
Real-Time Characterization of Clonal Fate Decisions in Complex Leukemia Samples by Fluorescent Genetic Barcoding

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation and Cultivation of H9M Cell Lines
2.2. Nucleofection
2.3. Lentiviral Vector Cloning and Production
2.4. Lentiviral Transductions
2.5. Flow Cytometric Analysis
2.6. DNA Barcode Amplification and Sequencing
2.7. Integration Site Analysis
2.8. Exome Sequencing and Mutation Calling
2.9. RNA-Sequencing and Raw Data Processing
2.10. Animal Experiments
3. Results
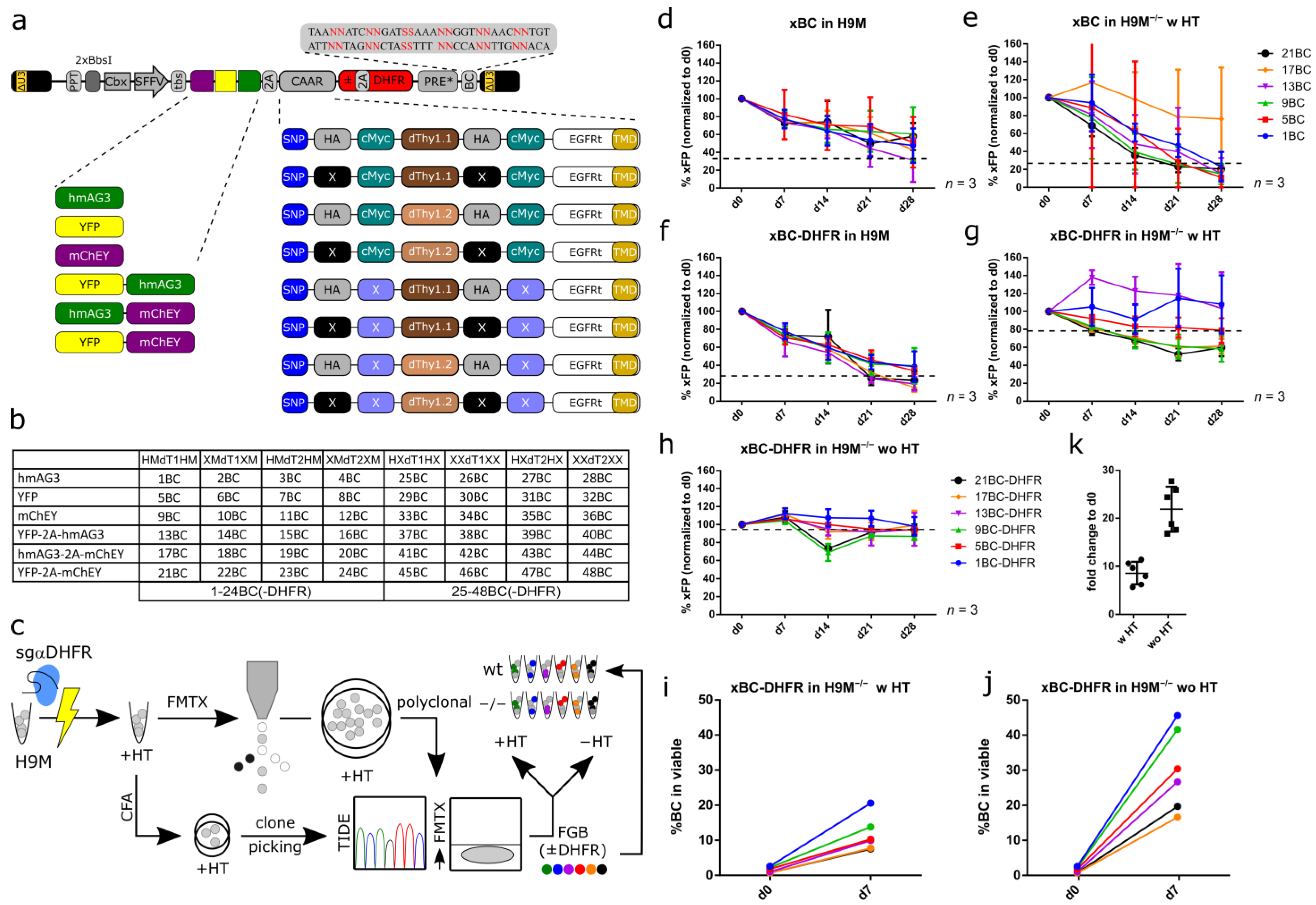
3.1. Design Considerations for a Complex Lentiviral Fluorescent Genetic Barcoding Vector System
3.2. Stable FGB-D Vector Expression in H9M−/− Cells
3.3. FGB-D Vectors Produce Highly Specific Color Codes in Flow Cytometric Detection
3.4. FGB-D Enables the Tracking and Identification of Growth-Enhanced H9M Clones In Vitro
3.5. Color-Coded H9M Clones Follow Pre-Determined Behavior in Short-Term Assays
3.6. FGB-D Labeled H9M−/− Clones Differentially Respond to Cytokine Stimulation and Drug Challenge
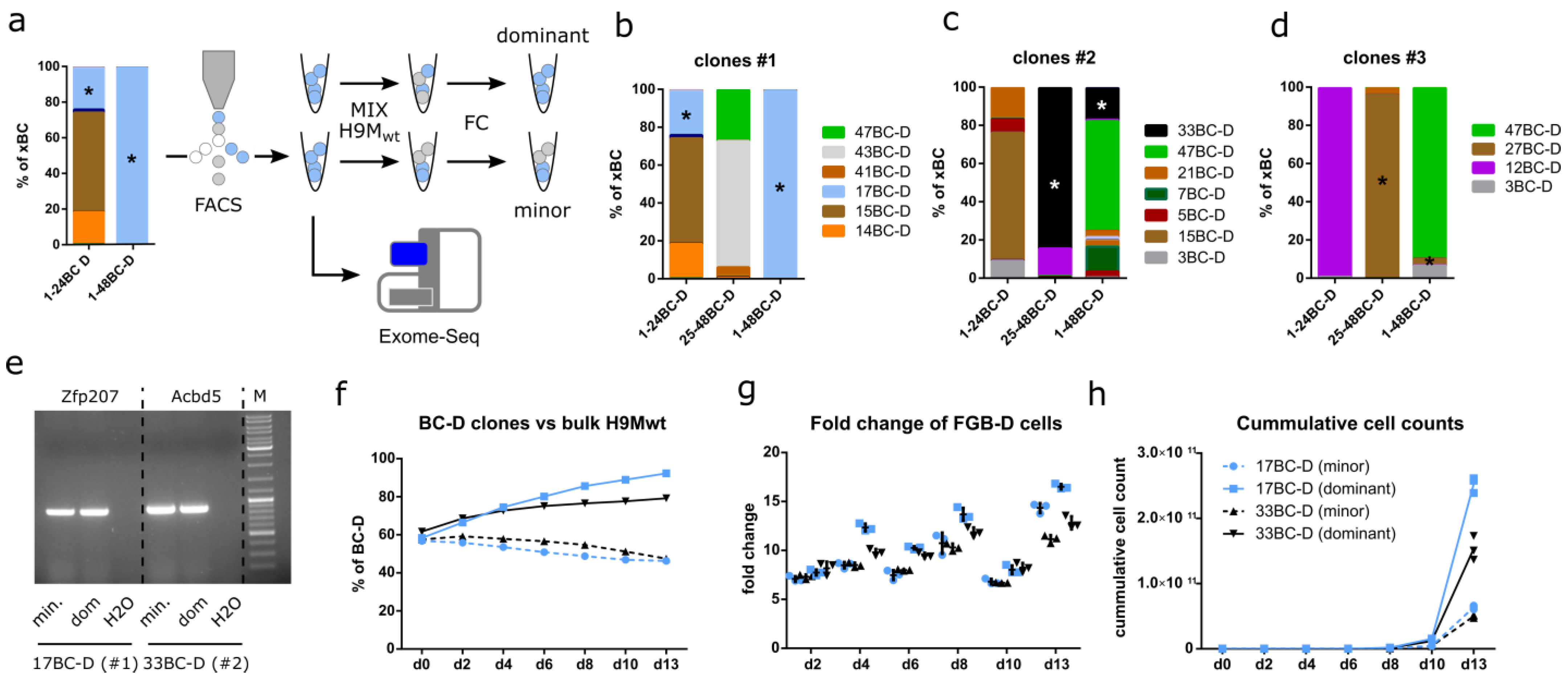
3.7. Growth Characteristics Are Stably Maintained in Purified Clones
3.8. Exome Sequencing Identifies Unique Differences between Paired Clones
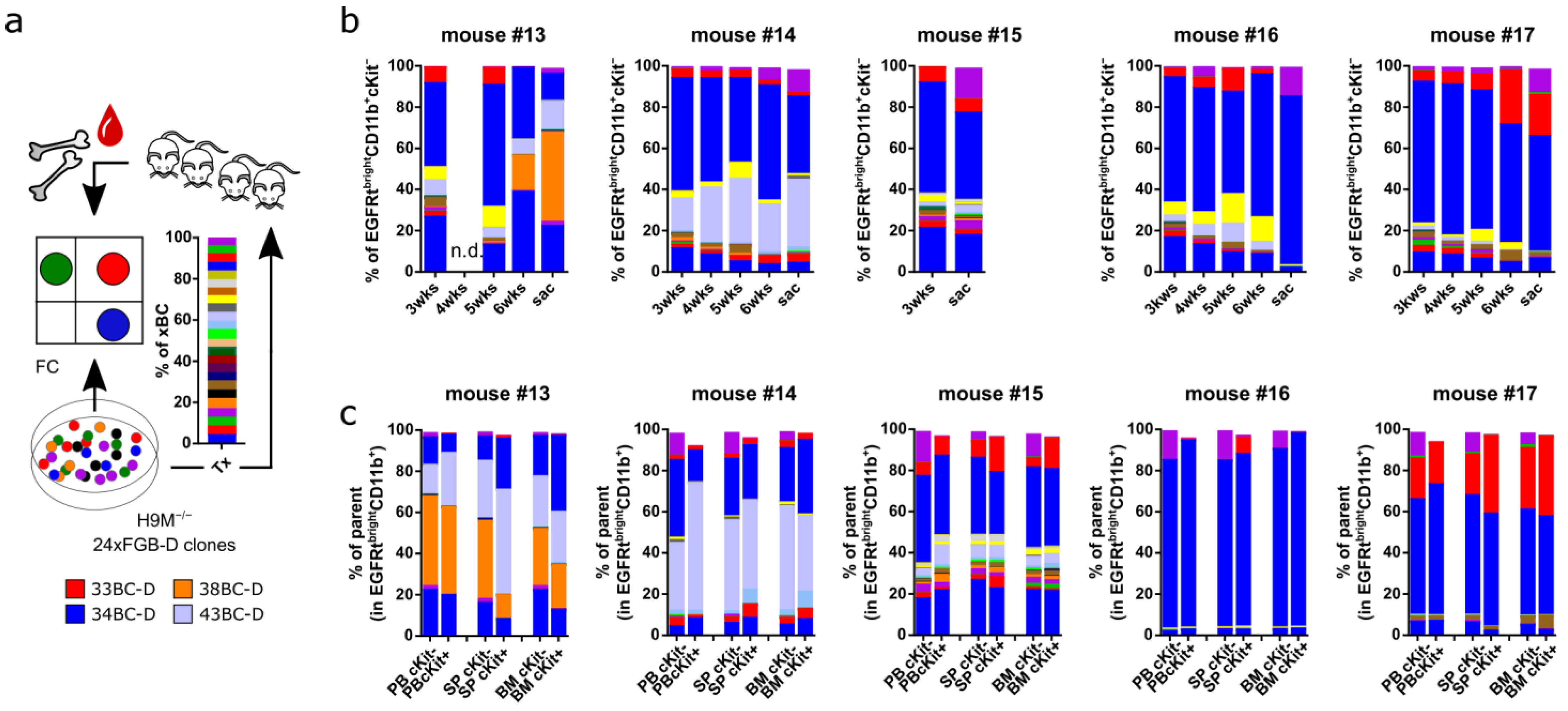
3.9. In Vivo Leukemogenesis Combines Stochastic and Pre-Determined Clonal Selection Mechanisms
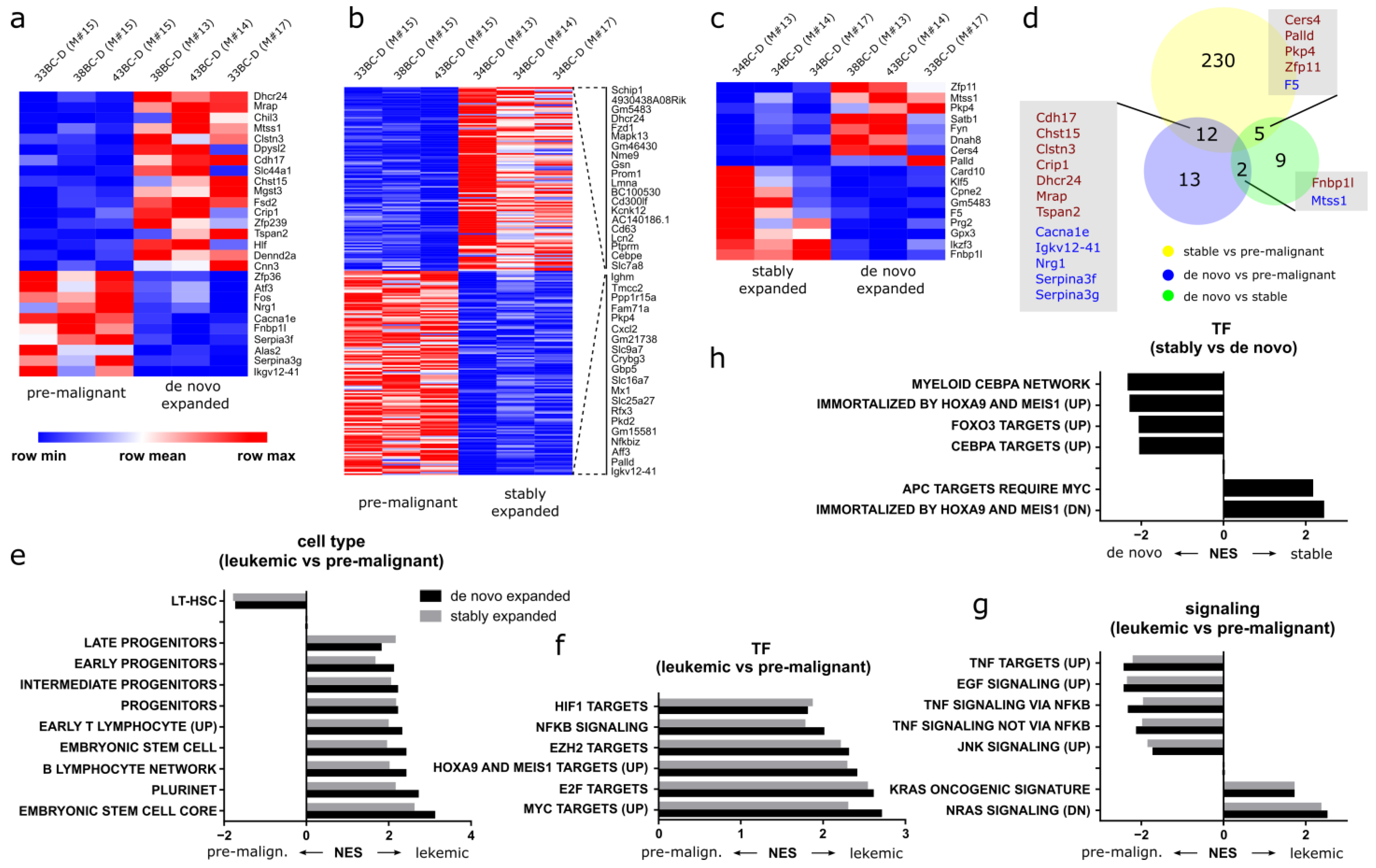
3.10. Distinct Gene Expression Programs Underlie Clonal Expansion and Maintenance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The Origin and Evolution of Mutations in Acute Myeloid Leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Clonal Evolution of Acute Myeloid Leukemia Revealed by High-Throughput Single-Cell Genomics. Nat. Commun. 2020, 11, 5327. [Google Scholar] [CrossRef] [PubMed]
- Corces-Zimmerman, M.R.; Hong, W.J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic Mutations in Human Acute Myeloid Leukemia Affect Epigenetic Regulators and Persist in Remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal Evolution of Preleukemic Hematopoietic Stem Cells Precedes Human Acute Myeloid Leukemia. Sci. Transl. Med. 2012, 4, 149ra118. [Google Scholar] [CrossRef] [Green Version]
