
SCAND1 Reverses Epithelial-to-Mesenchymal Transition (EMT) and Suppresses Prostate Cancer Growth and Migration
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. cDNA Transfection and Stable Cell Cloning
2.3. Lentiviral Infection and Cell Cloning
2.4. siRNA
2.5. qRT-PCR
2.6. Western Blotting
2.7. Post-translational modifications (PTM) and hotspot mutation
2.8. Immunocytochemistry and Confocal Microscopy
2.9. Xenograft
2.10. Immunohistochemistry (IHC)
2.11. Tissue-Specific Gene Expression
2.12. Co-Expression Analysis
2.13. Kaplan–Meier Analysis
2.14. Statistics
3. Results
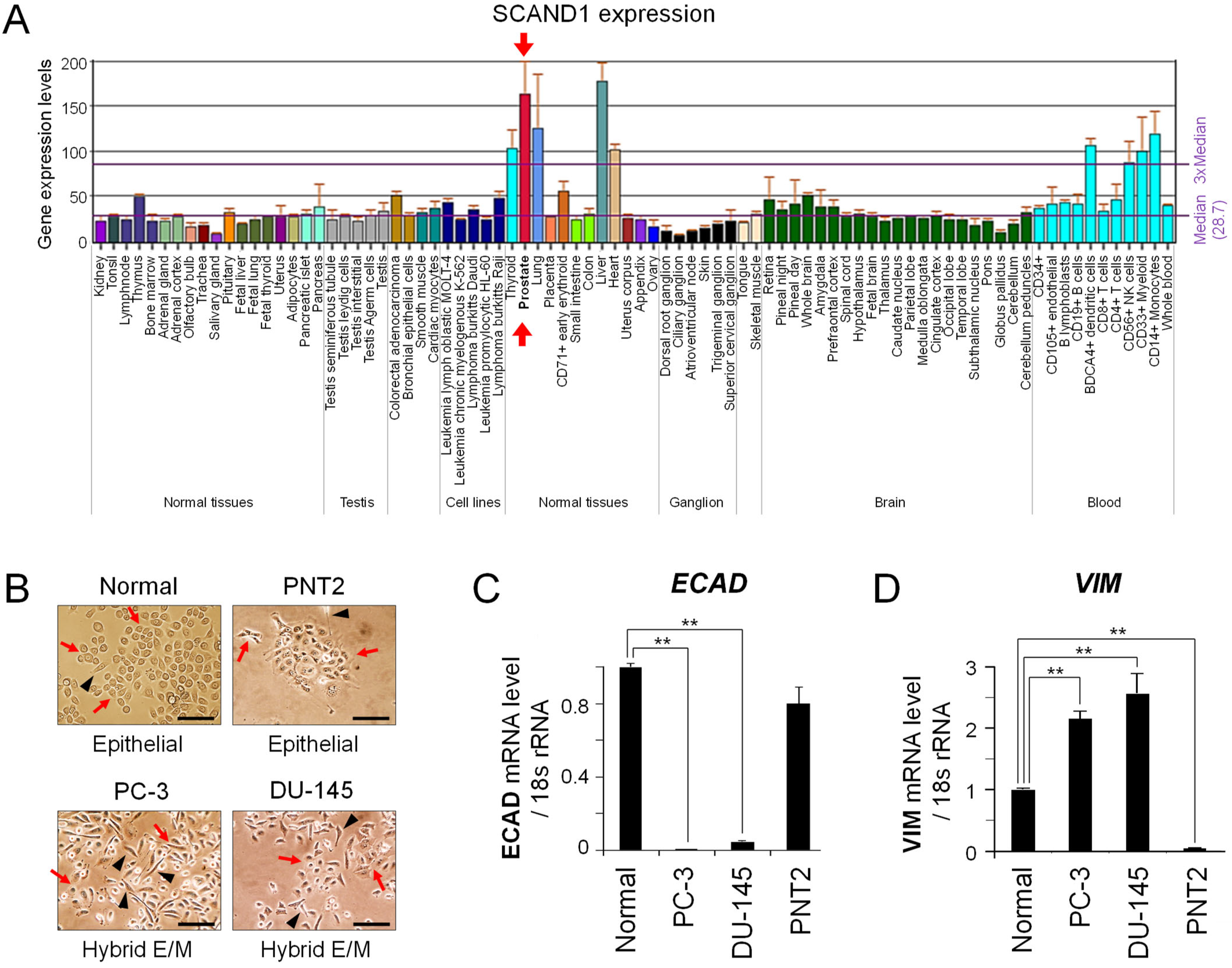
3.1. SCAND1 Expression Correlated with an Epithelial Phenotype
3.2. SCAND1 Is Mutually Inducible with MZF1(ZSCAN6) and Localized in Hetero-Chromatin
3.3. SCAND1 Inhibits Tumor Cell Proliferation and Reverses EMT
3.4. SCAND1 and MZF1 Expression Negatively Correlates with Gene Expression of MAP3Ks (MEKKs), MAPKs, and EMT Drivers
3.5. SCAND1 Overexpression Inhibited Collective Migration and Lymph-Node Metastasis of Prostate Cancer
3.6. SCAND1 and MZF1 Expression Correlated with the Prognosis of Patients Suffering from Cancers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nieto, M.A. Epithelial-Mesenchymal Transitions in development and disease: Old views and new perspectives. Int. J. Dev. Biol. 2009, 53, 1541–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, T.; Sogawa, C.; Ono, K.; Matsumoto, M.; Tran, M.T.; Okusha, Y.; Lang, B.J.; Okamoto, K.; Calderwood, S.K. Cell Stress Induced Stressome Release Including Damaged Membrane Vesicles and Extracellular HSP90 by Prostate Cancer Cells. Cells 2020, 9, 755. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Eguchi, T.; Sogawa, C.; Ono, K.; Murakami, J.; Ibaragi, S.; Asaumi, J.; Calderwood, S.K.; Okamoto, K.; Kozaki, K. Carcinogenic epithelial-mesenchymal transition initiated by oral cancer exosomes is inhibited by anti-EGFR antibody cetuximab. Oral Oncol. 2018, 86, 251–257. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Sogawa, C.; Kawai, H.; Tran, M.T.; Taha, E.A.; Lu, Y.; Oo, M.W.; Okusha, Y.; Okamura, H.; Ibaragi, S.; et al. Triple knockdown of CDC37, HSP90-alpha and HSP90-beta diminishes extracellular vesicles-driven malignancy events and macrophage M2 polarization in oral cancer. J. Extracell. Vesicles 2020, 9, 1769373. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Sogawa, C.; Okusha, Y.; Uchibe, K.; Iinuma, R.; Ono, K.; Nakano, K.; Murakami, J.; Itoh, M.; Arai, K.; et al. Organoids with Cancer Stem Cell-like Properties Secrete Exosomes and HSP90 in a 3D NanoEnvironment. PLoS ONE 2018, 13, e0191109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Mauri, F.; Song, Y.; de Cock, F.; Meeusen, B.; Swedlund, B.; Impens, F.; Van Haver, D.; Opitz, M.; Thery, M.; et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 2021, 589, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhan, H. Communication between EMT and PD-L1 signaling: New insights into tumor immune evasion. Cancer Lett. 2020, 468, 72–81. [Google Scholar] [CrossRef]
