
Towards a Better Vision of Retinoic Acid Signaling during Eye Development


Development, Aging, and Regeneration Program, Sanford Burnham Prebys Medical Discovery Institute, 10901 N. Torrey Pines Road, La Jolla, CA 92037, USA
Cells 2022, 11(3), 322; https://doi.org/10.3390/cells11030322
Submission received: 24 December 2021
/
Revised: 11 January 2022
/
Accepted: 17 January 2022
/
Published: 19 January 2022
(This article belongs to the Special Issue Retinoic Acid and Retinoid X Receptors)
Abstract
:Retinoic acid (RA) functions as an essential signal for development of the vertebrate eye by controlling the transcriptional regulatory activity of RA receptors (RARs). During eye development, the optic vesicles and later the retina generate RA as a metabolite of vitamin A (retinol). Retinol is first converted to retinaldehyde by retinol dehydrogenase 10 (RDH10) and then to RA by all three retinaldehyde dehydrogenases (ALDH1A1, ALDH1A2, and ALDH1A3). In early mouse embryos, RA diffuses to tissues throughout the optic placode, optic vesicle, and adjacent mesenchyme to stimulate folding of the optic vesicle to form the optic cup. RA later generated by the retina is needed for further morphogenesis of the optic cup and surrounding perioptic mesenchyme; loss of RA at this stage leads to microphthalmia and cornea plus eyelid defects. RA functions by binding to nuclear RARs at RA response elements (RAREs) that either activate or repress transcription of key genes. Binding of RA to RARs regulates recruitment of transcriptional coregulators such as nuclear receptor coactivator (NCOA) or nuclear receptor corepressor (NCOR), which in turn control binding of the generic coactivator p300 or the generic corepressor PRC2. No genes have been identified as direct targets of RA signaling during eye development, so future studies need to focus on identifying such genes and their RAREs. Studies designed to learn how RA normally controls eye development in vivo will provide basic knowledge valuable for determining how developmental eye defects occur and for improving strategies to treat eye defects.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Retinol, carried in the bloodstream by retinol-binding protein 4 (RBP4), is widely distributed in adult and embryonic tissues, and the membrane protein STRA6 facilitates cellular uptake of retinol carried by RBP4 [1]. However, the mechanism of RA signaling is dependent upon conversion of retinol to RA specifically in cells that possess RA-generating enzymes whose genes are expressed in a tissue-specific manner (Figure 1). During mouse eye development, retinol is oxidized to retinaldehyde by retinol dehydrogenase 10 (RDH10) expressed by Rdh10 [2], and retinaldehyde is oxidized to RA by retinaldehyde dehydrogenases (RALDHs, i.e., ALDH1A1, ALDH1A2, and ALDH1A3) expressed by the Aldh1a1, Aldh1a2, and Aldh1a3 genes [3,4,5]. RA released by RA-generating cells is taken up by surrounding target cells. RA controls transcription of key genes by regulating the activity of nuclear RA receptors (RARs), i.e., RARA, RARB, RARG encoded by Rara, Rarb, and Rarg in mouse. In order to regulate genes, RA binds to RARs that are bound to RA response elements (RAREs) as a heterodimer with retinoid X receptors (RXRs) [6,7]. Binding of RA to the RAR portion of the RAR-RXR heterodimer alters recruitment of nuclear receptor coactivators (NCOAs) known to activate transcription, or nuclear receptor corepressors (NCORs) known to repress transcription; also altered is recruitment of the p300 general coactivator or the PRC2 general corepressor [8,9,10]. Thus, RA regulates transcriptional activation or repression through RARE enhancers or RARE silencers [11].