- Eagle, K.; Harada, T.; Kalfon, J.; Perez, M.W.; Heshmati, Y.; Ewers, J.; Koren, J.V.; Dempster, J.M.; Kugener, G.; Paralkar, V.R.; et al. Transcriptional Plasticity Drives Leukemia Immune Escape. Blood Cancer Discov. 2022, 3, 394–409. [Google Scholar] [CrossRef]
- Zeng, A.G.X.; Bansal, S.; Jin, L.; Mitchell, A.; Chen, W.C.; Abbas, H.A.; Chan-Seng-Yue, M.; Voisin, V.; van Galen, P.; Tierens, A.; et al. A Cellular Hierarchy Framework for Understanding Heterogeneity and Predicting Drug Response in Acute Myeloid Leukemia. Nat. Med. 2022, 28, 1212–1223. [Google Scholar] [CrossRef]
- De Boer, B.; Prick, J.; Pruis, M.G.; Keane, P.; Imperato, M.R.; Jaques, J.; Brouwers-Vos, A.Z.; Hogeling, S.M.; Woolthuis, C.M.; Nijk, M.T.; et al. Prospective Isolation and Characterization of Genetically and Functionally Distinct AML Subclones. Cancer Cell 2018, 34, 674–689.e8. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal Evolution in Relapsed Acute Myeloid Leukaemia Revealed by Whole-Genome Sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [Green Version]
- Klco, J.M.; Spencer, D.H.; Miller, C.A.; Griffith, M.; Lamprecht, T.L.; O’Laughlin, M.; Fronick, C.; Magrini, V.; Demeter, R.T.; Fulton, R.S.; et al. Functional Heterogeneity of Genetically Defined Subclones in Acute Myeloid Leukemia. Cancer Cell 2014, 25, 379–392. [Google Scholar] [CrossRef] [Green Version]
- van Galen, P.; Hovestadt, V.; Wadsworth II, M.H.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281.e24. [Google Scholar] [CrossRef]
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single-Cell Mutation Analysis of Clonal Evolution in Myeloid Malignancies. Nature 2020, 587, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Velten, L.; Story, B.A.; Hernández-Malmierca, P.; Raffel, S.; Leonce, D.R.; Milbank, J.; Paulsen, M.; Demir, A.; Szu-Tu, C.; Frömel, R.; et al. Identification of Leukemic and Pre-Leukemic Stem Cells by Clonal Tracking from Single-Cell Transcriptomics. Nat. Commun. 2021, 12, 1366. [Google Scholar] [CrossRef] [PubMed]