- Tadepally, H.D.; Burger, G.; Aubry, M. Evolution of C2H2-zinc finger genes and subfamilies in mammals: Species-specific duplication and loss of clusters, genes and effector domains. BMC Evol. Biol. 2008, 8, 176. [Google Scholar] [CrossRef]
- Edelstein, L.C.; Collins, T. The SCAN domain family of zinc finger transcription factors. Gene 2005, 359, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.L.; Stringer, K.F.; Maki, J.L.; Szauter, P.; Stone, J.R.; Collins, T. The SCAN domain defines a large family of zinc finger transcription factors. Gene 2003, 310, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, C.; Wang, H.; Honer, C.; Ding, W.; Koehn, J.; Lawrence, Q.; Coulis, C.M.; Wang, L.L.; Ballinger, D.; Bowen, B.R.; et al. The SCAN domain mediates selective oligomerization. J. Biol. Chem. 2000, 275, 17173–17179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.J.; Blacklow, S.C.; Collins, T. The zinc finger-associated SCAN box is a conserved oligomerization domain. Mol. Cell. Biol. 1999, 19, 8526–8535. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, T.; Prince, T.; Wegiel, B.; Calderwood, S.K. Role and Regulation of Myeloid Zinc Finger Protein 1 in Cancer. J. Cell. Biochem. 2015, 116, 2146–2154. [Google Scholar] [CrossRef] [Green Version]
- Sander, T.L.; Haas, A.L.; Peterson, M.J.; Morris, J.F. Identification of a novel SCAN box-related protein that interacts with MZF1B. The leucine-rich SCAN box mediates hetero- and homoprotein associations. J. Biol. Chem. 2000, 275, 12857–12867. [Google Scholar] [CrossRef] [Green Version]
- Perrotti, D.; Melotti, P.; Skorski, T.; Casella, I.; Peschle, C.; Calabretta, B. Overexpression of the zinc finger protein MZF1 inhibits hematopoietic development from embryonic stem cells: Correlation with negative regulation of CD34 and c-myb promoter activity. Mol. Cell. Biol. 1995, 15, 6075–6087. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, T.; Prince, T.L.; Tran, M.T.; Sogawa, C.; Lang, B.J.; Calderwood, S.K. MZF1 and SCAND1 Reciprocally Regulate CDC37 Gene Expression in Prostate Cancer. Cancers 2019, 11, 792. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Jiao, W.; Mei, H.; Song, H.; Li, D.; Xiang, X.; Chen, Y.; Yang, F.; Li, H.; Huang, K.; et al. miRNA-337-3p inhibits gastric cancer progression through repressing myeloid zinc finger 1-facilitated expression of matrix metalloproteinase 14. Oncotarget 2016, 7, 40314–40328. [Google Scholar] [CrossRef] [Green Version]
- Ko, H.; Kim, S.; Yang, K.; Kim, K. Phosphorylation-dependent stabilization of MZF1 upregulates N-cadherin expression during protein kinase CK2-mediated epithelial-mesenchymal transition. Oncogenesis 2018, 7, 27. [Google Scholar] [CrossRef]
