
The GABA Polarity Shift and Bumetanide Treatment: Making Sense Requires Unbiased and Undogmatic Analysis



Abstract
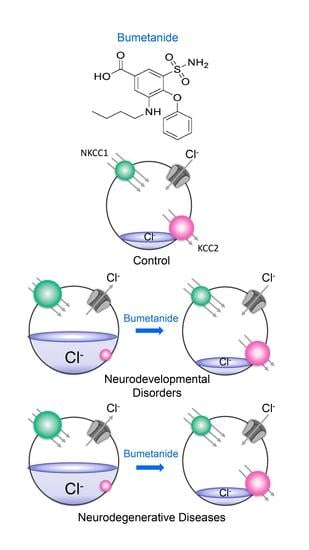
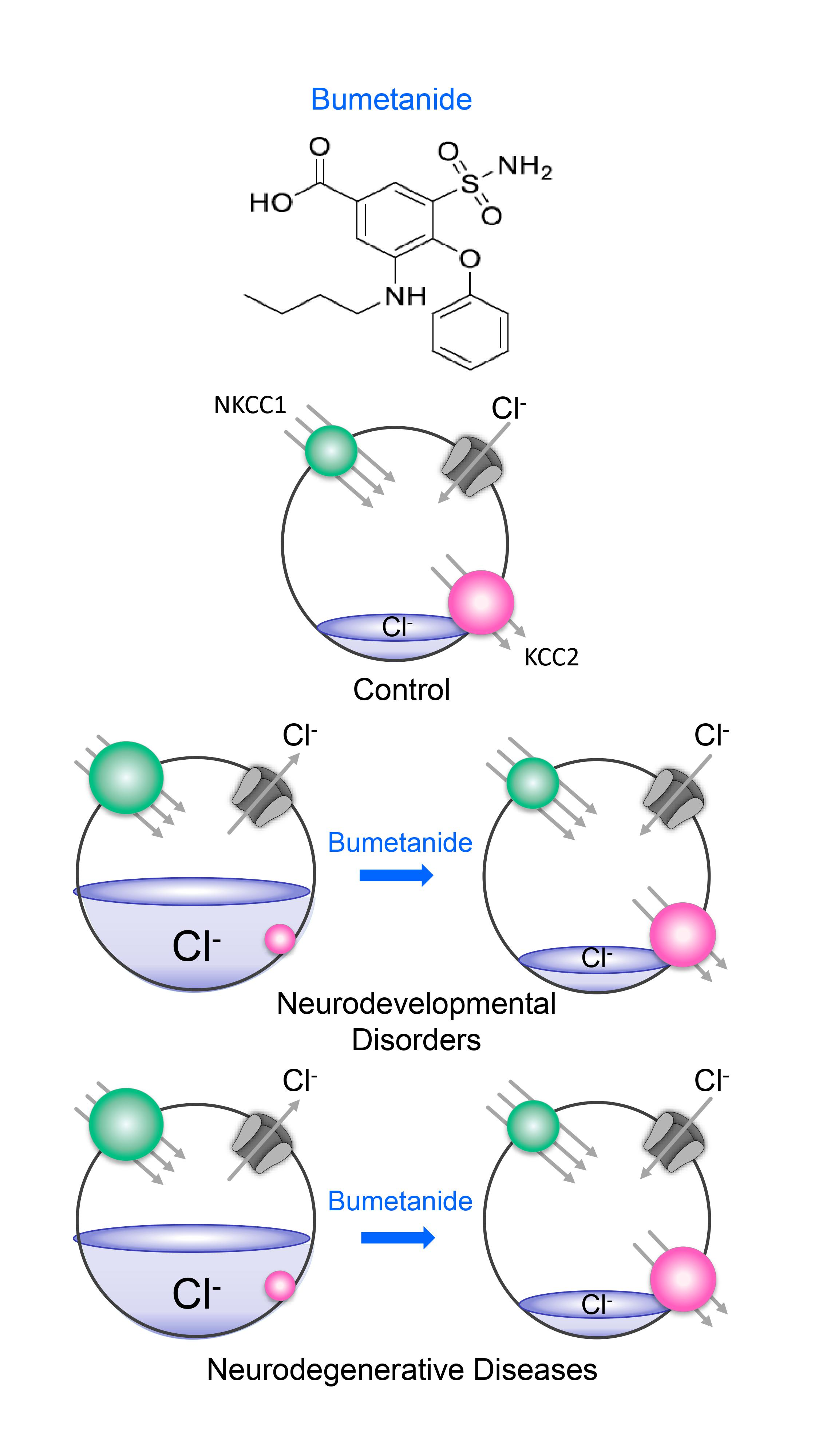
:
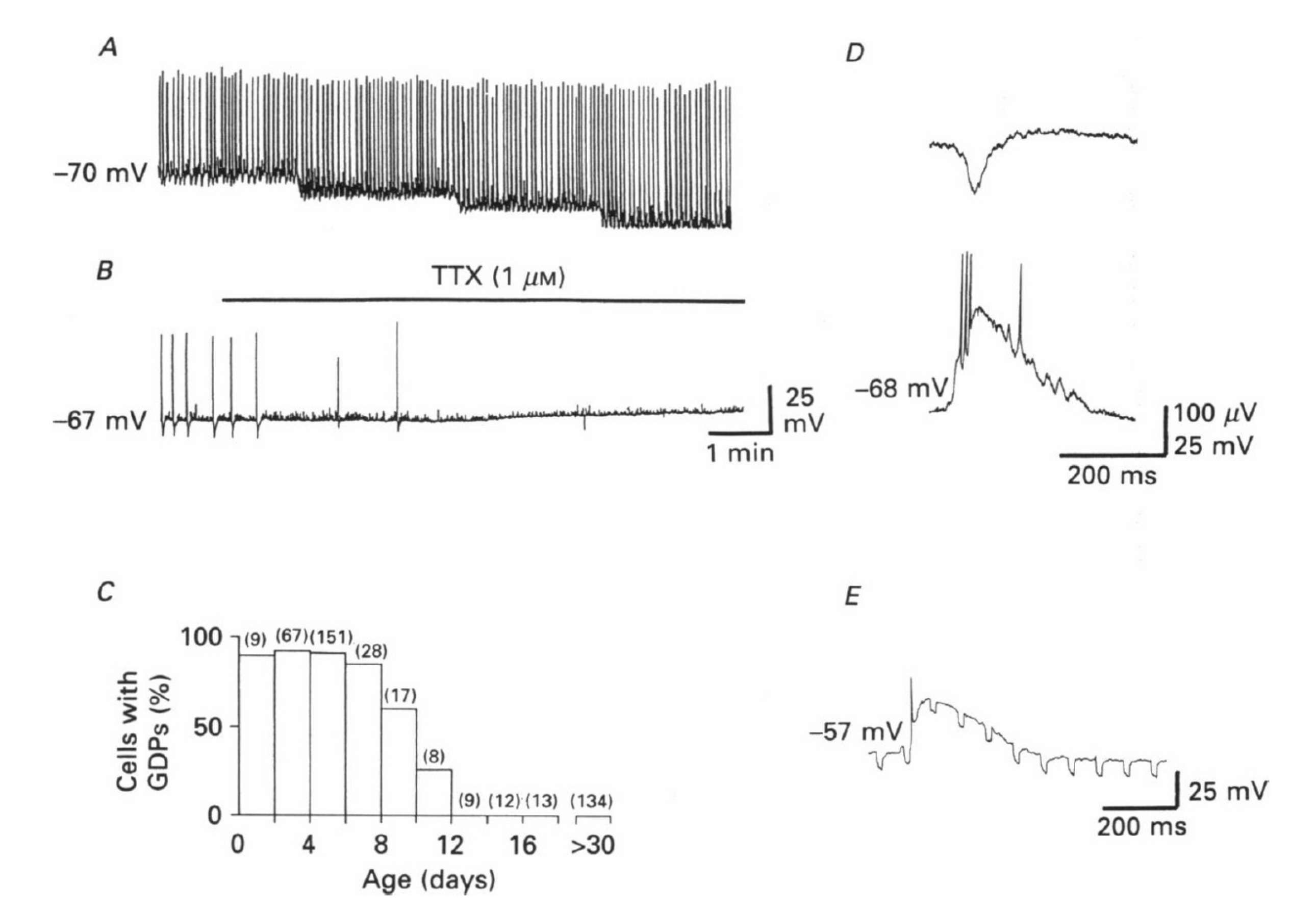
1. GABA Polarity Shift: A Brief Historical Perspective
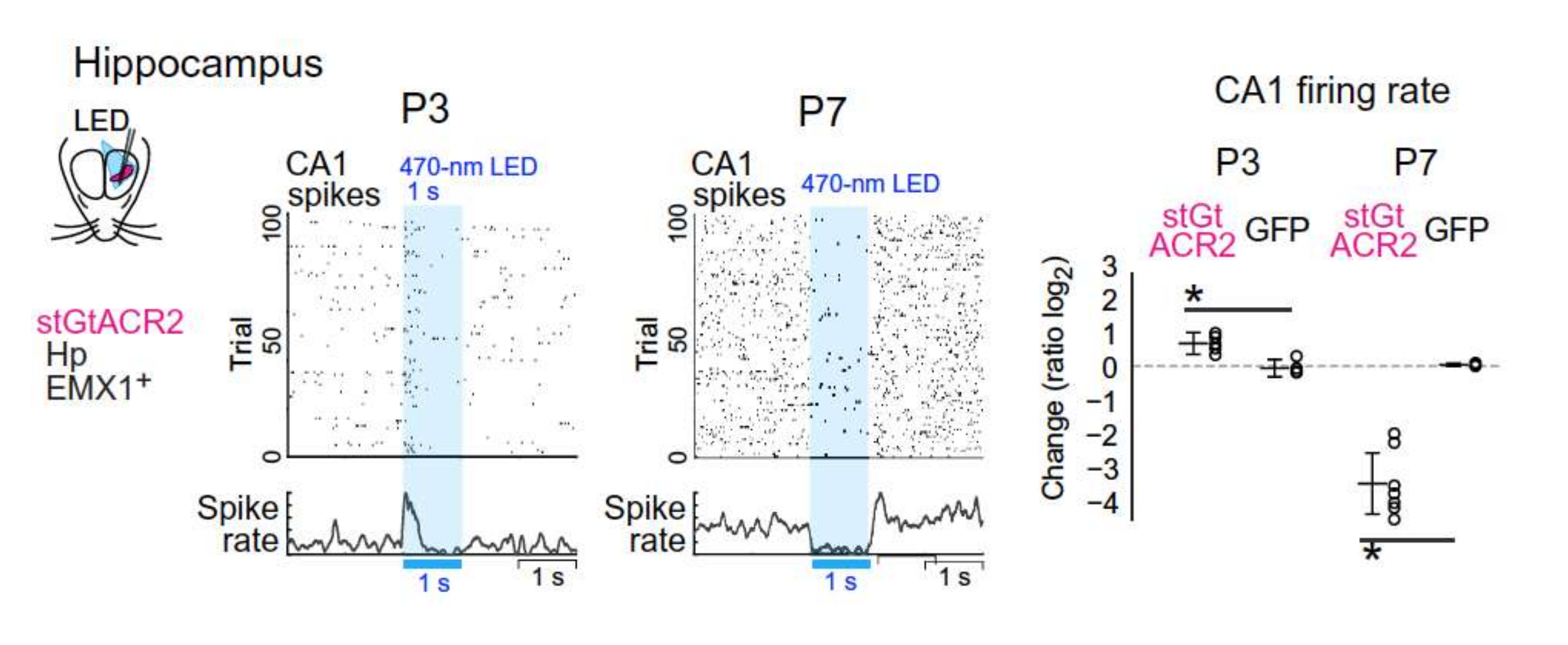
2. Fact 1: The Polarity Shift Is Not Anymore Debated
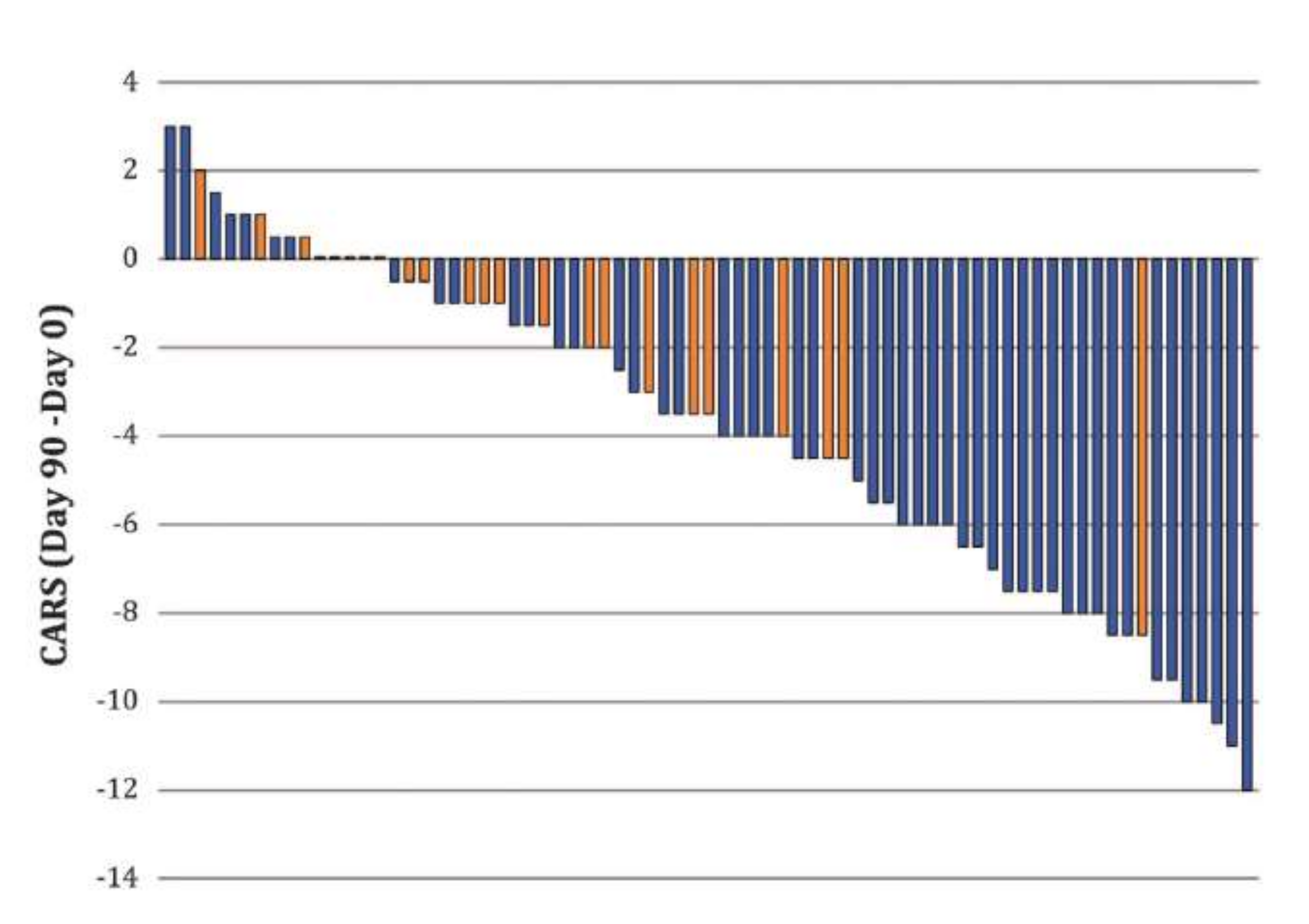
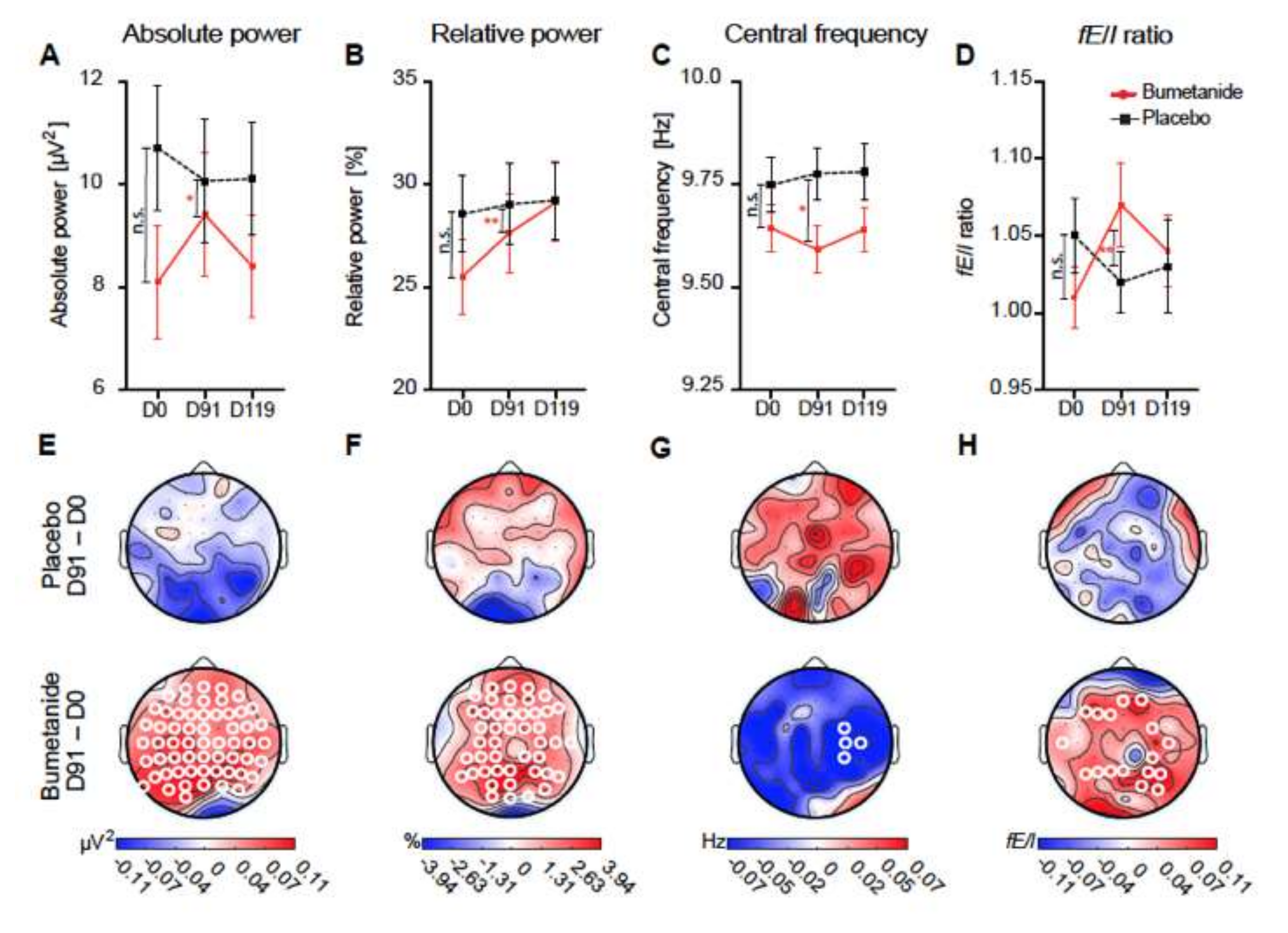
3. Fact 2: Reducing High [Cl−]i Levels with Bumetanide Has Beneficial Effects in Several Neurological and Psychiatric Disorders
4. Fact 3: The Side Effects of Bumetanide Are Well Controlled and Limited
5. Conclusions: Future Directions Will Have to Rely on Many Avenues
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kursan, S.; McMillen, T.; Beesetty, P.; Dias-Junior, E.; Almutairi, M.M.; Sajib, A.A.; Kozak, J.A.; Aguilar-Bryan, L.; Di Fulvio, M. The neuronal K+Cl− co-transporter 2 (Slc12a5) modulates insulin secretion. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, A.; Taube, J.; Schwartzkroin, P. Development of hyperpolarizing inhibitory postsynaptic potentials and hyperpolarizing response to gamma-aminobutyric acid in rabbit hippocampus studied in vitro. J. Neurosci. 1984, 4, 860–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, K.M.; Teyler, T.J. Evidence for late development of inhibition in area CA1 of the rat hippocampus. Brain Res. 1983, 268, 339–343. [Google Scholar] [CrossRef]
- Dunwiddie, T.V. Age-Related Differences in the in vitro Rat Hippocampus. Dev. Neurosci. 1981, 4, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Gaiarsa, J.-L.; Tyzio, R.; Khazipov, R. GABA: A Pioneer Transmitter That Excites Immature Neurons and Generates Primitive Oscillations. Physiol. Rev. 2007, 87, 1215–1284. [Google Scholar] [CrossRef]
- Kasyanov, A.M.; Safiulina, V.F.; Voronin, L.L.; Cherubini, E. From The Cover: GABA-mediated giant depolarizing potentials as coincidence detectors for enhancing synaptic efficacy in the developing hippocampus. Proc. Natl. Acad. Sci. USA 2004, 101, 3967–3972. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Kriegstein, A.R. A GABAergic projection from the zona incerta to cortex promotes cortical neuron development. Science 2015, 350, 554–558. [Google Scholar] [CrossRef] [Green Version]
- Ben-Ari, Y. Excitatory actions of gaba during development: The nature of the nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef]
