
Ursolic Acid Impairs Cellular Lipid Homeostasis and Lysosomal Membrane Integrity in Breast Carcinoma Cells
 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
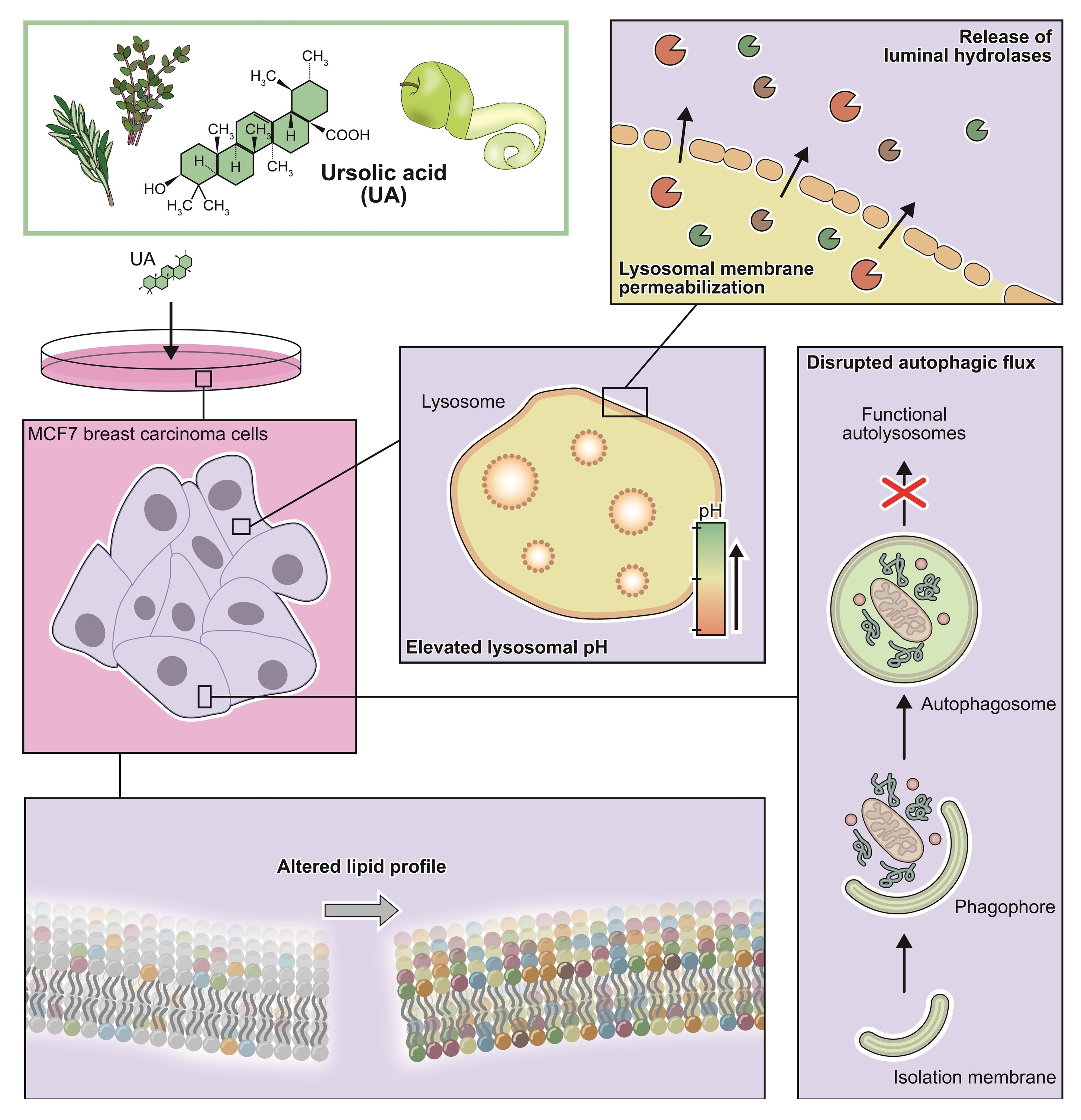
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Treatments
2.3. Cell Death Assays
2.4. Luciferase-Based Autophagic Flux Assay
2.5. Immunocytochemistry
2.6. LC3-Puncta Formation Assays
2.7. Measuring Nuclear Proximity of LAMP2 and EEA1
2.8. Lysosomal pH Measurements
2.9. Cystein Cathepsin and NAG Activities
2.10. Protein Concentration Measurements
2.11. Western Blotting
2.12. Galectin-3 Puncta Assay
2.13. Lipid Extraction and Lipidomics
2.14. Lipidomics Data Analysis
2.15. Statistical Analysis
3. Results
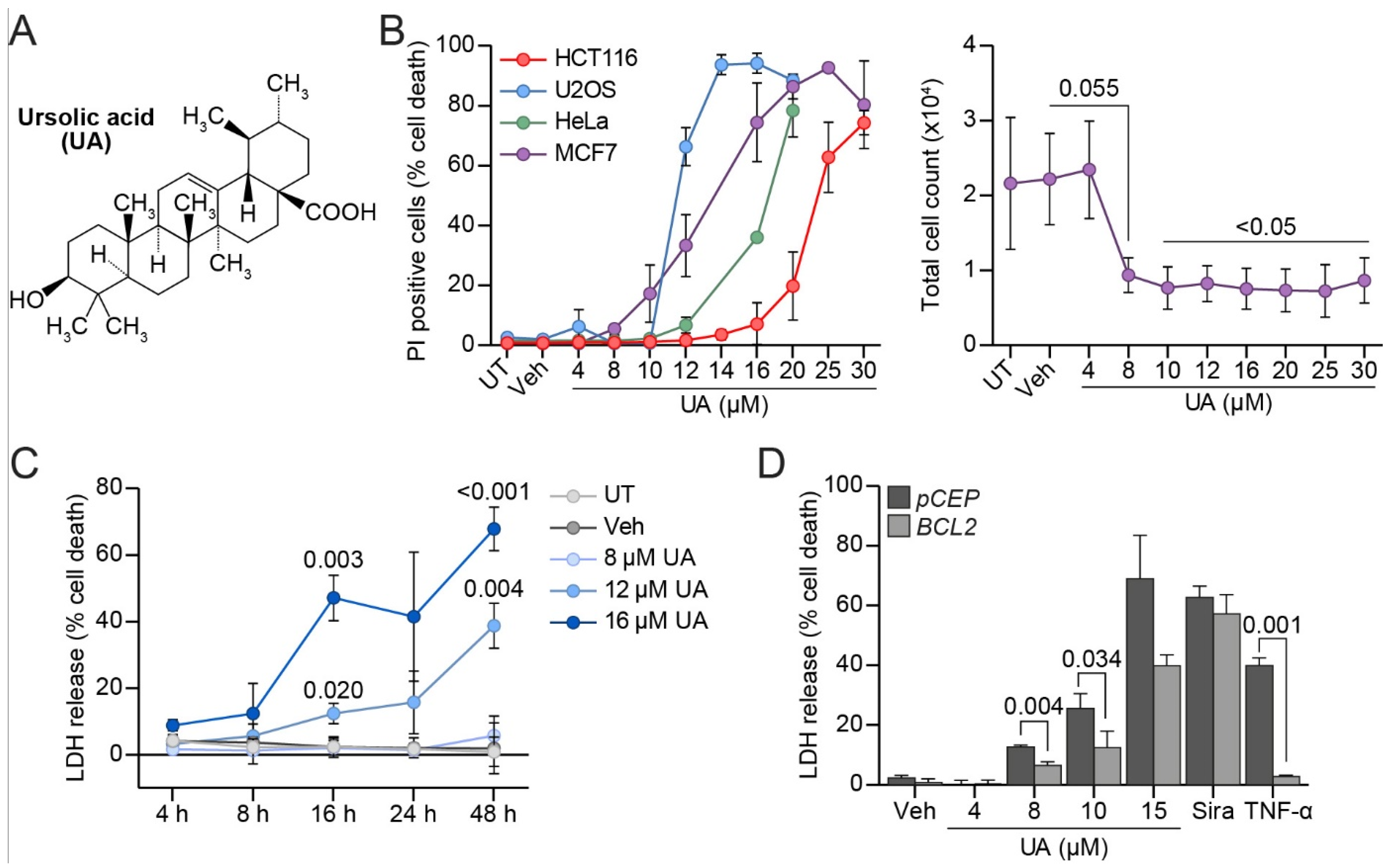
3.1. UA Kills MCF Breast Cancer Cells Partly via Apoptosis
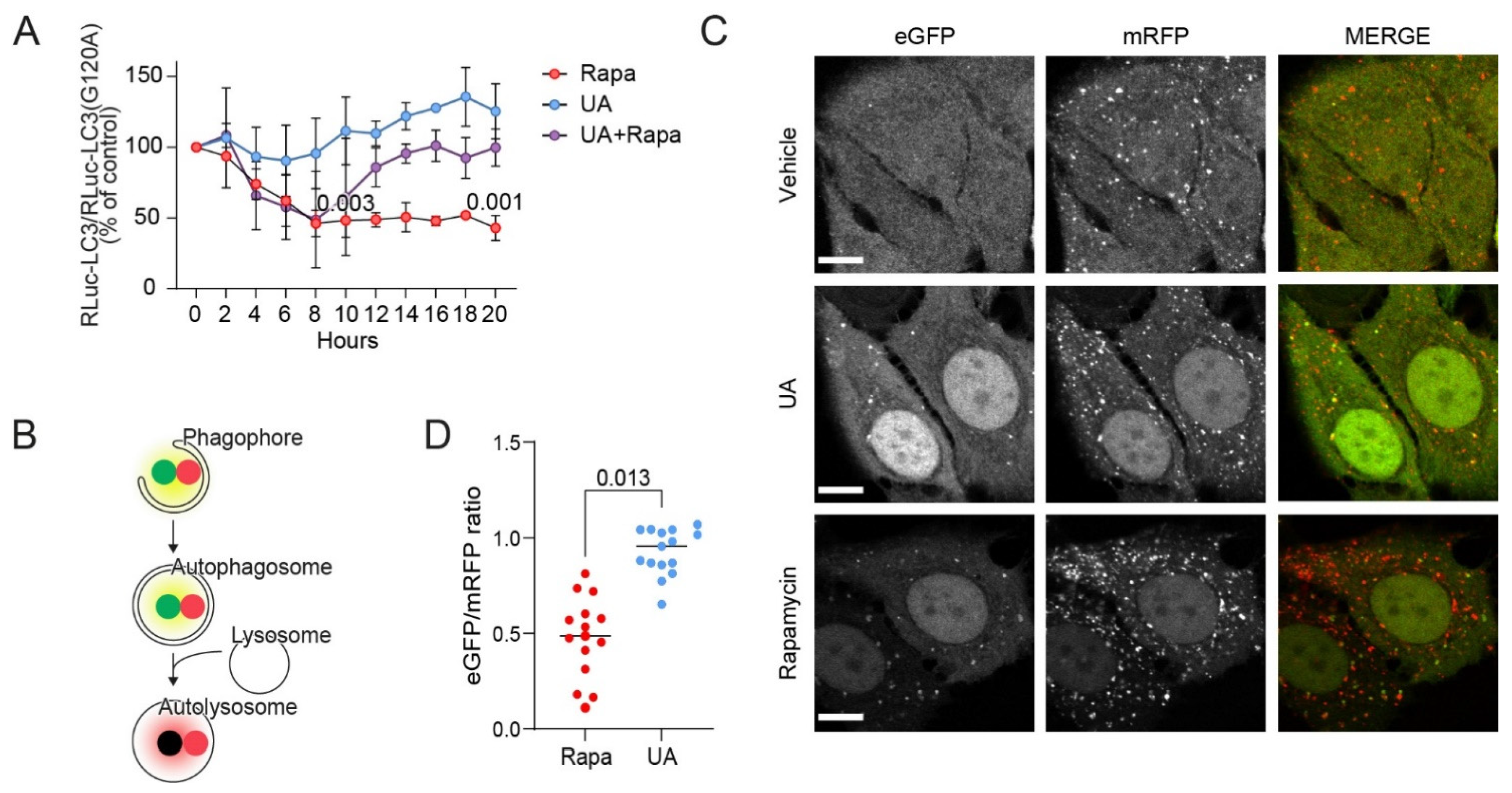
3.2. UA Inhibits Autophagosome Maturation
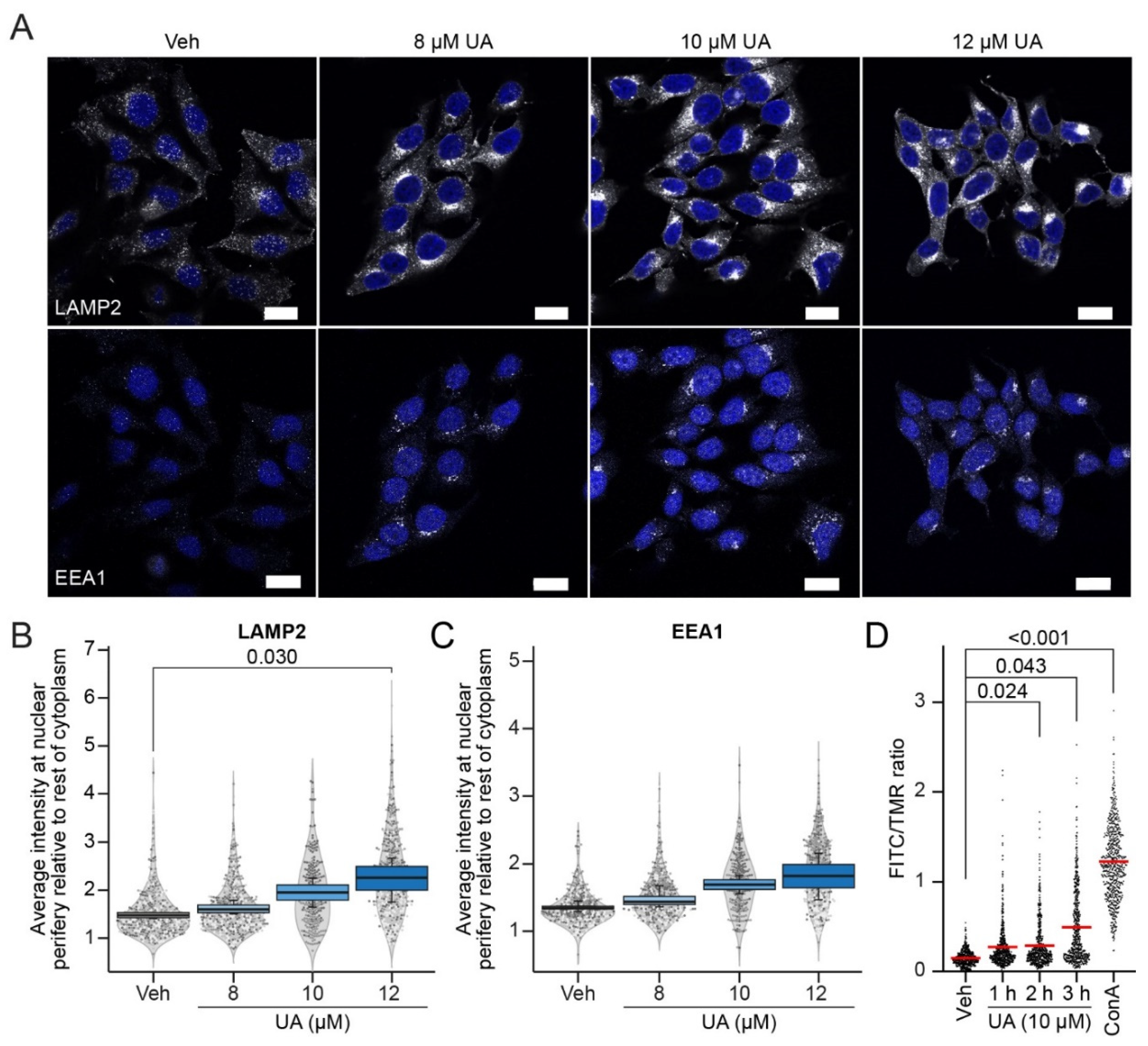
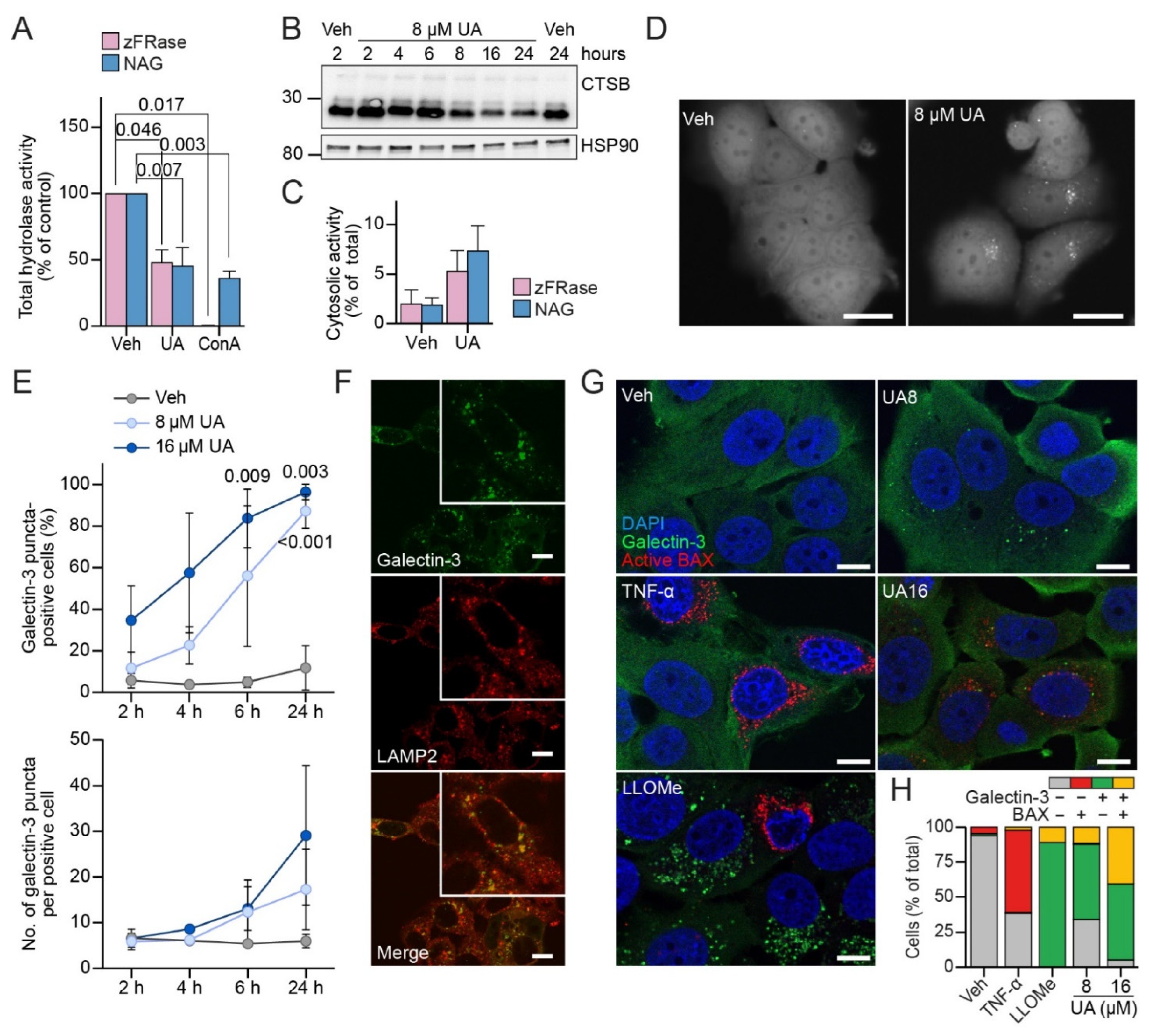
3.3. UA Alters Lysosomal Trafficking and pH
3.4. UA Causes Lysosomal Membrane Permeabilization Prior to BAX Activation
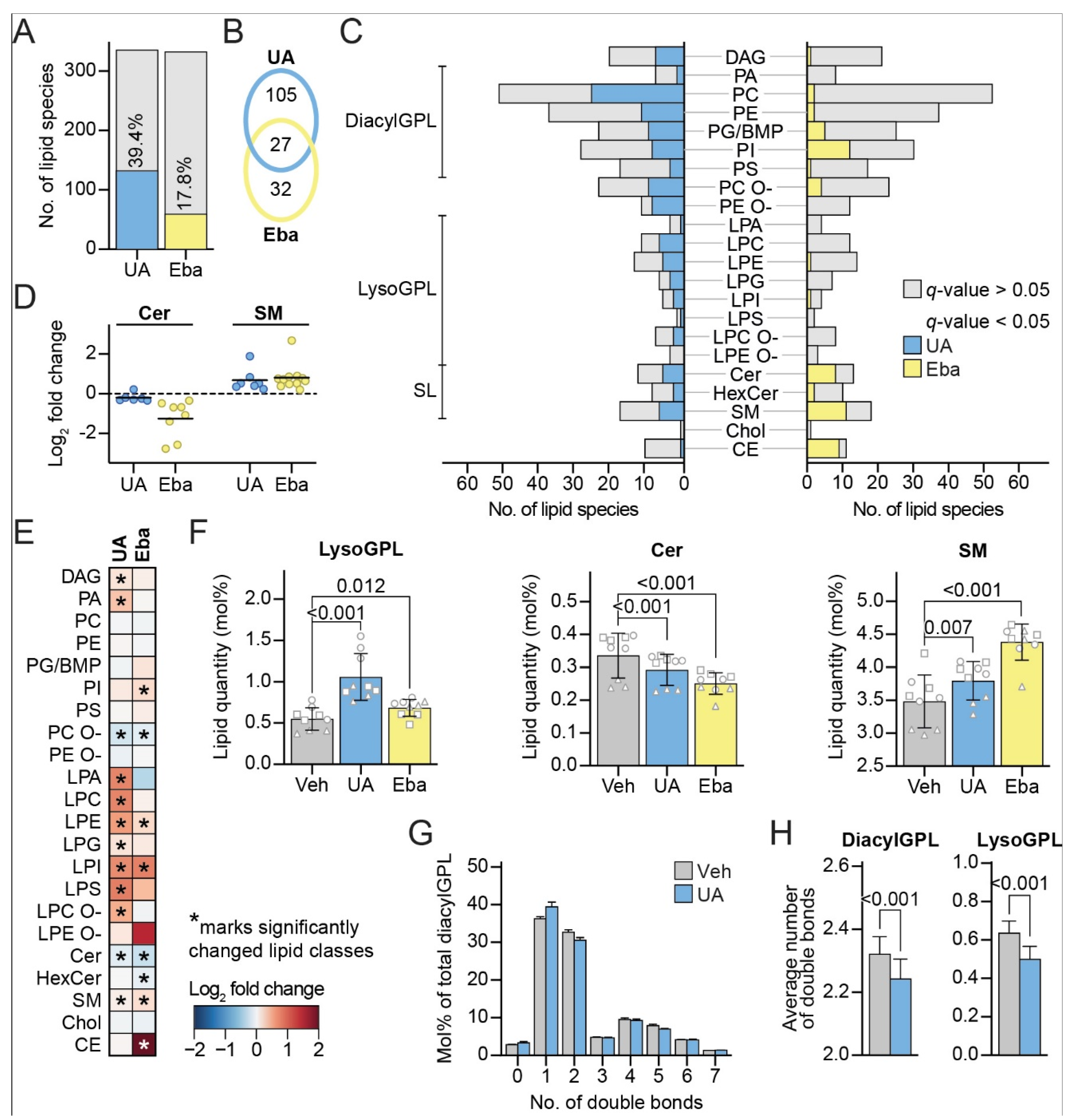
3.5. UA Induces Changes in the Lipid Profile of MCF7 Cells
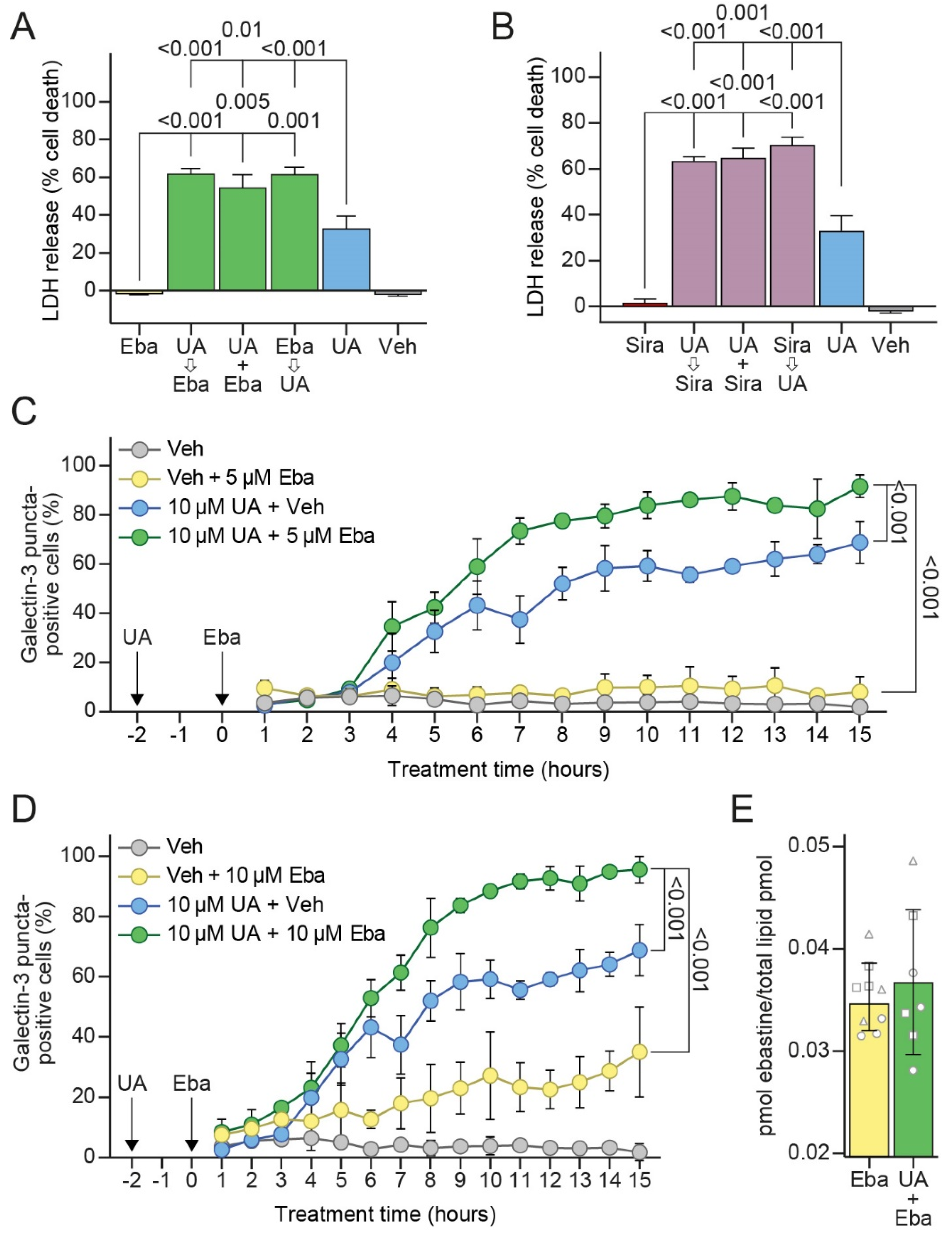
3.6. Sublethal Concentrations of CADs Sensitize MCF7 Cells to UA-Induced LMP and Cell Death
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jäger, S.; Trojan, H.; Kopp, T.; Laszczyk, M.N.; Scheffler, A. Pentacyclic Triterpene Distribution in Various Plants–Rich Sources for a New Group of Multi-Potent Plant Extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos, J.-L. Effects of triterpenes on the immune system. J. Ethnopharmacol. 2010, 128, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chang, X.; Zhang, J.; Li, J.; Wang, N.; Yang, B.; Pan, B.; Zheng, Y.; Wang, X.; Ou, H.; et al. Ursolic Acid Inhibits Breast Cancer Metastasis by Suppressing Glycolytic Metabolism via Activating SP1/Caveolin-1 Signaling. Front. Oncol. 2021, 11, 745584. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.Y.; Lee, S.R.; Heo, J.-W.; No, M.-H.; Rhee, B.D.; Ko, K.S.; Kwak, H.-B.; Han, J. Ursolic acid in health and disease. Korean J. Physiol. Pharmacol. 2018, 22, 235–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manu, K.A.; Kuttan, G. Ursolic acid induces apoptosis by activating p53 and caspase-3 gene expressions and suppressing NF-kappaB mediated activation of bcl-2 in B16F-10 melanoma cells. Int. Immunopharmacol. 2008, 8, 974–981. [Google Scholar] [CrossRef]