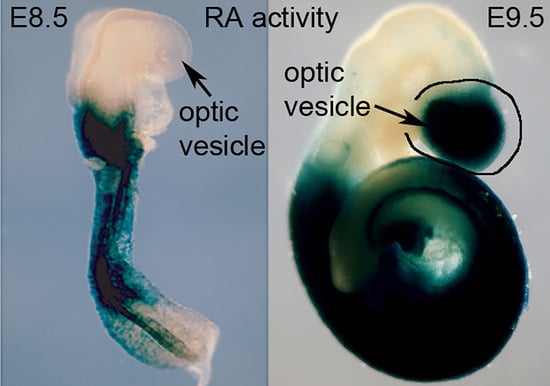
RA controls critical functions during eye development in humans, mice, and zebrafish [12,13,14,15,16]. Human studies have associated mutations in four components of the RA signaling pathway (RBP4, STRA6, ALDH1A3, RARB) and two genes up-regulated by RA (PITX2, FOXC1) with anophthalmia/microphthalmia [12,13,14,15,16,17]. However, studies on mouse embryos will likely identify additional RA target genes. In mouse embryos, Rdh10 is expressed at E8.5 in optic mesenchyme and at E9.5 onwards in the optic vesicle/cup [2]. Aldh1a1 (Raldh1) is expressed in the dorsal retina from E9.5 onwards, Aldh1a2 (Raldh2) is expressed in the optic mesenchyme from only E8.5 to E9.5, and Aldh1a3 (Raldh3) is expressed in the ventral retina from E8.5 onwards [18]. RA is required for folding of the optic vesicle to form the optic cup and ventral retina as shown in Rdh10-/- embryos [2] and in Aldh1a1/ldh1a2/Aldh1a3 triple knockouts [18]. Aldh1a1/Aldh1a3 double knockouts form the optic cup (due to early Aldh1a2 RA synthesis), but they exhibit excessive neural crest-derived perioptic mesenchyme growth and anterior segment defects (cornea and eyelid), and Pitx2 and Foxc1 were found to be down-regulated in perioptic mesenchyme [18,19]. Pitx2 and Foxc1 knockouts exhibit anterior segment eye defects, but as they still generate the optic cup [20,21], it is likely that RA regulates other genes very early needed for optic cup formation.
2. Requirement of Retinoic Acid for Optic Cup Formation
During the earliest stage of eye development which begins at approximately E8.5 in mouse, the optic vesicles are formed as out-pocketings of the forebrain. Soon after that, the portion of the optic vesicle nearest to the head surface ectoderm invaginates back towards the forebrain to form an optic cup with a connection to the forebrain that will become the optic stalk. Early studies designed to reduce RA signaling, such as vitamin A deficiency in rat [22] and RAR double knockouts in mouse [23], showed that although the optic vesicle forms and undergoes optic cup formation which is complete at E10.5 in mouse, RA is required afterwards for further optic cup morphogenesis and proper formation of anterior eye structures adjacent to the optic cup such as cornea and eyelids. Although three RAR genes exist, which are all expressed in the early eye, triple RAR knockout studies have not been reported. However, RA function in mouse can also be examined genetically by knockout studies targeting RA-generating enzymes. At E9.5, when the optic vesicle begins to undergo optic cup formation, all three ALDH1A RA-generating enzymes are expressed, with Aldh1a1 in the dorsal optic vesicle, Aldh1a2 in the optic mesenchyme, and Aldh1a3 in the ventral optic vesicle [18]. This triple redundancy, with Aldh1a2-/- embryos being stunted and not surviving beyond E8.5 [24], and with Aldh1a3-/- embryos not surviving past birth [4], thus hampers genetic studies to examine eye RA function. However, as Aldh1a1-/- mice survive as adults and are fertile [3], it was possible to generate adult mice that are Aldh1a1-/-/Aldh1a2+/-/Aldh1a3+/- and perform matings to generate E10.5 Aldh1a1/Aldh1a2/Aldh1a3 triple knockout embryos (ratio of 1 in 16) to study optic cup formation in the absence of RA activity. In order to generate E10.5 embryos to study optic cup formation, this required treatment of the mother with one low dose of RA at E7.5 to prevent early lethality at E8.5 due to Aldh1a2-/- [25]; at E10.5 it was observed that all RA activity in the optic field was missing in the triple knockout and the optic cup did not form in the triple knockout while the eye was normal in control Aldh1a2-/- embryos due to the exogenous RA rescue performed at E7.5 plus the endogenous RA generated in the optic vesicles by Aldh1a1 and Aldh1a3 [18] (Figure 2). Thus, RA is required for invagination of the optic vesicle to form the optic cup; in particular, invagination of the ventral portion of the optic vesicle is inhibited by loss of RA signaling while some degree of dorsal invagination is still observed (Figure 2).