- Mohme, M.; Maire, C.L.; Riecken, K.; Zapf, S.; Aranyossy, T.; Westphal, M.; Lamszus, K.; Fehse, B. Optical Barcoding for Single-Clone Tracking to Study Tumor Heterogeneity. Mol. Ther. 2017, 25, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Maire, C.L.; Mohme, M.; Bockmayr, M.; Fita, K.D.; Riecken, K.; Börnigen, D.; Alawi, M.; Failla, A.; Kolbe, K.; Zapf, S.; et al. Glioma Escape Signature and Clonal Development under Immune Pressure. J. Clin. Investig. 2020, 130, 5257–5271. [Google Scholar] [CrossRef] [PubMed]
- Maetzig, T.; Ruschmann, J.; Sanchez Milde, L.; Lai, C.K.; von Krosigk, N.; Humphries, R.K. Lentiviral Fluorescent Genetic Barcoding for Multiplex Fate Tracking of Leukemic Cells. Mol. Ther.—Methods Clin. Dev. 2017, 6, 54–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maetzig, T.; Ruschmann, J.; Lai, C.K.; Ngom, M.; Imren, S.; Rosten, P.; Norddahl, G.L.; von Krosigk, N.; Sanchez Milde, L.; May, C.; et al. A Lentiviral Fluorescent Genetic Barcoding System for Flow Cytometry-Based Multiplex Tracking. Mol. Ther. 2017, 25, 606–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohr, S.; Doebele, C.; Comoglio, F.; Berg, T.; Beck, J.; Bohnenberger, H.; Alexe, G.; Corso, J.; Strobel, P.; Wachter, A.; et al. Hoxa9 and Meis1 Cooperatively Induce Addiction to Syk Signaling by Suppressing MiR-146a in Acute Myeloid Leukemia. Cancer Cell 2017, 31, 549–562.e11. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, J.M.; Kettyle, L.M.; Sharpe, D.J.; Mulgrew, N.M.; Dickson, G.J.; Bijl, J.J.; Austin, P.; Mayotte, N.; Cellot, S.; Lappin, T.R.; et al. Entinostat Prevents Leukemia Maintenance in a Collaborating Oncogene-Dependent Model of Cytogenetically Normal Acute Myeloid Leukemia. Stem Cells 2013, 31, 1434–1445. [Google Scholar] [CrossRef]
- Fennell, K.A.; Vassiliadis, D.; Lam, E.Y.N.; Martelotto, L.G.; Balic, J.J.; Hollizeck, S.; Weber, T.S.; Semple, T.; Wang, Q.; Miles, D.C.; et al. Non-Genetic Determinants of Malignant Clonal Fitness at Single-Cell Resolution. Nature 2022, 601, 125–131. [Google Scholar] [CrossRef]