- Park, J.H.; Kwon, H.Y.; Sohn, E.J.; Kim, K.A.; Kim, B.; Jeong, S.J.; Koo, J.S.; Kim, S.H. Inhibition of Wnt/beta-catenin signaling mediates ursolic acid-induced apoptosis in PC-3 prostate cancer cells. Pharmacol. Rep. 2013, 65, 1366–1374. [Google Scholar] [CrossRef]
- Xavier, C.P.; Lima, C.F.; Pedro, D.F.; Wilson, J.M.; Kristiansen, K.; Pereira-Wilson, C. Ursolic acid induces cell death and modulates autophagy through JNK pathway in apoptosis-resistant colorectal cancer cells. J. Nutr. Biochem. 2013, 24, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Conway, G.; Zizyte, D.; Mondala, J.; He, Z.; Lynam, L.; Lecourt, M.; Barcia, C.; Howe, O.; Curtin, J. Ursolic Acid Inhibits Collective Cell Migration and Promotes JNK-Dependent Lysosomal Associated Cell Death in Glioblastoma Multiforme Cells. Pharmaceuticals 2021, 14, 91. [Google Scholar] [CrossRef]
- Leng, S.; Hao, Y.; Du, D.; Xie, S.; Hong, L.; Gu, H.; Zhu, X.; Zhang, J.; Fan, D.; Kung, H.-F. Ursolic acid promotes cancer cell death by inducing Atg5-dependent autophagy. Int. J. Cancer 2013, 133, 2781–2790. [Google Scholar] [CrossRef]
- Shishodia, S.; Majumdar, S.; Banerjee, S.; Aggarwal, B.B. Ursolic acid inhibits nuclear factor-kappaB activation induced by carcinogenic agents through suppression of IkappaBalpha kinase and p65 phosphorylation: Correlation with down-regulation of cyclooxygenase 2, matrix metalloproteinase 9, and cyclin D1. Cancer Res. 2003, 63, 4375–4383. [Google Scholar]
- Kroemer, G.; Jäättelä, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim. Biophys. Acta Rev. Cancer 2014, 1845, 136–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.-T.; Wu, C.-H.; Yen, G.-C. Erratum: Ursolic acid, a naturally occurring triterpenoid, suppresses migration and invasion of human breast cancer cells by modulating c-Jun N-terminal kinase, Akt and mammalian target of rapamycin signaling. Mol. Nutr. Food Res. 2010, 54, 1696. [Google Scholar] [CrossRef]
- Zong, L.; Cheng, G.; Liu, S.; Pi, Z.; Liu, Z.; Song, F. Reversal of multidrug resistance in breast cancer cells by a combination of ursolic acid with doxorubicin. J. Pharm. Biomed. Anal. 2019, 165, 268–275. [Google Scholar] [CrossRef]