- Luan, H.; Mohapatra, B.; Bielecki, T.A.; Mushtaq, I.; Mirza, S.; Jennings, T.A.; Clubb, R.J.; An, W.; Ahmed, D.; El-Ansari, R.; et al. Loss of the Nuclear Pool of Ubiquitin Ligase CHIP/STUB1 in Breast Cancer Unleashes the MZF1-Cathepsin Pro-oncogenic Program. Cancer Res. 2018, 78, 2524–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, N.K.; Gadi, A.; Maurizi, G.; Roy, U.B.; Mansukhani, A.; Basilico, C. Myeloid Zinc Finger 1 and GA Binding Protein Co-Operate with Sox2 in Regulating the Expression of Yes-Associated Protein 1 in Cancer Cells. Stem Cells 2017, 35, 2340–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Han, L.; Zhou, C.; Wei, W.; Chen, X.; Yi, H.; Wu, X.; Bai, X.; Guo, S.; Yu, Y.; et al. TGF-beta1-induced CK17 enhances cancer stem cell-like properties rather than EMT in promoting cervical cancer metastasis via the ERK1/2-MZF1 signaling pathway. FEBS J. 2017, 284, 3000–3017. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.J.; Hwang, J.M.; Hsieh, S.C.; Ying, T.H.; Hsieh, Y.H. Overexpression of myeloid zinc finger 1 suppresses matrix metalloproteinase-2 expression and reduces invasiveness of SiHa human cervical cancer cells. Biochem. Biophys. Res. Commun. 2012, 425, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Tan, H.; Su, W.; Cheng, D.; Wang, G.; Wang, J.; Ma, D.A.; Dong, G.M.; Sun, P. MZF1 mediates oncogene-induced senescence by promoting the transcription of p16(INK4A). Oncogene 2022, 41, 414–426. [Google Scholar] [CrossRef]
- Weber, C.E.; Kothari, A.N.; Wai, P.Y.; Li, N.Y.; Driver, J.; Zapf, M.A.; Franzen, C.A.; Gupta, G.N.; Osipo, C.; Zlobin, A.; et al. Osteopontin mediates an MZF1-TGF-beta1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 2015, 34, 4821–4833. [Google Scholar] [CrossRef] [Green Version]
- Krüger, M.; Metzger, C.; Al-Nawas, B.; Kämmerer, P.W.; Brieger, J. Cigarette smoke modulates binding of the transcription factor MZF1 to the VEGF promoter and regulates VEGF expression in dependence of genetic variation SNP 405. J. Oral Pathol. Med. 2020, 49, 780–786. [Google Scholar] [CrossRef]
- Noll, L.; Peterson, F.C.; Hayes, P.L.; Volkman, B.F.; Sander, T. Heterodimer formation of the myeloid zinc finger 1 SCAN domain and association with promyelocytic leukemia nuclear bodies. Leuk. Res. 2008, 32, 1582–1592. [Google Scholar] [CrossRef]
- Eguchi, T.; Calderwood, S.K.; Takigawa, M.; Kubota, S.; Kozaki, K. Intracellular MMP3 Promotes HSP Gene Expression in Collaboration With Chromobox Proteins. J. Cell. Biochem. 2017, 118, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, T.; Kubota, S.; Kawata, K.; Mukudai, Y.; Uehara, J.; Ohgawara, T.; Ibaragi, S.; Sasaki, A.; Kuboki, T.; Takigawa, M. Novel transcription-factor-like function of human matrix metalloproteinase 3 regulating the CTGF/CCN2 gene. Mol. Cell. Biol. 2008, 28, 2391–2413. [Google Scholar] [CrossRef]