- Hassan, J.J.; Lieske, A.; Dörpmund, N.; Klatt, D.; Hoffmann, D.; Kleppa, M.-J.; Kustikova, O.S.; Stahlhut, M.; Schwarzer, A.; Schambach, A.; et al. A Multiplex CRISPR-Screen Identifies PLA2G4A as Prognostic Marker and Druggable Target for HOXA9 and MEIS1 Dependent AML. Int. J. Mol. Sci. 2021, 22, 9411. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy Quantitative Assessment of Genome Editing by Sequence Trace Decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chang, W.-C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A Transgene-Encoded Cell Surface Polypeptide for Selection, in Vivo Tracking, and Ablation of Engineered Cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, S.; Araki, T.; Yamamoto-Hino, M.; Miyawaki, A. A Green-Emitting Fluorescent Protein from Galaxeidae Coral and Its Monomeric Version for Use in Fluorescent Labeling. J. Biol. Chem. 2003, 278, 34167–34171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maunder, H.E.; Wright, J.; Kolli, B.R.; Vieira, C.R.; Mkandawire, T.T.; Tatoris, S.; Kennedy, V.; Iqball, S.; Devarajan, G.; Ellis, S.; et al. Enhancing Titres of Therapeutic Viral Vectors Using the Transgene Repression in Vector Production (TRiP) System. Nat. Commun. 2017, 8, 14834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutner, R.H.; Zhang, X.-Y.; Reiser, J. Production, Concentration and Titration of Pseudotyped HIV-1-Based Lentiviral Vectors. Nat. Protoc. 2009, 4, 495–505. [Google Scholar] [CrossRef]
- Santiago, Y.; Chan, E.; Liu, P.-Q.; Orlando, S.; Zhang, L.; Urnov, F.D.; Holmes, M.C.; Guschin, D.; Waite, A.; Miller, J.C.; et al. Targeted Gene Knockout in Mammalian Cells by Using Engineered Zinc-Finger Nucleases. Proc. Natl. Acad. Sci. USA 2008, 105, 5809–5814. [Google Scholar] [CrossRef] [Green Version]