- Shan, J.-Z.; Xuan, Y.-Y.; Ruan, S.-Q.; Sun, M. Proliferation-inhibiting and apoptosis-inducing effects of ursolic acid and oleanolic acid on multi-drug resistance cancer cells in vitro. Chin. J. Integr. Med. 2011, 17, 607–611. [Google Scholar] [CrossRef]
- Gou, W.; Luo, N.; Yu, B.; Wu, H.; Wu, S.; Tian, C.; Guo, J.; Ning, H.; Bi, C.; Wei, H.; et al. Ursolic Acid Derivative UA232 Promotes Tumor Cell Apoptosis by Inducing Endoplasmic Reticulum Stress and Lysosomal Dysfunction. Int. J. Biol. Sci. 2022, 18, 2639–2651. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Fehrenbacher, N.; Gyrd-Hansen, M.; Poulsen, B.; Felbor, U.; Kallunki, T.; Boes, M.; Weber, E.; Leist, M.; Jäättelä, M. Sensitization to the Lysosomal Cell Death Pathway upon Immortalization and Transformation. Cancer Res 2004, 64, 5301–5310. [Google Scholar] [CrossRef] [Green Version]
- Kågedal, K.; Zhao, M.; Svensson, I.; Brunk, U.T. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem. J. 2001, 359 (Pt 2), 335–343. [Google Scholar] [CrossRef]
- Kirkegaard, T.; Jäättelä, M. Lysosomal involvement in cell death and cancer. Biochim. Et Biophys. Acta BBA Mol. Cell Res. 2009, 1793, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Groth-Pedersen, L.; Jäättelä, M. Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett. 2013, 332, 265–274. [Google Scholar] [CrossRef]
- Appelqvist, H.; Wäster, P.; Kågedal, K.; Öllinger, K. The lysosome: From waste bag to potential therapeutic target. J. Mol. Cell Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Petersen, N.H.; Olsen, O.D.; Groth-Pedersen, L.; Ellegaard, A.-M.; Bilgin, M.; Redmer, S.; Ostenfeld, M.S.; Ulanet, D.; Dovmark, T.H.; Lønborg, A.; et al. Transformation-Associated Changes in Sphingolipid Metabolism Sensitize Cells to Lysosomal Cell Death Induced by Inhibitors of Acid Sphingomyelinase. Cancer Cell 2013, 24, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadanaciva, S.; Lu, S.; Gebhard, D.F.; Jessen, B.A.; Pennie, W.D.; Will, Y. A high content screening assay for identifying lysosomotropic compounds. Toxicol. Vitr. 2011, 25, 715–723. [Google Scholar] [CrossRef]