Subsequent to these mouse genetic loss-of-function studies, not much progress has been made on the mechanism through which RA controls optic cup formation. No direct RA target genes have been identified in the optic vesicle due to the difficulty of obtaining Aldh1a1/Aldh1a2/Aldh1a3 triple knockouts [18]. Fortunately, RDH10 was found to be the only retinol dehydrogenase for the first step of RA synthesis in the eye (conversion of retinol to retinaldehyde); Rdh10-/- embryos survive to E10.5, have no RA activity detected in the optic field, and fail to undergo optic cup formation with loss of ventral invagination [2], similar to that observed in Aldh1a1/Aldh1a2/Aldh1a3 triple knockouts. Further studies on Rdh10-/- embryos should allow progress to be made on the mechanism through which RA controls optic cup formation.
3. Requirement of Retinoic Acid for Morphogenesis of Anterior Eye Structures
Following optic cup formation at E10.5 in mouse, RA continues to be required for several stages of eye morphogenesis that result in maintenance of optic cup morphology and formation of anterior eye structures, particularly the cornea and eyelids. This later function of RA in eye morphogenesis was initially discovered by analysis of RAR double knockouts [23] and also by analysis of the RAR-alpha/RXR-alpha double knockout, thus revealing a requirement for RXR in eye RA signaling [26]. During these later stages, RA is generated only by Aldh1a1 expressed in the dorsal retina and Aldh1a3 expressed in the ventral retina as Aldh1a2 expression is no longer observed after E9.5 [18]. Whereas Aldh1a1 knockouts revealed no obvious eye defects due to compensation by Aldh1a3, and Aldh1a3 knockouts exhibited only mild eye defects due to compensation by Aldh1a1, Aldh1a1/Aldh1a3 double knockout embryos examined at E14.5 exhibited severe eye defects [18,19]. Interestingly, the Aldh1a1/Aldh1a3 double knockout revealed that RA signaling is not required for formation of the retina or establishment or maintenance of dorsoventral patterning in the retina which had originally been proposed based on the dorsal and ventral expression patterns of Aldh1a1 and Aldh1a3 [18]. Instead, Aldh1a1/Aldh1a3 double knockouts revealed that RA generated in the retina acts by diffusing outside of the optic cup to the nearby neural crest-derived perioptic mesenchyme to maintain optic cup shape and to control anterior eye formation [18,19]. RA was found to limit anterior invasion of perioptic mesenchyme during formation of corneal mesenchyme and eyelids which both show mesenchymal overgrowth in the Aldh1a1/Aldh1a3 double knockout; such mesenchymal overgrowth also exerts mechanical force that results in abnormal optic cup morphology and position within the head [18,19]. These defects are consistent with the observation that loss of RA signaling reduced expression of Pitx2 and Foxc1 in perioptic mesenchyme [19]. These studies demonstrated that RA-generating enzymes function cell-nonautonomously to generate paracrine RA signals that guide eye morphogenetic movements in neighboring cells.
As the Aldh1a3 knockout dies at birth, no studies were possible in the adult eye [4]. The initial studies on Aldh1a1 knockout mice that survive to adulthood found no major eye defects in embryos and adults likely due to compensating expression of Aldh1a3 in the ventral retina [3]. However, subsequent studies on the adult Aldh1a1 knockout eye demonstrated a reduction in dorsal choroidal vascular development [27]; ventral choroidal development was normal in the Aldh1a1 knockout presumably due to the action of Aldh1a3 in the ventral retina.
RA also controls zebrafish eye development by activating Pitx2 in neural crest-derived perioptic mesenchyme [28]. Additional mouse knockout studies confirmed that Pitx2-/- eyes exhibit excessive perioptic mesenchyme growth, plus Pitx2 up-regulates Dkk2 (encoding a WNT antagonist) in perioptic mesenchyme and Dkk2-/- eyes have a similar eye defect [20,29]. Further Aldh1a1/Aldh1a3 double knockout studies showed that loss of RA generated in the retina results in loss of both Pitx2 and Dkk2 expression in perioptic mesenchyme plus increased expression of Wnt5a, thus demonstrating that RA activates Pitx2 which then activates Dkk2 to suppress WNT signaling and thus limit perioptic mesenchyme growth [30] (Figure 3).