- Mohajerani, M.H.; Sivakumaran, S.; Zacchi, P.; Aguilera, P.; Cherubini, E. Correlated network activity enhances synaptic efficacy via BDNF and the ERK pathway at immature CA3 CA1 connections in the hippocampus. Proc. Natl. Acad. Sci. USA 2007, 104, 13176–13181. [Google Scholar] [CrossRef] [Green Version]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Cherubini, E.; Corradetti, R.; Gaiarsa, J.-L. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 1989, 416, 303–325. [Google Scholar] [CrossRef] [PubMed]
- Taubes, A.; Nova, P.; Zalocusky, K.A.; Kosti, I.; Bicak, M.; Zilberter, M.Y.; Hao, Y.; Yoon, S.Y.; Oskotsky, T.; Pineda, S.; et al. Experimental and real-world evidence supporting the computational repurposing of bumetanide for APOE4-related Alzheimer’s disease. Nat. Aging 2021, 1, 932–947. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Woodin, M.A.; Sernagor, E.; Cancedda, L.; Vinay, L.; Rivera, C.; Legendre, P.; Luhmann, H.J.; Bordey, A.; Wenner, P.; et al. Refuting the challenges of the developmental shift of polarity of GABA actions: GABA more exciting than ever! Front. Cell Neurosci. 2012, 6, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruusuvuori, E.; Kirilkin, I.; Pandya, N.; Kaila, K. Spontaneous Network Events Driven by Depolarizing GABA Action in Neonatal Hippocampal Slices are Not Attributable to Deficient Mitochondrial Energy Metabolism. J. Neurosci. 2010, 30, 15638–15642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valeeva, G.; Tressard, T.; Mukhtarov, M.; Baude, A.; Khazipov, R. An Optogenetic Approach for Investigation of Excitatory and Inhibitory Network GABA Actions in Mice Expressing Channelrhodopsin-2 in GABAergic Neurons. J. Neurosci. 2016, 36, 5961–5973. [Google Scholar] [CrossRef]
- Kirmse, K.; Kummer, M.; Kovalchuk, Y.; Witte, O.W.; Garaschuk, O.; Holthoff, K. GABA depolarizes immature neurons and inhibits network activity in the neonatal neocortex in vivo. Nat. Commun. 2015, 6, 7750. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.S.; Artoni, P.; Landi, S.; Cozzolino, O.; Parra, R.; Pracucci, E.; Trovato, F.; Szczurkowska, J.; Luin, S.; Arosio, D.; et al. Simultaneous two-photon imaging of intracellular chloride concentration and pH in mouse pyramidal neurons in vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E8770–E8779. [Google Scholar] [CrossRef] [Green Version]