- Kazmi, F.; Hensley, T.; Pope, C.; Funk, R.S.; Loewen, G.J.; Buckley, D.B.; Parkinson, A. Lysosomal Sequestration (Trapping) of Lipophilic Amine (Cationic Amphiphilic) Drugs in Immortalized Human Hepatocytes (Fa2N-4 Cells). Drug Metab. Dispos. 2013, 41, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Kornhuber, J.; Tripal, P.; Reichel, M.; Mühle, C.; Rhein, C.; Muehlbacher, M.; Groemer, T.; Gulbins, E. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): A Novel Pharmacological Group of Drugs with Broad Clinical Applications. Cell. Physiol. Biochem. 2010, 26, 9–20. [Google Scholar] [CrossRef]
- Kolter, T.; Sandhoff, K. Lysosomal degradation of membrane lipids. FEBS Lett. 2010, 584, 1700–1712. [Google Scholar] [CrossRef] [Green Version]
- Appelqvist, H.; Sandin, L.; Björnström, K.; Saftig, P.; Garner, B.; Öllinger, K.; Kågedal, K. Sensitivity to Lysosome-Dependent Cell Death Is Directly Regulated by Lysosomal Cholesterol Content. PLoS ONE 2012, 7, e50262. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.-S.; Li, Y.-B.; Wang, J.-W.; Sun, L.; Zhang, G.-J. Mechanism of Lysophosphatidylcholine-Induced Lysosome Destabilization. J. Membr. Biol. 2007, 215, 27–35. [Google Scholar] [CrossRef]
- Nielsen, I.; Groth-Pedersen, L.; Dicroce-Giacobini, J.; Jonassen, A.S.H.; Mortensen, M.; Bilgin, M.; Schmiegelow, K.; Jäättelä, M.; Maeda, K. Cationic amphiphilic drugs induce elevation in lysoglycerophospholipid levels and cell death in leukemia cells. Metabolomics 2020, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ellegaard, A.-M.; Bach, P.; Jäättelä, M. Targeting Cancer Lysosomes with Good Old Cationic Amphiphilic Drugs. Rev. Physiol. Biochem. Pharmacol. 2020, 1–46. [Google Scholar] [CrossRef]
- Jaattela, M.; Benedict, M.; Tewari, M.; Shayman, J.A.; Dixit, V.M. Bcl-x and Bcl-2 inhibit TNF and Fas-induced apoptosis and activation of phospholipase A2 in breast carcinoma cells. Oncogene 1995, 10, 2297–2305. [Google Scholar] [PubMed]
- Høyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Szyniarowski, P.; Corcelle-Termeau, E.; Farkas, T.; Høyer-Hansen, M.; Nylandsted, J.; Kallunki, T.; Jäättelä, M. A comprehensive siRNA screen for kinases that suppress macroautophagy in optimal growth conditions. Autophagy 2011, 7, 892–903. [Google Scholar] [CrossRef] [Green Version]
- Farkas, T.; Høyer-Hansen, M.; Jäättelä, M. Identification of novel autophagy regulators by a luciferase-based assay for the kinetics of autophagic flux. Autophagy 2009, 5, 1018–1025. [Google Scholar] [CrossRef] [Green Version]