- Berry, C.C.; Nobles, C.; Six, E.; Wu, Y.; Malani, N.; Sherman, E.; Dryga, A.; Everett, J.K.; Male, F.; Bailey, A.; et al. INSPIIRED: Quantification and Visualization Tools for Analyzing Integration Site Distributions. Mol. Ther.—Methods Clin. Dev. 2017, 4, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Sherman, E.; Nobles, C.; Berry, C.C.; Six, E.; Wu, Y.; Dryga, A.; Malani, N.; Male, F.; Reddy, S.; Bailey, A.; et al. INSPIIRED: A Pipeline for Quantitative Analysis of Sites of New DNA Integration in Cellular Genomes. Mol. Ther.—Methods Clin. Dev. 2017, 4, 39–49. [Google Scholar] [CrossRef]
- Ha, T.-C.; Stahlhut, M.; Rothe, M.; Paul, G.; Dziadek, V.; Morgan, M.; Brugman, M.; Fehse, B.; Kustikova, O.; Schambach, A.; et al. Multiple Genes Surrounding Bcl-x L, a Common Retroviral Insertion Site, Can Influence Hematopoiesis Individually or in Concert. Hum. Gene Ther. 2021, 32, 458–472. [Google Scholar] [CrossRef]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [CrossRef]
- Lieske, A.; Ha, T.-C.; Schambach, A.; Maetzig, T. An Improved Lentiviral Fluorescent Genetic Barcoding Approach Distinguishes Hematopoietic Stem Cell Properties in Multiplexed in Vivo Experiments. Hum. Gene Ther. 2021, 32, 1280–1294. [Google Scholar] [CrossRef] [PubMed]
- Mulgrew, N.M.; Kettyle, L.M.; Ramsey, J.M.; Cull, S.; Smyth, L.J.; Mervyn, D.M.; Bijl, J.J.; Thompson, A. C-Met Inhibition in a HOXA9/Meis1 Model of CN-AML. Dev. Dyn. 2014, 243, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Caiado, F.; Maia-Silva, D.; Jardim, C.; Schmolka, N.; Carvalho, T.; Reforço, C.; Faria, R.; Kolundzija, B.; Simões, A.E.; Baubec, T.; et al. Lineage Tracing of Acute Myeloid Leukemia Reveals the Impact of Hypomethylating Agents on Chemoresistance Selection. Nat. Commun. 2019, 10, 4986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, S.; Jackson, T.; Crump, N.T.; Fordham, N.; Elliott, N.; O’Byrne, S.; Fanego, M.; del Mar Lara Fanego, L.; Addy, D.; Crabb, T.; et al. A Human Fetal Liver-Derived Infant MLL-AF4 Acute Lymphoblastic Leukemia Model Reveals a Distinct Fetal Gene Expression Program. Nat. Commun. 2021, 12, 6905. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Yun, H.; Berg, T.; Yung, E.; Argiropoulos, B.; Kuchenbauer, F.; Park, G.; Hamwi, I.; Palmqvist, L.; Lai, C.K.; et al. Cell of Origin in AML: Susceptibility to MN1-Induced Transformation Is Regulated by the MEIS1/AbdB-like HOX Protein Complex. Cancer Cell 2011, 20, 39–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basilico, S.; Wang, X.; Kennedy, A.; Tzelepis, K.; Giotopoulos, G.; Kinston, S.J.; Quiros, P.M.; Wong, K.; Adams, D.J.; Carnevalli, L.S.; et al. Dissecting the Early Steps of MLL Induced Leukaemogenic Transformation Using a Mouse Model of AML. Nat. Commun. 2020, 11, 1407. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting Cancer Stem Cell Pathways for Cancer Therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Bystrykh, L.V.; Verovskaya, E.; Zwart, E.; Broekhuis, M.; de Haan, G. Counting Stem Cells: Methodological Constraints. Nat. Methods 2012, 9, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Herault, O.; Hope, K.J.; Deneault, E.; Mayotte, N.; Chagraoui, J.; Wilhelm, B.T.; Cellot, S.; Sauvageau, M.; Andrade-Navarro, M.A.; Hébert, J.; et al. A Role for GPx3 in Activity of Normal and Leukemia Stem Cells. J. Exp. Med. 2012, 209, 895–901. [Google Scholar] [CrossRef]
- Kagoya, Y.; Yoshimi, A.; Kataoka, K.; Nakagawa, M.; Kumano, K.; Arai, S.; Kobayashi, H.; Saito, T.; Iwakura, Y.; Kurokawa, M. Positive Feedback between NF-ΚB and TNF-α Promotes Leukemia-Initiating Cell Capacity. J. Clin. Investig. 2014, 124, 528–542. [Google Scholar] [CrossRef] [Green Version]
- Volk, A.; Li, J.; Xin, J.; You, D.; Zhang, J.; Liu, X.; Xiao, Y.; Breslin, P.; Li, Z.; Wei, W.; et al. Co-Inhibition of NF-ΚB and JNK Is Synergistic in TNF-Expressing Human AML. J. Exp. Med. 2014, 211, 1093–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapellier, M.; Peña-Martínez, P.; Ramakrishnan, R.; Eriksson, M.; Talkhoncheh, M.S.; Orsmark-Pietras, C.; Lilljebjörn, H.; Högberg, C.; Hagström-Andersson, A.; Fioretos, T.; et al. Arrayed Molecular Barcoding Identifies TNFSF13 as a Positive Regulator of Acute Myeloid Leukemia-Initiating Cells. Haematologica 2019, 104, 2006–2016. [Google Scholar] [CrossRef]