- Potez, S.; Larkum, M. Effect of Common Anesthetics on Dendritic Properties in Layer 5 Neocortical Pyramidal Neurons. J. Neurophysiol. 2008, 99, 1394–1407. [Google Scholar] [CrossRef]
- Murata, Y.; Colonnese, M.T. GABAergic interneurons excite neonatal hippocampus in vivo. Sci. Adv. 2020, 6, eaba1430. [Google Scholar] [CrossRef]
- Tyzio, R.; Cossart, R.; Khalilov, I.; Minlebaev, M.; Hübner, C.A.; Represa, A.; Ben-Ari, Y.; Khazipov, R. Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science 2006, 314, 1788–1792. [Google Scholar] [CrossRef] [Green Version]
- Spoljaric, A.; Seja, P.; Spoljaric, I.; Virtanen, M.A.; Lindfors, J.; Uvarov, P.; Summanen, M.; Crow, A.K.; Hsueh, B.; Puskarjov, M.; et al. Vasopressin excites interneurons to suppress hippocampal network activity across a broad span of brain maturity at birth. Proc. Natl. Acad. Sci. USA 2017, 114, E10819–E10828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Ari, Y. Oxytocin and Vasopressin, and the GABA Developmental Shift during Labor and Birth: Friends or Foes? Front Cell Neurosci. 2018, 12, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyzio, R.; Represa, A.; Jorquera, I.; Ben-Ari, Y.; Gozlan, H.; Aniksztejn, L. The Establishment of GABAergic and Glutamatergic Synapses on CA1 Pyramidal Neurons is Sequential and Correlates with the Development of the Apical Dendrite. J. Neurosci. 1999, 19, 10372–10382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyzio, R.; Ivanov, A.; Bernard, C.; Holmes, G.L.; Ben-Ari, Y.; Khazipov, R. Membrane Potential of CA3 Hippocampal Pyramidal Cells During Postnatal Development. J. Neurophysiol. 2003, 90, 2964–2972. [Google Scholar] [CrossRef] [Green Version]
- Lozovaya, N.; Nardou, R.; Tyzio, R.; Chiesa, M.; Pons-Bennaceur, A.; Eftekhari, S.; Bui, T.-T.; Billon-Grand, M.; Rasero, J.; Bonifazi, P.; et al. Early alterations in a mouse model of Rett syndrome: The GABA developmental shift is abolished at birth. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Tyzio, R.; Nardou, R.; Ferrari, D.C.; Tsintsadze, T.; Shahrokhi, A.; Eftekhari, S.; Khalilov, I.; Tsintsadze, V.; Brouchoud, C.; Chazal, G.; et al. Oxytocin-Mediated GABA Inhibition during Delivery Attenuates Autism Pathogenesis in Rodent Offspring. Science 2014, 343, 675–679. [Google Scholar] [CrossRef]