- Aits, S.; Kricker, J.; Liu, B.; Ellegaard, A.-M.; Hämälistö, S.; Tvingsholm, S.; Corcelle-Termeau, E.; Høgh, S.; Farkas, T.; Jonassen, A.H.; et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 2015, 11, 1408–1424. [Google Scholar] [CrossRef] [Green Version]
- Shirasawa, S.; Furuse, M.; Yokoyama, N.; Sasazuki, T. Altered Growth of Human Colon Cancer Cell Lines Disrupted at Activated Ki-ras. Science 1993, 260, 85–88. [Google Scholar] [CrossRef]
- Lord, S.J.; Velle, K.; Mullins, R.D.; Fritz-Laylin, L.K. SuperPlots: Communicating reproducibility and variability in cell biology. J. Cell Biol. 2020, 219, e202001064. [Google Scholar] [CrossRef]
- Nylandsted, J.; Gyrd-Hansen, M.; Danielewicz, A.; Fehrenbacher, N.; Lademann, U.; Høyer-Hansen, M.; Weber, E.; Multhoff, G.; Rohde, M.; Jaattela, M. Heat Shock Protein 70 Promotes Cell Survival by Inhibiting Lysosomal Membrane Permeabilization. J. Exp. Med. 2004, 200, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Aits, S.; Jäättelä, M.; Nylandsted, J. Methods for the quantification of lysosomal membrane permeabilization: A hallmark of lysosomal cell death. Methods Cell Biol. 2015, 126, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, I.; Maeda, K.; Bilgin, M. Global Monitoring of the Mammalian Lipidome by Quantitative Shotgun Lipidomics. Methods Mol. Biol. 2017, 1609, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Herzog, R.; Schuhmann, K.; Schwudke, D.; Sampaio, J.; Bornstein, S.R.; Schroeder, M.; Shevchenko, A. LipidXplorer: A Software for Consensual Cross-Platform Lipidomics. PLoS ONE 2012, 7, e29851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, I.O.; Olsen, A.V.; Dicroce-Giacobini, J.; Papaleo, E.; Andersen, K.K.; Jäättelä, M.; Maeda, K.; Bilgin, M. Comprehensive Evaluation of a Quantitative Shotgun Lipidomics Platform for Mammalian Sample Analysis on a High-Resolution Mass Spectrometer. J. Am. Soc. Mass Spectrom. 2020, 31, 894–907. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Jung, J.; Seo, J.; Kim, J.; Kim, J.H. Ursolic Acid Causes Cell Death in PC-12 Cells by Inducing Apoptosis and Impairing Autophagy. Anticancer. Res. 2018, 38, 847–853. [Google Scholar]
- Kang, D.Y.; Sp, N.; Jang, K.-J.; Jo, E.S.; Bae, S.W.; Yang, Y.M. Antitumor Effects of Natural Bioactive Ursolic Acid in Embryonic Cancer Stem Cells. J. Oncol. 2022, 2022, 6737248. [Google Scholar] [CrossRef]