- Eguchi, T.; Lu, Y.; Taha, E.A.; Okusha, Y. Transfection, Spinfection, Exofection, and Luciferase Assays for Analysis of CCN Genes Expression Mechanism. In CCN Proteins: Methods and Protocols; Takigawa, M., Ed.; Humana: New York, NY, USA, 2023; Volume 2582, pp. 103–126. [Google Scholar]
- Eguchi, T.; Watanabe, K.; Hara, E.S.; Ono, M.; Kuboki, T.; Calderwood, S.K. OstemiR: A novel panel of microRNA biomarkers in osteoblastic and osteocytic differentiation from mesencymal stem cells. PLoS ONE 2013, 8, e58796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okusha, Y.; Eguchi, T.; Tran, M.T.; Sogawa, C.; Yoshida, K.; Itagaki, M.; Taha, E.A.; Ono, K.; Aoyama, E.; Okamura, H.; et al. Extracellular Vesicles Enriched with Moonlighting Metalloproteinase Are Highly Transmissive, Pro-Tumorigenic, and Trans-Activates Cellular Communication Network Factor (CCN2/CTGF): CRISPR against Cancer. Cancers 2020, 12, 881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Tang, Z.; Zhang, W.; Ye, Z.; Liu, F. GEPIA2021: Integrating multiple deconvolution-based analysis into GEPIA. Nucleic Acids Res. 2021, 49, W242–W246. [Google Scholar] [CrossRef]
- Lánczky, A.; Győrffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Jongsma, J.; Oomen, M.H.; Noordzij, M.A.; Romijn, J.C.; van Der Kwast, T.H.; Schroder, F.H.; van Steenbrugge, G.J. Androgen-independent growth is induced by neuropeptides in human prostate cancer cell lines. Prostate 2000, 42, 34–44. [Google Scholar] [CrossRef]
- Li, C.F.; Chen, J.Y.; Ho, Y.H.; Hsu, W.H.; Wu, L.C.; Lan, H.Y.; Hsu, D.S.; Tai, S.K.; Chang, Y.C.; Yang, M.H. Snail-induced claudin-11 prompts collective migration for tumour progression. Nat. Cell Biol. 2019, 21, 251–262. [Google Scholar] [CrossRef]
- Chen, G.; Shukeir, N.; Potti, A.; Sircar, K.; Aprikian, A.; Goltzman, D.; Rabbani, S.A. Up-regulation of Wnt-1 and beta-catenin production in patients with advanced metastatic prostate carcinoma: Potential pathogenetic and prognostic implications. Cancer 2004, 101, 1345–1356. [Google Scholar] [CrossRef]
- Rau, T.T.; Bettschen, E.; Buchi, C.; Christe, L.; Rohner, A.; Muller, M.D.; Carlson, J.W.; Imboden, S.; Zlobec, I. Prognostic impact of tumor budding in endometrial carcinoma within distinct molecular subgroups. Mod. Pathol. 2021, 34, 222–232. [Google Scholar] [CrossRef]
- Mitrovic, B.; Schaeffer, D.F.; Riddell, R.H.; Kirsch, R. Tumor budding in colorectal carcinoma: Time to take notice. Mod. Pathol. 2012, 25, 1315–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.Q.; He, Q.; Yang, L.; Wang, S.B.; Yu, D.D.; He, Y.Q.; Hu, J.; Pan, Y.M.; Wu, Y. Clinical Significance of Myeloid Zinc Finger 1 Expression in the Progression of Gastric Tumourigenesis. Cell Physiol. Biochem. 