Pitx2 was found to possess a nearby RARE that is able to recruit RARs in eye ChIP studies, suggesting it may be a direct RA target gene important for anterior eye morphogenesis [30]. However, there has been no success in identifying any RA target gene for optic cup formation, and it remains unclear if Pitx2 and Foxc1 are direct RA target genes for later eye development or whether other late targets exist. Identification of direct RA target genes is difficult as thousands of RAREs are observed in the mouse and human genomes [32,33] plus the expression of thousands of genes is altered when RA is lost or added for any tissue or cell line examined [34,35]. In order to fully understand eye RA signaling, it will be essential to identify direct RA target genes that are activated or repressed in specific regions of the developing eye.
4. Identification of RA Direct Target Genes and Essential RAREs during Eye Formation
Identification of direct transcriptional targets of RA is difficult as loss or gain of RA signaling activity alters the expression of thousands of genes in cell lines or animals [34,35], with most genes probably being indirect targets of RA or regulated post-transcriptionally. As RA transcriptional control is associated with RAREs, identification of RAREs has been employed to identify direct RA target genes. A common approach is to find DNA sequences near RA-activated genes matching the RARE consensus that can activate transcription in reporter transgenes either in cell lines or transgenic animals. However, RAR chromatin immunoprecipitation (ChIP) analysis of mouse embryoid bodies found ~14,000 potential RAREs in the mouse genome [32,33]. Thus, it remains unclear which of these RAREs are needed to regulate genes during development since just a few of these RAREs have been shown to have specific enhancer activity in transgenic animals [7], and only three RAREs have been shown to result in developmental defects when deleted in mouse, i.e., the RARE enhancer activating Hoxa1 in the hindbrain [36], a RARE enhancer activating Cdx1 in the spinal cord [37], and a RARE silencer that represses caudal Fgf8 in the developing trunk [38]. In one case, a RARE reported within intron 2 of Tbx5 was reported to play a role in activation of Tbx5 in the forelimb based on enhancer reporter transgene activity and RAR binding studies [39]; however, this RARE was shown to be unnecessary for Tbx5 expression and forelimb budding when subjected to CRISPR deletion analysis by our laboratory [40]. A potentially redundant Tbx5 forelimb enhancer was identified by others using an enhancer reporter transgene [41], but CRISPR studies showed that this enhancer is also unnecessary, plus a double knockout also had no affect showing the two enhancers are not redundant [40]. Thus, knockouts of RAREs and other types of DNA control elements sometimes result in validation of a required function; however, in many cases, presumed enhancers are nonessential. The disconnect is due to the fact that enhancer reporter transgenes are generated by joining a potential enhancer to a basal promoter upstream of a marker gene, then randomly inserting this into the genome; in this case, the enhancer is removed from its endogenous location and inserted into a foreign location close to a basal promoter, instead of within its normal location containing the gene and promoter it is proposed to control. It remains to be determined whether nonessential enhancers identified in transgene studies are redundant with yet more enhancers or whether they are pseudoenhancers not able to regulate expression of nearby genes [42].