- Hu, M.; Carraway, K.L.I., 3rd. Repurposing Cationic Amphiphilic Drugs and Derivatives to Engage Lysosomal Cell Death in Cancer Treatment. Front. Oncol. 2020, 10, 605361. [Google Scholar] [CrossRef]
- Ellegaard, A.-M.; Dehlendorff, C.; Vind, A.C.; Anand, A.; Cederkvist, L.; Petersen, N.H.; Nylandsted, J.; Stenvang, J.; Mellemgaard, A.; Østerlind, K.; et al. Repurposing Cationic Amphiphilic Antihistamines for Cancer Treatment. eBioMedicine 2016, 9, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal Turnover, but Not a Cellular Level, of Endogenous LC3 is a Marker for Autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decuypere, J.P.; Kindt, D.; Luyten, T.; Welkenhuyzen, K.; Missiaen, L.; De Smedt, H.; Bultynck, G.; Parys, J.B. mTOR-Controlled Autophagy Requires Intracellular Ca(2+) Signaling. PLoS ONE 2013, 8, e61020. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Fujita, N.; Noda, T.; Yoshimori, T. Monitoring Autophagy in Mammalian Cultured Cells through the Dynamics of LC3. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 452, pp. 1–12. [Google Scholar]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Rout, A.K.; Strub, M.-P.; Piszczek, G.; Tjandra, N. Structure of Transmembrane Domain of Lysosome-associated Membrane Protein Type 2a (LAMP-2A) Reveals Key Features for Substrate Specificity in Chaperone-mediated Autophagy. J. Biol. Chem. 2014, 289, 35111–35123. [Google Scholar] [CrossRef] [Green Version]
- Ostenfeld, M.S.; Høyer-Hansen, M.; Bastholm, L.; Fehrenbacher, N.; Olsen, O.D.; Groth-Pedersen, L.; Puustinen, P.; Kirkegaard-Sørensen, T.; Nylandsted, J.; Farkas, T.; et al. Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy 2008, 4, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The Ins and Outs of Cathepsins: Physiological Function and Role in Disease Management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef]
- Lüllmann, H.; Lüllmann-Rauch, R.; Wassermann, O. Lipidosis induced by amphiphilic cationic drugs. Biochem. Pharmacol. 1978, 27, 1103–1108. [Google Scholar] [CrossRef]
- Henriksen, J.R.; Andresen, T.L.; Feldborg, L.N.; Duelund, L.; Ipsen, J.H. Understanding Detergent Effects on Lipid Membranes: A Model Study of Lysolipids. Biophys. J. 2010, 98, 2199–2205. [Google Scholar] [CrossRef] [Green Version]
- Billen, L.P.; Shamas-Din, A.; Andrews, D.W. Bid: A Bax-like BH3 protein. Oncogene 2008, 27, S93–S104. [Google Scholar] [CrossRef] [Green Version]