- Fernandez, A.; Dumon, C.; Guimond, D.; Tyzio, R.; Bonifazi, P.; Lozovaya, N.; Burnashev, N.; Ferrari, D.C.; Ben-Ari, Y. The GABA Developmental Shift Is Abolished by Maternal Immune Activation Already at Birth. Cereb. Cortex 2018, 29, 3982–3992. [Google Scholar] [CrossRef]
- Leonzino, M.; Busnelli, M.; Antonucci, F.; Verderio, C.; Mazzanti, M.; Chini, B. The timing of the excitatory-to-inhibitory GABA switch is regulated by the oxytocin receptor via KCC2. Cell Rep. 2016, 15, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Modahl, C.; Green, L.A.; Fein, D.; Morris, M.; Waterhouse, L.; Feinstein, C.; Levin, H. Plasma oxytocin levels in autistic children. Biol. Psychiatry 1998, 43, 270–277. [Google Scholar] [CrossRef]
- Feldman, R.; Zagoory-Sharon, O.; Weisman, O.; Schneiderman, I.; Gordon, I.; Maoz, R.; Shalev, I.; Ebstein, R. Sensitive Parenting Is Associated with Plasma Oxytocin and Polymorphisms in the OXTR and CD38 Genes. Biol. Psychiatry 2012, 72, 175–181. [Google Scholar] [CrossRef]
- Parker, K.J.; Oztan, O.; Libove, R.A.; Sumiyoshi, R.D.; Jackson, L.P.; Karhson, D.S.; Summers, J.E.; Hinman, K.E.; Motonaga, K.S.; Phillips, J.M.; et al. Intranasal oxytocin treatment for social deficits and biomarkers of response in children with autism. Proc. Natl. Acad. Sci. USA 2017, 114, 8119–8124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardou, R.; Yamamoto, S.; Chazal, G.; Bhar, A.; Ferrand, N.; Dulac, O.; Ben-Ari, Y.; Khalilov, I. Neuronal chloride accumulation and excitatory GABA underlie aggravation of neonatal epileptiform activities by phenobarbital. Brain 2011, 134, 987–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huberfeld, G.; Wittner, L.; Clemenceau, S.; Baulac, M.; Kaila, K.; Miles, R.; Rivera, C. Perturbed Chloride Homeostasis and GABAergic Signaling in Human Temporal Lobe Epilepsy. J. Neurosci. 2007, 27, 9866–9873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallud, J.; Le Van Quyen, M.; Bielle, F.; Pellegrino, C.; Varlet, P.; Labussiere, M.; Cresto, N.; Dieme, M.-J.; Baulac, M.; Duyckaerts, C.; et al. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci. Transl. Med. 2014, 6, 244ra89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 2017, 40, 536–554. [Google Scholar] [CrossRef] [PubMed]
- Kaila, K.; Price, T.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef] [Green Version]
- Soul, J.S.; Bergin, A.M.; Stopp, C.; Hayes, B.; Singh, A.; Fortuno, C.R.; O’Reilly, D.; Krishnamoorthy, K.; Jensen, F.E.; Rofeberg, V.; et al. A pilot randomized, controlled, double-blind trial of bumetanide to treat neonatal seizures. Ann. Neurol. 2021, 89, 327–340. [Google Scholar] [CrossRef]