These recent studies show that methods other than traditional transgenes are required to predict enhancers. Additionally, as transgenes are not generally useful to predict silencers, other methods are required to identify silencers. Genomic and epigenetic methods would be superior to the one gene at a time approach in order to allow global identification of important control elements. Advances in epigenetics have revealed chromatin modifications (such as H3K27ac associated with gene activation) and H3K27me3 (associated with gene repression) near transcription factor binding sites required for either gene activation (enhancers) or gene repression (silencers) [43,44,45]. The distances between genes and enhancers/silencers can be large, but these interactions occur within constrained chromatin domains known as topologically associated domains (TADs) that contain an average of 880 kb of DNA for ~2200 TADs [46]. TAD locations are conserved across cell types and mammalian species [46] and they are required for normal gene-enhancer interactions [47,48]. Therefore, RARE enhancers/silencers identified during optic cup formation and later eye morphogenesis will most likely control genes within the same TAD. Such RARE enhancers/silencers may be associated with H3K27ac or H3K27me3 marks controlled by RA signaling. In fact, recent studies have shown that identification of RA direct target genes can be accomplished by identifying genes with significant decreases or increases in expression when RA is missing that also have nearby RA-regulated deposition of H3K27ac (gene activation mark) or H3K27me3 (gene repression mark) associated with RAREs [11]. Such studies performed on trunk tissue from mouse E8.5 wild-type and Aldh1a2-/- embryos lacking RA synthesis resulted in the ability to take an RNA-seq list of 4298 genes shown to have significantly altered expression when RA is lost and reduce this down to 93 genes that also have RA-regulated deposition of nearby H3K27ac or H3K27me3 marks discovered using ChIP-seq, thus suggesting RA regulates their transcription. DNA sequence analysis of such RA-regulated H3K27ac/H3K27me3 ChIP-seq peaks revealed that 45 contain RAREs, providing evidence for the nearby genes being directly regulated by RA at the transcriptional level. This approach was validated by finding RAREs already known to be needed for development by previous knockout studies, i.e., deletions of Hoxa1, Cdx1, and Fgf8 RAREs as described above. Many new candidates for direct RA target genes were found and knockouts of some (Nr2f1, Nr2f2, Meis1, Meis2) showed they are required for body axis or limb formation [11]. With this proof-of principle, similar studies on wild-type vs. RA-deficient eyes can be used to identify direct target genes for RA during eye formation. Such studies will provide vital information on the mechanisms utilized by RA to control transcription in the eye and will identify gene regulatory networks during eye formation. This knowledge will help determine how eye defects occur, identify new genes or enhancers/silencers that may be mutational targets causing human eye defects, and improve strategies to treat eye defects.
Funding
This work was funded by the National Institutes of Health (National Eye Institute) grant R01 EY031745 (G.D.).
Acknowledgments
This work was supported by the Sanford Burnham Prebys Medical Discovery Institute.
Conflicts of Interest
The author declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.L.; Sanderson, B.W.; Moiseyev, G.; Johnson, T.; Mushegian, A.; Young, K.; Rey, J.P.; Ma, J.X.; Staehling-Hampton, K.; Trainor, P.A. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 2007, 21, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Molotkov, A.; Manabe, S.-I.; Donmoyer, C.M.; Deltour, L.; Foglio, M.H.; Cuenca, A.E.; Blaner, W.S.; Lipton, S.A.; Duester, G. Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol. Cell. Biol. 2003, 23, 4637–4648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupé, V.; Matt, N.; Garnier, J.-M.; Chambon, P.; Mark, M.; Ghyselinck, N.B. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc. Natl. Acad. Sci. USA 2003, 100, 14036–14041. [Google Scholar] [CrossRef] [Green Version]
- Mic, F.A.; Molotkov, A.; Molotkova, N.; Duester, G. Raldh2 expression in optic vesicle generates a retinoic acid signal needed for invagination of retina during optic cup formation. Dev. Dyn. 2004, 231, 270–277. [Google Scholar] [CrossRef]
- Rhinn, M.; Dolle, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef] [Green Version]