2017, 44, 1242–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, K.; Eguchi, T.; Rahman, M.M.; Sakamoto, R.; Masuda, N.; Nakatsura, T.; Calderwood, S.K.; Kozaki, K.; Itoh, M. A Novel High-Throughput 3D Screening System for EMT Inhibitors: A Pilot Screening Discovered the EMT Inhibitory Activity of CDK2 Inhibitor SU9516. PLoS ONE 2016, 11, e0162394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sogawa, C.; Eguchi, T.; Namba, Y.; Okusha, Y.; Aoyama, E.; Ohyama, K.; Okamoto, K. Gel-Free 3D Tumoroids with Stem Cell Properties Modeling Drug Resistance to Cisplatin and Imatinib in Metastatic Colorectal Cancer. Cells 2021, 10, 344. [Google Scholar] [CrossRef]
- Taha, E.A.; Sogawa, C.; Okusha, Y.; Kawai, H.; Oo, M.W.; Elseoudi, A.; Lu, Y.; Nagatsuka, H.; Kubota, S.; Satoh, A.; et al. Knockout of MMP3 Weakens Solid Tumor Organoids and Cancer Extracellular Vesicles. Cancers 2020, 12, 1260. [Google Scholar] [CrossRef] [PubMed]
- Namba, Y.; Sogawa, C.; Okusha, Y.; Kawai, H.; Itagaki, M.; Ono, K.; Murakami, J.; Aoyama, E.; Ohyama, K.; Asaumi, J.; et al. Depletion of Lipid Efflux Pump ABCG1 Triggers the Intracellular Accumulation of Extracellular Vesicles and Reduces Aggregation and Tumorigenesis of Metastatic Cancer Cells. Front. Oncol. 2018, 8, 376. [Google Scholar] [CrossRef] [Green Version]
- Ferber, E.C.; Kajita, M.; Wadlow, A.; Tobiansky, L.; Niessen, C.; Ariga, H.; Daniel, J.; Fujita, Y. A role for the cleaved cytoplasmic domain of E-cadherin in the nucleus. J. Biol. Chem. 2008, 283, 12691–12700. [Google Scholar] [CrossRef] [Green Version]
- Cespedes, M.V.; Larriba, M.J.; Pavon, M.A.; Alamo, P.; Casanova, I.; Parreno, M.; Feliu, A.; Sancho, F.J.; Munoz, A.; Mangues, R. Site-dependent E-cadherin cleavage and nuclear translocation in a metastatic colorectal cancer model. Am. J. Pathol. 2010, 177, 2067–2079. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.J.; Chang, Y.W.; Lin, W.H.; Liang, C.L.; Lee, J.L. An aberrant nuclear localization of E-cadherin is a potent inhibitor of Wnt/beta-catenin-elicited promotion of the cancer stem cell phenotype. Oncogenesis 2015, 4, e157. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. TGF-beta and cancer. Cytokine Growth Factor Rev. 2006, 17, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Eguchi, T.; Sogawa, C.; Ono, K.; Murakami, J.; Ibaragi, S.; Asaumi, J.; Okamoto, K.; Calderwood, S.K.; Kozaki, K. Anti-EGFR antibody cetuximab is secreted by oral squamous cell carcinoma and alters EGF-driven mesenchymal transition. Biochem. Biophys. Res. Commun. 2018, 503, 1267–1272. [Google Scholar] [CrossRef]
- Wang, Q.; Liao, C.; Tan, Z.; Li, X.; Guan, X.; Li, H.; Tian, Z.; Liu, J.; An, J. FUT6 inhibits the proliferation, migration, invasion, and EGF-induced EMT of head and neck squamous cell carcinoma (HNSCC) by regulating EGFR/ERK/STAT signaling pathway. Cancer Gene Ther. 2022. [Google Scholar] [CrossRef] [PubMed]