- Lemonnier, E.; Degrez, C.; Phelep, M.; Tyzio, R.; Josse, F.; Grandgeorge, M.; Hadjikhani, N.; Ben-Ari, Y. A randomised controlled trial of bumetanide in the treatment of autism in children. Transl. Psychiatry 2012, 2, e202. [Google Scholar] [CrossRef]
- Lemonnier, E.; Ben-Ari, Y. The diuretic bumetanide decreases autistic behaviour in five infants treated during 3 months with no side effects. Acta Paediatratica 2010, 99, 1885–1888. [Google Scholar] [CrossRef]
- Dai, Y.; Zhang, L.; Yu, J.; Zhou, X.; He, H.; Ji, Y.; Wang, K.; Du, X.; Liu, X.; Tang, Y.; et al. Improved symptoms following bumetanide treatment in children aged 3−6 years with autism spectrum disorder: A randomized, double-blind, placebo-controlled trial. Sci. Bull. 2021, 66, 1591–1598. [Google Scholar] [CrossRef]
- Lemonnier, E.; Villeneuve, N.; Sonie, S.; Serret, S.; Rosier, A.; Roué, J.-M.; Brosset, P.; Viellard, M.; Bernoux, D.; Rondeau, S.; et al. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 2017, 7, e1056. [Google Scholar] [CrossRef]
- Fernell, E.; Gustafsson, P.; Gillberg, C. Bumetanide for autism: Open-label trial in six children. Acta Paediatr. 2020, 110, 1548–1553. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Shan, L.; Wang, B.; Li, H.; Xu, Z.; Staal, W.G.; Jia, F. A Pilot Study on the Combination of Applied Behavior Analysis and Bumetanide Treatment for Children with Autism. J. Child Adolesc. Psychopharmacol. 2015, 25, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, C.-C.; Dai, Y.; Luo, Q.; Ji, Y.; Wang, K.; Deng, S.; Yu, J.; Xu, M.; Du, X.; et al. Symptom improvement in children with autism spectrum disorder following bumetanide administration is associated with decreased GABA/glutamate ratios. Transl. Psychiatry 2020, 10, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprengers, J.J.; van Andel, D.M.; Zuithoff, N.P.A.; Keijzer-Veen, M.G.; Schulp, A.J.A.; Scheepers, F.E.; Lilien, M.R.; Oranje, B.; Bruining, H. Bumetanide for Core Symptoms of Autism Spectrum Disorder (BAMBI): A Single Center, Double-Blinded, Participant-Randomized, Placebo-Controlled, Phase-2 Superiority Trial. J. Am. Acad. Child Adolesc. Psychiatry 2020, 60, 865–876. [Google Scholar] [CrossRef]