- Birkenkamp, K.U.; Geugien, M.; Schepers, H.; Westra, J.; Lemmink, H.H.; Vellenga, E. Constitutive NF-ΚB DNA-Binding Activity in AML Is Frequently Mediated by a Ras/PI3-K/PKB-Dependent Pathway. Leukemia 2004, 18, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, H.; Yoshida, K.; Nakatani, K.; Harata, Y.; Higashitani, M.; Ito, Y.; Kamikubo, Y.; Shiozawa, Y.; Shiraishi, Y.; Chiba, K.; et al. Fusion Partner–Specific Mutation Profiles and KRAS Mutations as Adverse Prognostic Factors in MLL-Rearranged AML. Blood Adv. 2020, 4, 4623–4631. [Google Scholar] [CrossRef]
- Trentin, L.; Bresolin, S.; Giarin, E.; Bardini, M.; Serafin, V.; Accordi, B.; Fais, F.; Tenca, C.; De Lorenzo, P.; Valsecchi, M.G.; et al. Deciphering KRAS and NRAS Mutated Clone Dynamics in MLL-AF4 Paediatric Leukaemia by Ultra Deep Sequencing Analysis. Sci. Rep. 2016, 6, 34449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driessen, E.M.C.; van Roon, E.H.J.; Spijkers-Hagelstein, J.A.P.; Schneider, P.; de Lorenzo, P.; Valsecchi, M.G.; Pieters, R.; Stam, R.W. Frequencies and Prognostic Impact of RAS Mutations in MLL-Rearranged Acute Lymphoblastic Leukemia in Infants. Haematologica 2013, 98, 937–944. [Google Scholar] [CrossRef]
- Lavallee, V.P.; Baccelli, I.; Krosl, J.; Wilhelm, B.; Barabe, F.; Gendron, P.; Boucher, G.; Lemieux, S.; Marinier, A.; Meloche, S.; et al. The Transcriptomic Landscape and Directed Chemical Interrogation of MLL-Rearranged Acute Myeloid Leukemias. Nat. Genet. 2015, 47, 1030–1037. [Google Scholar] [CrossRef]
- Daniluk, J.; Liu, Y.; Deng, D.; Chu, J.; Huang, H.; Gaiser, S.; Cruz-Monserrate, Z.; Wang, H.; Ji, B.; Logsdon, C.D. An NF-ΚB Pathway–Mediated Positive Feedback Loop Amplifies Ras Activity to Pathological Levels in Mice. J. Clin. Investig. 2012, 122, 1519–1528. [Google Scholar] [CrossRef] [Green Version]
- Corces-Zimmerman, M.R.; Majeti, R. Pre-Leukemic Evolution of Hematopoietic Stem Cells: The Importance of Early Mutations in Leukemogenesis. Leukemia 2014, 28, 2276–2282. [Google Scholar] [CrossRef]
- Dobson, S.M.; García-Prat, L.; Vanner, R.J.; Wintersinger, J.; Waanders, E.; Gu, Z.; McLeod, J.; Gan, O.I.; Grandal, I.; Payne-Turner, D.; et al. Relapse-Fated Latent Diagnosis Subclones in Acute B Lineage Leukemia Are Drug Tolerant and Possess Distinct Metabolic Programs. Cancer Discov. 2020, 10, 568–587. [Google Scholar] [CrossRef]