- Germain, P.; Iyer, J.; Zechel, C.; Gronemeyer, H. Co-regulator recruitment and the mechanism of retinoic acid receptor synergy. Nature 2002, 415, 187–192. [Google Scholar] [CrossRef]
- Perissi, V.; Rosenfeld, M.G. Controlling nuclear receptors: The circular logic of cofactor cycles. Nat. Rev. Mol. Cell Biol. 2005, 6, 542–554. [Google Scholar] [CrossRef]
- Lonard, D.M.; O’Malley, B.W. Nuclear receptor coregulators: Modulators of pathology and therapeutic targets. Nat. Rev. Endocrinol. 2012, 8, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Berenguer, M.; Meyer, K.F.; Yin, J.; Duester, G. Discovery of genes required for body axis and limb formation by global identification of retinoic acid-regulated epigenetic marks. PLoS Biol. 2020, 18, e3000719. [Google Scholar] [CrossRef]
- Nedelec, B.; Rozet, J.M.; Fares Taie, L. Genetic architecture of retinoic-acid signaling-associated ocular developmental defects. Hum. Genet. 2019, 138, 937–955. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A. Genetics of anophthalmia and microphthalmia. Part 2: Syndromes associated with anophthalmia-microphthalmia. Hum. Genet. 2019, 138, 831–846. [Google Scholar] [CrossRef]
- Mory, A.; Ruiz, F.X.; Dagan, E.; Yakovtseva, E.A.; Kurolap, A.; Pares, X.; Farres, J.; Gershoni-Baruch, R. A missense mutation in ALDH1A3 causes isolated microphthalmia/anophthalmia in nine individuals from an inbred Muslim kindred. Eur. J. Hum. Genet. 2014, 22, 419–422. [Google Scholar] [CrossRef] [Green Version]
- Plaisancie, J.; Bremond-Gignac, D.; Demeer, B.; Gaston, V.; Verloes, A.; Fares-Taie, L.; Gerber, S.; Rozet, J.M.; Calvas, P.; Chassaing, N. Incomplete penetrance of biallelic ALDH1A3 mutations. Eur. J. Med. Gen. 2016, 59, 215–218. [Google Scholar] [CrossRef]
- Williams, A.L.; Bohnsack, B.L. What’s retinoic acid got to do with it? Retinoic acid regulation of the neural crest in craniofacial and ocular development. Genesis 2019, 57, e23308. [Google Scholar] [CrossRef] [PubMed]
- Weisschuh, N.; Dressler, P.; Schuettauf, F.; Wolf, C.; Wissinger, B.; Gramer, E. Novel mutations of FOXC1 and PITX2 in patients with Axenfeld-Rieger malformations. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3846–3852. [Google Scholar] [CrossRef] [Green Version]
- Molotkov, A.; Molotkova, N.; Duester, G. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development 2006, 133, 1901–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matt, N.; Dupé, V.; Garnier, J.-M.; Dennefeld, C.; Chambon, P.; Mark, M.; Ghyselinck, N.B. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development 2005, 132, 4789–4800. [Google Scholar] [CrossRef] [Green Version]
- Evans, A.L.; Gage, P.J. Expression of the homeobox gene Pitx2 in neural crest is required for optic stalk and ocular anterior segment development. Hum. Mol. Genet. 2005, 14, 3347–3359. [Google Scholar] [CrossRef]
- Kidson, S.H.; Kume, T.; Deng, K.; Winfrey, V.; Hogan, B.L. The forkhead/winged-helix gene, Mf1, is necessary for the normal development of the cornea and formation of the anterior chamber in the mouse eye. Dev. Biol. 1999, 211, 306–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warkany, J.; Schraffenberger, S. Congenital malformations induced in rats by maternal vitamin A deficiency. I. Defects of the eye. Arch. Ophthalmol. 1946, 35, 150–169. [Google Scholar] [CrossRef]
- Lohnes, D.; Mark, M.; Mendelsohn, C.; Dollé, P.; Dierich, A.; Gorry, P.; Gansmuller, A.; Chambon, P. Function of the retinoic acid receptors (RARs) during development. (I) Craniofacial and skeletal abnormalities in RAR double mutants. Development 1994, 120, 2723–2748. [Google Scholar] [CrossRef]
- Niederreither, K.; Subbarayan, V.; Dollé, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 1999, 21, 444–448. [Google Scholar] [CrossRef]
- Mic, F.A.; Haselbeck, R.J.; Cuenca, A.E.; Duester, G. Novel retinoic acid generating activities in the neural tube and heart identified by conditional rescue of Raldh2 null mutant mice. Development 2002, 129, 2271–2282. [Google Scholar] [CrossRef] [PubMed]
- Kastner, P.; Grondona, J.M.; Mark, M.; Gansmuller, A.; LeMeur, M.; Decimo, D.; Vonesch, J.-L.; Dollé, P.; Chambon, P. Genetic analysis of RXRa developmental function: Convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell 1994, 78, 987–1003. [Google Scholar] [CrossRef]