- Hajri, M.; Amor, A.B.; Abbes, Z.; Dhouib, S.; Ouanes, S.; Mrabet, A.; Daghfous, R.; Bouden, A. Bumetanide in the management of autism. Tunisian experience in Razi Hospital | Le bumétanide dans l’autisme. Expérience pilote du service de pédopsychiatrie de l’hôpital Razi Tunisie. Tuinisie Med. 2019, 97, 971–977. [Google Scholar]
- James, B.; Gales, M.A.; Gales, B.J. Bumetanide for autism spectrum Disorder in children: A review of Randomized controlled trials. Annals of pharmacol. Ann. Pharmacother. 2019, 53, 537–544. [Google Scholar] [CrossRef]
- Wang, T.; Shan, L.; Miao, C.; Xu, Z.; Jia, F. Treatment effect of bumetanide in children with autism spectrum disorder: A systematic review and meta-analysis. Front. Psychiatry 2021, 12, 751575. [Google Scholar] [CrossRef]
- Juarez-Martinez, E.L.; Sprengers, J.J.; Cristian, G.; Oranje, B.; van Andel, D.M.; Avramiea, A.E.; Simpraga, S.; Houtman, S.J.; Hardstone, R.; Gerver, C.; et al. Prediction of Behavioral Improvement through Resting-State Electroencephalography and Clinical Severity in a Randomized Controlled Trial Testing Bumetanide in Autism Spectrum Disorder. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2021. [Google Scholar] [CrossRef]
- Van Andel, D.M.; Sprengers, J.J.; Oranje, B.; Scheepers, F.E.; Jansen, F.E.; Bruining, H. Effects of bumetanide on neurodevelopmental impairments in patients with tuberous sclerosis complex: An open-label pilot study. Mol. Autism 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Hadjikhani, N.; Johnels, J.; Zürcher, N.R.; Lassalle, A.; Guillon, Q.; Hippolyte, L.; Billstedt, E.; Ward, N.; Lemonnier, E.; Gillberg, C. Look me in the eyes: Constraining gaze in the eye-region provokes abnormally high subcortical activation in autism. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hadjikhani, N.; Johnels, J.Å.; Lassalle, A.; Zürcher, N.R.; Hippolyte, L.; Gillberg, C.; Lemonnier, E.; Ben-Ari, Y. Bumetanide for autism: More eye contact, less amygdala activation. Sci. Rep. 2018, 8, 3602. [Google Scholar] [CrossRef] [PubMed]
- Bruining, H.; Passtoors, L.; Goriounova, N.; Jansen, F.; Hakvoort, B.; de Jonge, M.; Poil, S.-S. Paradoxical benzodiazepine response: A rationale for bumetanide in neurodevelopmental disorders? Pediatrics 2015, 136, e539–e543. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W.; Kaila, K. Reply to the commentary by Ben-Ari and Delpire: Bumetanide and neonatal seizures: Fiction versus reality. Epilepsia 2021, 62, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.; Seja, P.; Töllner, K.; Römermann, K.; Hampel, P.; Kalesse, M.; Kipper, A.; Feit, P.W.; Lykke, K.; Toft-Bertelsen, T.L.; et al. Bumepamine, a brain-permeant benzylamine derivative of bumetanide, does not inhibit NKCC1 but is more potent to enhance phenobarbital’s anti-seizure efficacy. Neuropharmacology 2018, 143, 186–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löscher, W.; Puskarjov, M.; Kaila, K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology 2013, 69, 62–74. [Google Scholar] [CrossRef]
- Smit, E.; Liu, X.; Gill, H.; Sabir, H.; Jary, S.; Thoresen, M. Factors Associated with Permanent Hearing Impairment in Infants Treated with Therapeutic Hypothermia. J. Pediatr. 2013, 163, 995–1000. [Google Scholar] [CrossRef]
- Pressler, R.M.; Boylan, G.; Marlow, N.; Blennow, M.; Chiron, C.; Cross, J.H.; de Vries, L.S.; Hallberg, B.; Hellstrom-Westas, L.; Jullien, V.; et al. Bumetanide for the treatment of seizures in newborn babies with hypoxic ischaemic encephalopathy (NEMO): An open-label, dose finding, and feasibility phase 1/2 trial. Lancet Neurol. 2015, 14, 469–477. [Google Scholar] [CrossRef]
- Kealy, J.; Greene, C.; Campbell, M. Blood-brain barrier regulation in psychiatric disorders. Neurosci. Lett. 2018, 726, 133664. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Prasad, S.; Sajja, R.K.; Naik, P.; Cucullo, L. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J. Pharmacovigil. 2014, 2, 125. [Google Scholar] [PubMed]
- Zemkova, H.W.; Bjelobaba, I.; Tomic, M.; Zemkova, H.; Stojilkovic, S.S. Molecular, pharmacological and functional properties of GABAA receptors in anterior pituitary cells. J. Physiol. 2018, 586, 3097–3111. [Google Scholar] [CrossRef] [PubMed]
- Iwata, K.; Matsuzaki, H.; Miyachi, T.; Shimmura, C.; Suda, S.; Tsuchiya, K.J.; Matsumoto, K.; Suzuki, K.; Iwata, Y.; Nakamura, K.; et al. Investigation of the serum levels of anterior pituitary hormones in male children with autism. Mol. Autism 2011, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caly, H.; Rabiei, H.; Coste-Mazeau, P.; Hantz, S.; Alain, S.; Eyraud, J.-L.; Chianea, T.; Caly, C.; Makowski, D.; Hadjikhani, N.; et al. Machine learning analysis of pregnancy data enables early identification of a subpopulation of newborns with ASD. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Neuro-archaeology: Pre-symptomatic architecture and signature of neurological disorders. Trends Neurosci. 2008, 31, 626–636. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country | N. | Age (y) | Rating Scale | Dose | Duration | End Points | Side Effects | References |