- Slezak, J.; Truong, M.; Huang, W.; Jarrard, D. HP1γ expression is elevated in prostate cancer and is superior to Gleason score as a predictor of biochemical recurrence after radical prostatectomy. BMC Cancer 2013, 13, 148. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Fu, X.; Su, X.; Yang, A. CBX3/HP1gamma is upregulated in tongue squamous cell carcinoma and is associated with an unfavorable prognosis. Exp. Ther. Med. 2018, 15, 4271–4276. [Google Scholar]
- Mao, G.; Zheng, Y.; Lin, S.; Ma, L.; Zhou, Z.; Zhang, S. Bioinformatic Analysis of Prognostic Value, Genetic Interaction, and Immune Infiltration of Chromobox Family Proteins in Breast Cancer. Int. J. Gen. Med. 2021, 14, 9181–9191. [Google Scholar] [CrossRef]
- Li, Q.; Pan, Y.; Cao, Z.; Zhao, S. Comprehensive Analysis of Prognostic Value and Immune Infiltration of Chromobox Family Members in Colorectal Cancer. Front. Oncol. 2020, 10, 582667. [Google Scholar] [CrossRef]
- Chen, L.Y.; Cheng, C.S.; Qu, C.; Wang, P.; Chen, H.; Meng, Z.Q.; Chen, Z. Overexpression of CBX3 in Pancreatic Adenocar-cinoma Promotes Cell Cycle Transition-Associated Tumor Progression. Int. J. Mol. Sci. 2018, 19. [Google Scholar]
- Grimaldi, A.M.; Affinito, O.; Salvatore, M.; Franzese, M. CBX Family Members in Two Major Subtypes of Renal Cell Carcinoma: A Comparative Bioinformatic Analysis. Diagnostics 2022, 12. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Sun, S.X.; Zhu, S.X.; Bu, J. Identification of the Roles of Chromobox Family Members in Gastric Cancer: A Study Based on Multiple Datasets. Biomed. Res. Int. 2020, 2020, 5306509. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.C.; Lai, Y.C.; Chen, Y.C.; Wang, N.K.; Wang, W.S.; Lai, J.I. CBX3/heterochromatin protein 1 gamma is significantly upregulated in patients with non-small cell lung cancer. Asia. Pac. J. Clin. Oncol. 2018, 14, e283–e288. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, L.; Fanti, L.; Berloco, M.; Perrini, B.; Pimpinelli, S. Heterochromatin protein 1 (HP1) is associated with induced gene expression in Drosophila euchromatin. J. Cell. Biol. 2003, 161, 707–714. [Google Scholar] [CrossRef]
- Hediger, F.; Gasser, S.M. Heterochromatin protein 1: Don’t judge the book by its cover! Curr. Opin. Genet. Dev. 2006, 16, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Kubota, S.; Kawata, K.; Mukudai, Y.; Uehara, J.; Ohgawara, T.; Ibaragi, S.; Sasaki, A.; Kuboki, T.; Takigawa, M. Novel transcriptional regulation of CCN2/CTGF by nuclear translocation of MMP3. In CCN Proteins in Health and Disease; Perbal, A., Takigawa, M., Perbal, B., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 255–264. [Google Scholar]
- Smallwood, A.; Hon, G.C.; Jin, F.; Henry, R.E.; Espinosa, J.M.; Ren, B. CBX3 regulates efficient RNA processing genome-wide. Genome Res. 2012, 22, 1426–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, T.; Taha, E.A. Extracellular Vesicle-associated Moonlighting Proteins: Heat Shock Proteins and Metalloproteinases. In Heat Shock Proteins; Asea, A.A.A., Kaur, P., Eds.; Springer Nature: Cham, Switzerland, 2021; Volume 22, pp. 1–18. [Google Scholar]