- Goto, S.; Onishi, A.; Misaki, K.; Yonemura, S.; Sugita, S.; Ito, H.; Ohigashi, Y.; Ema, M.; Sakaguchi, H.; Nishida, K.; et al. Neural retina-specific Aldh1a1 controls dorsal choroidal vascular development via Sox9 expression in retinal pigment epithelial cells. eLife 2018, 7, e32358. [Google Scholar] [CrossRef] [Green Version]
- Chawla, B.; Schley, E.; Williams, A.L.; Bohnsack, B.L. Retinoic Acid and Pitx2 Regulate Early Neural Crest Survival and Migration in Craniofacial and Ocular Development. Birth Defects Res. Part B 2016, 107, 126–135. [Google Scholar] [CrossRef]
- Gage, P.J.; Qian, M.; Wu, D.; Rosenberg, K.I. The canonical Wnt signaling antagonist DKK2 is an essential effector of PITX2 function during normal eye development. Dev. Biol. 2008, 317, 310–324. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Duester, G. Retinoic acid signaling in perioptic mesenchyme represses Wnt signaling via induction of Pitx2 and Dkk2. Dev. Biol. 2010, 340, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghyselinck, N.B.; Duester, G. Retinoic acid signaling pathways. Development 2019, 146, dev167502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutier, E.; Ye, T.; Choukrallah, M.A.; Urban, S.; Osz, J.; Chatagnon, A.; Delacroix, L.; Langer, D.; Rochel, N.; Moras, D.; et al. Retinoic Acid Receptors Recognize the Mouse Genome through Binding Elements with Diverse Spacing and Topology. J. Biol. Chem. 2012, 287, 26328–26341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalevee, S.; Anno, Y.N.; Chatagnon, A.; Samarut, E.; Poch, O.; Laudet, V.; Benoit, G.; Lecompte, O.; Rochette-Egly, C. Genome-wide in Silico Identification of New Conserved and Functional Retinoic Acid Receptor Response Elements (Direct Repeats Separated by 5 bp). J. Biol. Chem. 2011, 286, 33322–33334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschaki, M.; Schneider, C.; Rhinn, M.; Thibault-Carpentier, C.; Dembele, D.; Niederreither, K.; Dolle, P. Transcriptomic analysis of murine embryos lacking endogenous retinoic Acid signaling. PLoS ONE 2013, 8, e62274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, D.; Gudas, L.J. Gene expression profiling elucidates a specific role for RARgamma in the retinoic acid-induced differentiation of F9 teratocarcinoma stem cells. Biochem. Pharmacol. 2008, 75, 1129–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupé, V.; Davenne, M.; Brocard, J.; Dollé, P.; Mark, M.; Dierich, A.; Chambon, P.; Rijli, F.M. In vivo functional analysis of the Hoxa-1 3’ retinoic acid response element (3’RARE). Development 1997, 124, 399–410. [Google Scholar] [CrossRef]
- Houle, M.; Sylvestre, J.R.; Lohnes, D. Retinoic acid regulates a subset of Cdx1 function in vivo. Development 2003, 130, 6555–6567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Cunningham, T.J.; Duester, G. Nuclear receptor corepressors Ncor1 and Ncor2 (Smrt) are required for retinoic acid-dependent repression of Fgf8 during somitogenesis. Dev. Biol. 2016, 418, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, S.; Wilde, S.M.; Wood, S.; Logan, M.P. RA Acts in a Coherent Feed-Forward Mechanism with Tbx5 to Control Limb Bud Induction and Initiation. Cell Rep. 2015, 12, 879–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, T.J.; Lancman, J.J.; Berenguer, M.; Dong, P.D.S.; Duester, G. Genomic knockout of two presumed forelimb Tbx5 enhancers reveals they are nonessential for limb development. Cell Rep. 2018, 23, 3146–3151. [Google Scholar] [CrossRef]
- Adachi, N.; Robinson, M.; Goolsbee, A.; Shubin, N.H. Regulatory evolution of Tbx5 and the origin of paired appendages. Proc. Natl. Acad. Sci. USA 2016, 113, 10115–10120. [Google Scholar] [CrossRef] [Green Version]
- Duester, G. Knocking out enhancers to enhance epigenetic research. Trends Genet. 2019, 35, 89. [Google Scholar] [CrossRef]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Laugesen, A.; Helin, K. Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell 2014, 14, 735–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolovos, P.; Knoch, T.A.; Grosveld, F.G.; Cook, P.R.; Papantonis, A. Enhancers and silencers: An integrated and simple model for their function. Epigenetics Chromatin 2012, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibn-Salem, J.; Kohler, S.; Love, M.I.; Chung, H.R.; Huang, N.; Hurles, M.E.; Haendel, M.; Washington, N.L.; Smedley, D.; Mungall, C.J.; et al. Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol. 2014, 15, 423. [Google Scholar] [CrossRef] [Green Version]
- Lupianez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Generation of RA and control of transcriptional activation or repression of target genes.