- Guicciardi, M.E.; Leist, M.; Gores, G.J. Lysosomes in cell death. Oncogene 2004, 23, 2881–2890. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.J.; Zhang, X.; Guo, J.; Zhang, W.; Ai, S.; Zhang, F.; Wang, Y.; Liu, W.J. Lysosomal dysfunction–induced autophagic stress in diabetic kidney disease. J. Cell. Mol. Med. 2020, 24, 8276–8290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bai, J.; Hang, K.; Xu, J.; Zhou, C.; Li, L.; Wang, Z.; Wang, Y.; Wang, K.; Xue, D. Role of Lysosomal Acidification Dysfunction in Mesenchymal Stem Cell Senescence. Front. Cell Dev. Biol. 2022, 10, 817877. [Google Scholar] [CrossRef] [PubMed]
- Mauvezin, C.; Nagy, P.; Juhász, G.; Neufeld, T.P. Autophagosome–lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 2015, 6, 7007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, A.; Uchiyama, H.; Takano, S.; Nakamura, N.; Ohkuma, S. Autophagosome-Lysosome Fusion Depends on the pH in Acidic Compartments in CHO Cells. Autophagy 2007, 3, 154–157. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.; Rubinsztein, D.C. Regulation of autophagy by lysosomal positioning. Autophagy 2011, 7, 927–928. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef]
- Van Echteld, C.; De Kruijff, B.; Mandersloot, J.; De Gier, J. Effects of lysophosphatidylcholines on phosphatidylcholine and phosphatidylcholine/cholesterol liposome systems as revealed by 31P-NMR, electron microscopy and permeability studies. Biochim. Et Biophys. Acta BBA Biomembr. 1981, 649, 211–220. [Google Scholar] [CrossRef]
- Inoue, K.; Kitagawa, T. Effect of exogenous lysolecithin on liposomal membranes. Its relation to membrane fluidity. Biochim. Biophys. Acta 1974, 363, 361–372. [Google Scholar] [CrossRef]
- Zong, L.; Cheng, G.; Zhao, J.; Zhuang, X.; Zheng, Z.; Liu, Z.; Song, F. Inhibitory Effect of Ursolic Acid on the Migration and Invasion of Doxorubicin-Resistant Breast Cancer. Molecules 2022, 27, 1282. [Google Scholar] [CrossRef]
- Xiang, F.; Fan, Y.; Ni, Z.; Liu, Q.; Zhu, Z.; Chen, Z.; Hao, W.; Yue, H.; Wu, R.; Kang, X. Ursolic Acid Reverses the Chemoresistance of Breast Cancer Cells to Paclitaxel by Targeting MiRNA-149-5p/MyD88. Front. Oncol. 2019, 9, 501. [Google Scholar] [CrossRef] [Green Version]