- Okusha, Y.; Eguchi, T.; Sogawa, C.; Okui, T.; Nakano, K.; Okamoto, K.; Kozaki, K. The intranuclear PEX domain of MMP involves proliferation, migration, and metastasis of aggressive adenocarcinoma cells. J. Cell. Biochem. 2018, 119, 7363–7376. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, R.; Gonzales-Cope, M.; Chronis, C.; Bonora, G.; McKee, R.; Huang, C.; Patel, S.; Lopez, D.; Mishra, N.; Pellegrini, M.; et al. Proteomic and genomic approaches reveal critical functions of H3K9 methylation and heterochromatin protein-1γ in reprogramming to pluripotency. Nat. Cell. Biol. 2013, 15, 872–882. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequences (5′ to 3′) |
|---|---|
| h ECAD 1937F | AGG AAT CCA AAG CCT CAG GT |
| h ECAD 2065R | TTG GGT TGG GTC GTT GTA CT |
| h VIM 892F | AGG TGG ACC AGC TAA CCA AC |
| h VIM 1010R, | GGC TTC CTC TCT CTG AAG CA |
| h SCAND1 F#1 | CGC AGA GAA GCC AGA GAC TT |
| h SCAND1 R#10 | TCA GCA CTG CGT CTG CAC C |
| h MZF1-F785 | TGC AGG TGA AAG AGG AGT CA |
| h MZF1-R939 | AGT CTT GCT GTG GGG AAA GA |
| RPL32 F | CAG GGT TCG TAG AAG ATT CAA GGG |
| RPL32 R | CTT GGA AAC ATT GTG AGC GAT C |
| 18s rRNA-h1245F | GAC TCA ACA CGG GAA ACC TC |
| 18s rRNA-h1364R | AGA CAA ATC GCT CCA CCA AC |
| Cancer Type | Log-Rank P | Hazard Ratio | Correlation with SCAND1 and MZF1 High Expression | ||
|---|---|---|---|---|---|
| SCAND1 | MZF1 | SCAND1 | MZF1 | ||
| Pancreas DAC | 0.0041 ** | 0.0009 ** | 0.49 | 0.5 | Better prognosis |
| Head & Neck SCC, Stage III | 0.024 * | 0.018 * | 0.40 | 0.26 | Better prognosis |
| Renal Cell Carcinoma | 9.9 × 10−5 *** | 1.7 × 10−8 *** | 1.79 | 2.38 | Poorer prognosis |
| Prostate Adenocarcinoma | 0.15 | 0.002 ** | 0.21 | 0.0062 | MZF1: poorer prognosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eguchi, T.; Csizmadia, E.; Kawai, H.; Sheta, M.; Yoshida, K.; Prince, T.L.; Wegiel, B.; Calderwood, S.K. SCAND1 Reverses Epithelial-to-Mesenchymal Transition (EMT) and Suppresses Prostate Cancer Growth and Migration. Cells 2022, 11, 3993. https://doi.org/10.3390/cells11243993
Eguchi T, Csizmadia E, Kawai H, Sheta M, Yoshida K, Prince TL, Wegiel B, Calderwood SK. SCAND1 Reverses Epithelial-to-Mesenchymal Transition (EMT) and Suppresses Prostate Cancer Growth and Migration. Cells. 2022; 11(24):3993. https://doi.org/10.3390/cells11243993
Chicago/Turabian StyleEguchi, Takanori, Eva Csizmadia, Hotaka Kawai, Mona Sheta, Kunihiro Yoshida, Thomas L. Prince, Barbara Wegiel, and Stuart K. Calderwood. 2022. "SCAND1 Reverses Epithelial-to-Mesenchymal Transition (EMT) and Suppresses Prostate Cancer Growth and Migration" Cells 11, no. 24: 3993. https://doi.org/10.3390/cells11243993