Figure 2.
RA signaling is required during optic cup formation. (A) RA activity, detected by the RARE-lacZ transgene, and optic cup formation are both normal in E10.5 RA-rescued Aldh1a2-/- single knockout embryos (R2-/-); Aldh1a2-/- embryos do not develop beyond E8.5, but a single low-dose maternal treatment with exogenous RA at E7.5 results in growth to E10.5 and clearance of exogenous RA by E9.5 [25]; RA activity observed at E10.5 is due to expression of Aldh1a1 and Aldh1a3 in the optic vesicle [18]. (B) For E10.5 RA-rescued triple knockout embryos (R1:R2:R3-/-) one observes a lack of RA activity and a failure to form the optic cup; *, failure of ventral invagination of optic vesicle. (C,D) Another triple knockout (R1:R2:R3-/-) compared to wild type (WT) stained for Vax2 mRNA (a marker of the ventral retina) also shows a failure in optic cup formation primarily due to a failure in ventral invagination of the optic vesicle. Shown are dorsoventral sections; adapted from [18].
Figure 2.
RA signaling is required during optic cup formation. (A) RA activity, detected by the RARE-lacZ transgene, and optic cup formation are both normal in E10.5 RA-rescued Aldh1a2-/- single knockout embryos (R2-/-); Aldh1a2-/- embryos do not develop beyond E8.5, but a single low-dose maternal treatment with exogenous RA at E7.5 results in growth to E10.5 and clearance of exogenous RA by E9.5 [25]; RA activity observed at E10.5 is due to expression of Aldh1a1 and Aldh1a3 in the optic vesicle [18]. (B) For E10.5 RA-rescued triple knockout embryos (R1:R2:R3-/-) one observes a lack of RA activity and a failure to form the optic cup; *, failure of ventral invagination of optic vesicle. (C,D) Another triple knockout (R1:R2:R3-/-) compared to wild type (WT) stained for Vax2 mRNA (a marker of the ventral retina) also shows a failure in optic cup formation primarily due to a failure in ventral invagination of the optic vesicle. Shown are dorsoventral sections; adapted from [18].

Figure 3.
RA is generated in dorsal/ventral retina and diffuses to perioptic mesenchyme where it is required during anterior eye formation (cornea/eyelid) to activate Pitx2 which then activates Dkk2 that functions to repress WNT signaling to limit perioptic mesenchyme growth; adapted from [31].
Figure 3.
RA is generated in dorsal/ventral retina and diffuses to perioptic mesenchyme where it is required during anterior eye formation (cornea/eyelid) to activate Pitx2 which then activates Dkk2 that functions to repress WNT signaling to limit perioptic mesenchyme growth; adapted from [31].

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Duester, G. Towards a Better Vision of Retinoic Acid Signaling during Eye Development. Cells 2022, 11, 322. https://doi.org/10.3390/cells11030322
AMA Style
Duester G. Towards a Better Vision of Retinoic Acid Signaling during Eye Development. Cells. 2022; 11(3):322. https://doi.org/10.3390/cells11030322
Chicago/Turabian StyleDuester, Gregg. 2022. "Towards a Better Vision of Retinoic Acid Signaling during Eye Development" Cells 11, no. 3: 322. https://doi.org/10.3390/cells11030322
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.