- Halaby, R. Influence of lysosomal sequestration on multidrug resistance in cancer cells. Cancer Drug Resist. 2019, 2, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Linē, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updat. 2019, 46, 100645. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, A.C.; Richiardone, E.; Jorge, J.; Polónia, B.; Xavier, C.P.; Salaroglio, I.C.; Riganti, C.; Vasconcelos, M.H.; Corbet, C.; Sarmento-Ribeiro, A.B. Impact of cancer metabolism on therapy resistance—Clinical implications. Drug Resist. Updat. 2021, 59, 100797. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, N.; Akiyama, S.-I.; Kobayashi, M.; Kuwano, M. Lysosomotropic agents reverse multiple drug resistance in human cancer cells. Cancer Lett. 1986, 30, 251–259. [Google Scholar] [CrossRef]
- Ge, J.; Chen, Z.; Huang, J.; Chen, J.; Yuan, W.; Deng, Z.; Chen, Z. Upregulation of Autophagy-Related Gene-5 (ATG-5) Is Associated with Chemoresistance in Human Gastric Cancer. PLoS ONE 2014, 9, e110293. [Google Scholar] [CrossRef]
- Chittaranjan, S.; Bortnik, S.; Dragowska, W.H.; Xu, J.; Abeysundara, N.; Leung, A.; Go, N.E.; DeVorkin, L.; Weppler, S.A.; Gelmon, K.; et al. Autophagy Inhibition Augments the Anticancer Effects of Epirubicin Treatment in Anthracycline-Sensitive and -Resistant Triple-Negative Breast Cancer. Clin. Cancer Res. 2014, 20, 3159–3173. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fogde, D.L.; Xavier, C.P.R.; Balnytė, K.; Holland, L.K.K.; Stahl-Meyer, K.; Dinant, C.; Corcelle-Termeau, E.; Pereira-Wilson, C.; Maeda, K.; Jäättelä, M. Ursolic Acid Impairs Cellular Lipid Homeostasis and Lysosomal Membrane Integrity in Breast Carcinoma Cells. Cells 2022, 11, 4079. https://doi.org/10.3390/cells11244079
Fogde DL, Xavier CPR, Balnytė K, Holland LKK, Stahl-Meyer K, Dinant C, Corcelle-Termeau E, Pereira-Wilson C, Maeda K, Jäättelä M. Ursolic Acid Impairs Cellular Lipid Homeostasis and Lysosomal Membrane Integrity in Breast Carcinoma Cells. Cells. 2022; 11(24):4079. https://doi.org/10.3390/cells11244079
Chicago/Turabian StyleFogde, Ditte L., Cristina P. R. Xavier, Kristina Balnytė, Lya K. K. Holland, Kamilla Stahl-Meyer, Christoffel Dinant, Elisabeth Corcelle-Termeau, Cristina Pereira-Wilson, Kenji Maeda, and Marja Jäättelä. 2022. "Ursolic Acid Impairs Cellular Lipid Homeostasis and Lysosomal Membrane Integrity in Breast Carcinoma Cells" Cells 11, no. 24: 4079. https://doi.org/10.3390/cells11244079