|---|---|---|---|---|---|---|---|---|
| China | 119 | 3–6 | CARS, ADOS, CGI, SRS | 0.5 mg twice/day | 3 months | Improvement in CARS score | mild (polyuria, hypokalemia) | [40] |
| Sweden | 6 | 3–14 | CARS | 0.5 mg twice/day | 4-12 weeks | Improvement in CARS score | mild (polyuria) | [42] |
| Netherland | 92 | 7–15 | SRS2 | 0.5 mg twice/day | 3 months | Improvement in repetitive behavioral scale But not SRS2 | mild (hypokalemia) | [45] |
| China | 83 | 3–6 | CARS, ADOS, CGI | 0.5 mg twice/day | 3 months | Reduction in CARS score, CGI-I | mild (polyuria) | [44] |
| Netherland | 15 | 8–21 | ABC-I (TSC) | 0.5 mg twice/day | 3 months | Improvement in ABC-I score EEG | mild (hypokalemia) | [50] |
| Tunisia | 29 | Average 7.9 | ADI-R, CARS, CGI | 0.1 mg/day | 12 months | Improvement in CARS score | mild (hypokalemia) | [46] |
| France | 9 | Average 21.4 | Eye tracking | 1 mg/day | 10 months | fMRI | None | [51] |
| France | 88 | 2–18 | CARS, SRS, CGI | 0.5–2 mg twice/day | 3 months | Improvement in CARS, CGI, SRS score | mild (hypokalemia) | [41] |
| China | 60 | Average 4.5 | ABC, CARS, CGI | 0.5 mg twice/day | 3 months | Improvement in ABC, CARS, CGI score | None | [43] |
| France | 7 | Average 19.3 | ADOS, fMRI emotion recognition | 1 mg/day | 10 months | Improvement performance for emotion recognition | mild (polyuria) | [51] |
| France | 60 | 3–11 | CARS, SRS, ADOS | 1 mg/day | 3 months | Improvement in CARS, ADOS score | mild (hypokalemia) | [38] |
| France | 5 | 3–11 | CARS, ABC, CGI, RDEG, RRB | 1mg/day | 3months | Improvement in CARS, CGI, | None | [39] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben-Ari, Y.; Cherubini, E. The GABA Polarity Shift and Bumetanide Treatment: Making Sense Requires Unbiased and Undogmatic Analysis. Cells 2022, 11, 396. https://doi.org/10.3390/cells11030396
Ben-Ari Y, Cherubini E. The GABA Polarity Shift and Bumetanide Treatment: Making Sense Requires Unbiased and Undogmatic Analysis. Cells. 2022; 11(3):396. https://doi.org/10.3390/cells11030396
Chicago/Turabian StyleBen-Ari, Yehezkel, and Enrico Cherubini. 2022. "The GABA Polarity Shift and Bumetanide Treatment: Making Sense Requires Unbiased and Undogmatic Analysis" Cells 11, no. 3: 396. https://doi.org/10.3390/cells11030396