells in the adult brain. EMBO J. 2019, 38, e100481. [Google Scholar] [CrossRef] [PubMed]
- Sirko, S.; Behrendt, G.; Johansson, P.A.; Tripathi, P.; Costa, M.; Bek, S.; Heinrich, C.; Tiedt, S.; Colak, D.; Dichgans, M.; et al. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. [corrected]. Cell Stem Cell 2013, 12, 426–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]







PublisheR′s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez-Gonzalez, R.; Koupourtidou, C.; Lepko, T.; Zambusi, A.; Novoselc, K.T.; Durovic, T.; Aschenbroich, S.; Schwarz, V.; Breunig, C.T.; Straka, H.; et al. Innate Immune Pathways Promote Oligodendrocyte Progenitor Cell Recruitment to the Injury Site in Adult Zebrafish Brain. Cells 2022, 11, 520. https://doi.org/10.3390/cells11030520
Sanchez-Gonzalez R, Koupourtidou C, Lepko T, Zambusi A, Novoselc KT, Durovic T, Aschenbroich S, Schwarz V, Breunig CT, Straka H, et al. Innate Immune Pathways Promote Oligodendrocyte Progenitor Cell Recruitment to the Injury Site in Adult Zebrafish Brain. Cells. 2022; 11(3):520. https://doi.org/10.3390/cells11030520
Chicago/Turabian StyleSanchez-Gonzalez, Rosario, Christina Koupourtidou, Tjasa Lepko, Alessandro Zambusi, Klara Tereza Novoselc, Tamara Durovic, Sven Aschenbroich, Veronika Schwarz, Christopher T. Breunig, Hans Straka, and et al. 2022. "Innate Immune Pathways Promote Oligodendrocyte Progenitor Cell Recruitment to the Injury Site in Adult Zebrafish Brain" Cells 11, no. 3: 520. https://doi.org/10.3390/cells11030520