- Pilheden, M.; Ahlgren, L.; Hyrenius-Wittsten, A.; Gonzalez-Pena, V.; Sturesson, H.; Hansen Marquart, H.V.; Lausen, B.; Castor, A.; Pronk, C.J.; Barbany, G.; et al. Duplex Sequencing Uncovers Recurrent Low-Frequency Cancer-Associated Mutations in Infant and Childhood KMT2A-Rearranged Acute Leukemia. HemaSphere 2022, 6, e785. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Lee, K.L.; Luo, J. Control Dominating Subclones for Managing Cancer Progression and Posttreatment Recurrence by Subclonal Switchboard Signal: Implication for New Therapies. Stem Cells Dev. 2012, 21, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.X.; Li, S.C. Hunting down the Dominating Subclone of Cancer Stem Cells as a Potential New Therapeutic Target in Multiple Myeloma: An Artificial Intelligence Perspective. World J. Stem Cells 2020, 12, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous Epitope and Transcriptome Measurement in Single Cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maetzig, T.; Lieske, A.; Dörpmund, N.; Rothe, M.; Kleppa, M.-J.; Dziadek, V.; Hassan, J.J.; Dahlke, J.; Borchert, D.; Schambach, A. Real-Time Characterization of Clonal Fate Decisions in Complex Leukemia Samples by Fluorescent Genetic Barcoding. Cells 2022, 11, 4045. https://doi.org/10.3390/cells11244045
Maetzig T, Lieske A, Dörpmund N, Rothe M, Kleppa M-J, Dziadek V, Hassan JJ, Dahlke J, Borchert D, Schambach A. Real-Time Characterization of Clonal Fate Decisions in Complex Leukemia Samples by Fluorescent Genetic Barcoding. Cells. 2022; 11(24):4045. https://doi.org/10.3390/cells11244045
Chicago/Turabian StyleMaetzig, Tobias, Anna Lieske, Nicole Dörpmund, Michael Rothe, Marc-Jens Kleppa, Violetta Dziadek, Jacob Jalil Hassan, Julia Dahlke, Dorit Borchert, and Axel Schambach. 2022. "Real-Time Characterization of Clonal Fate Decisions in Complex Leukemia Samples by Fluorescent Genetic Barcoding" Cells 11, no. 24: 4045. https://doi.org/10.3390/cells11244045