
The N-Terminal Part of the 1A Domain of Desmin Is a Hot Spot Region for Putative Pathogenic DES Mutations Affecting Filament Assembly



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Generation and Site-Directed Mutagenesis
2.2. Cell Culture
2.3. Differentiation of Induced Pluripotent Stem Cells into Cardiomyocytes
2.4. Cell Transfection
2.5. Cell Fixation and Immunocytochemistry
2.6. Confocal Laser Scanning Microscopy
2.7. Molecular Visualization
2.8. Statistical Analysis
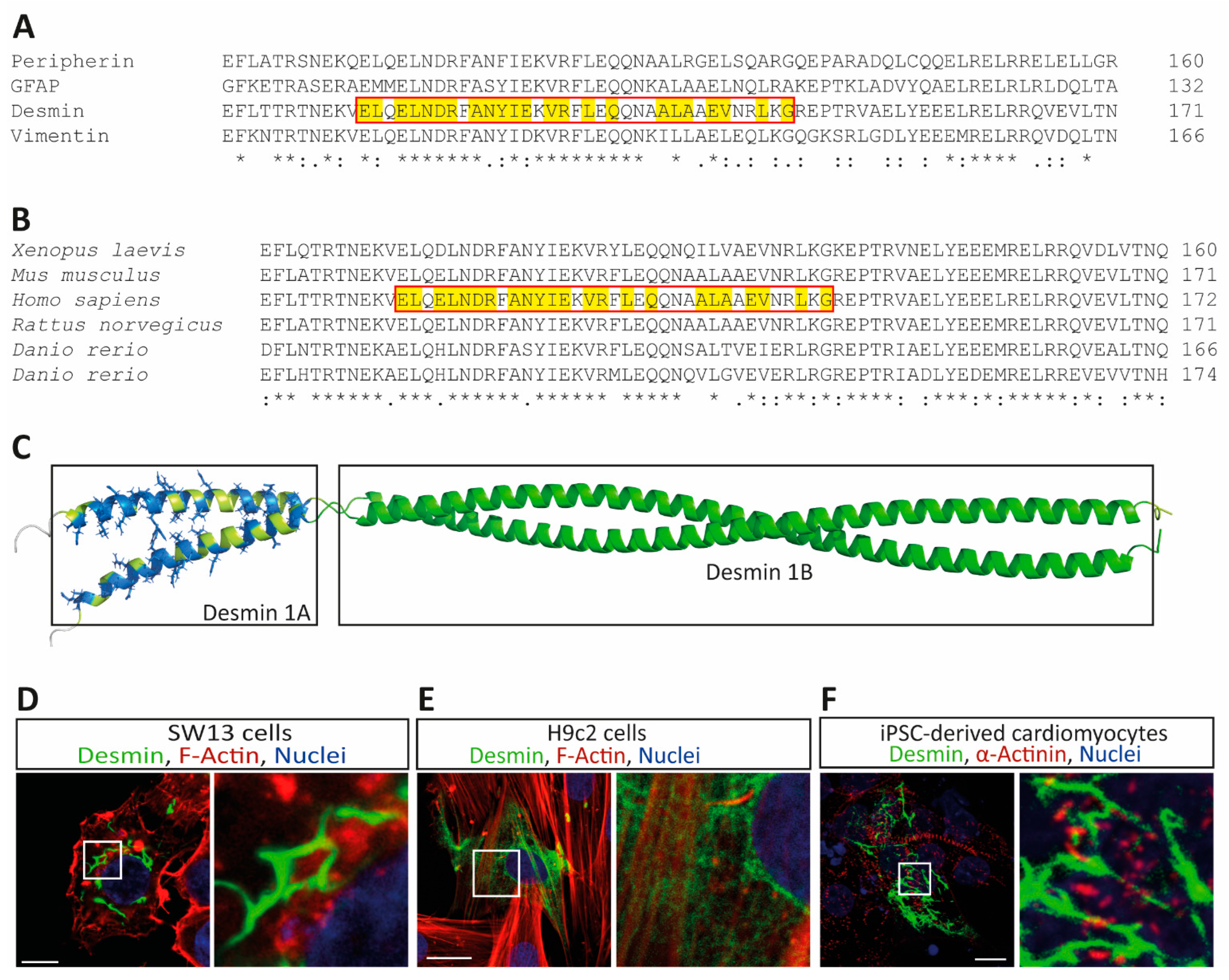
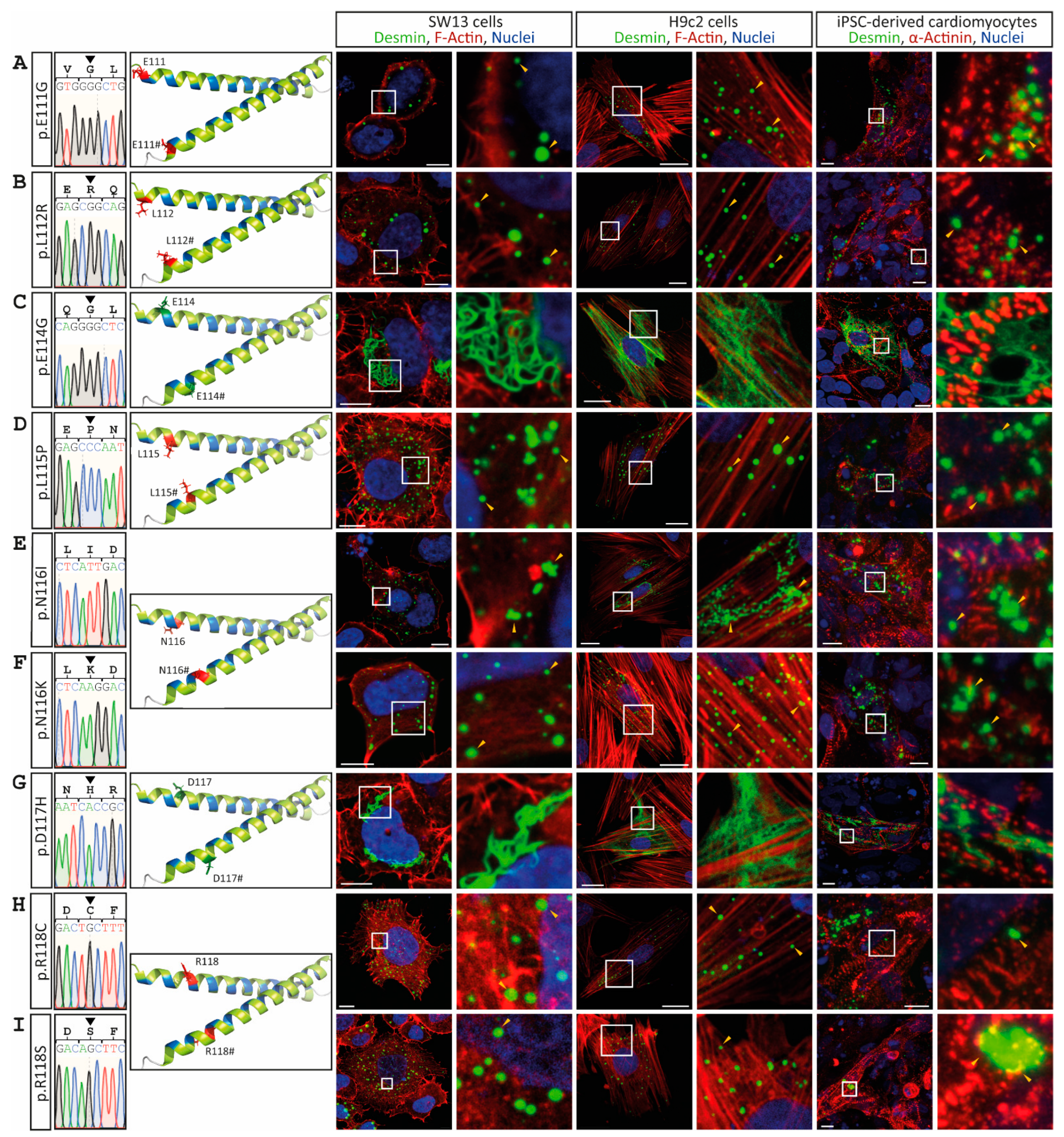
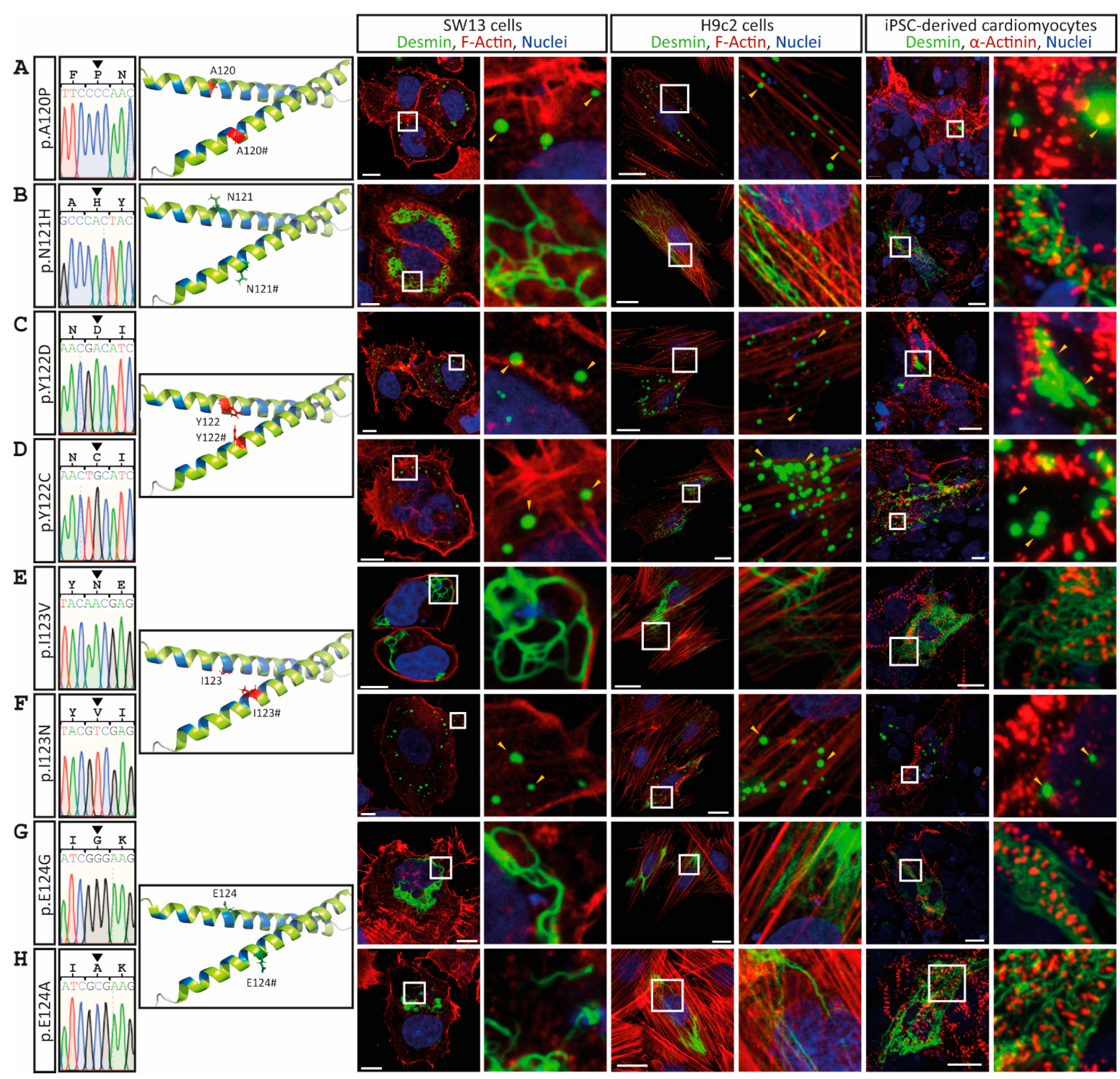
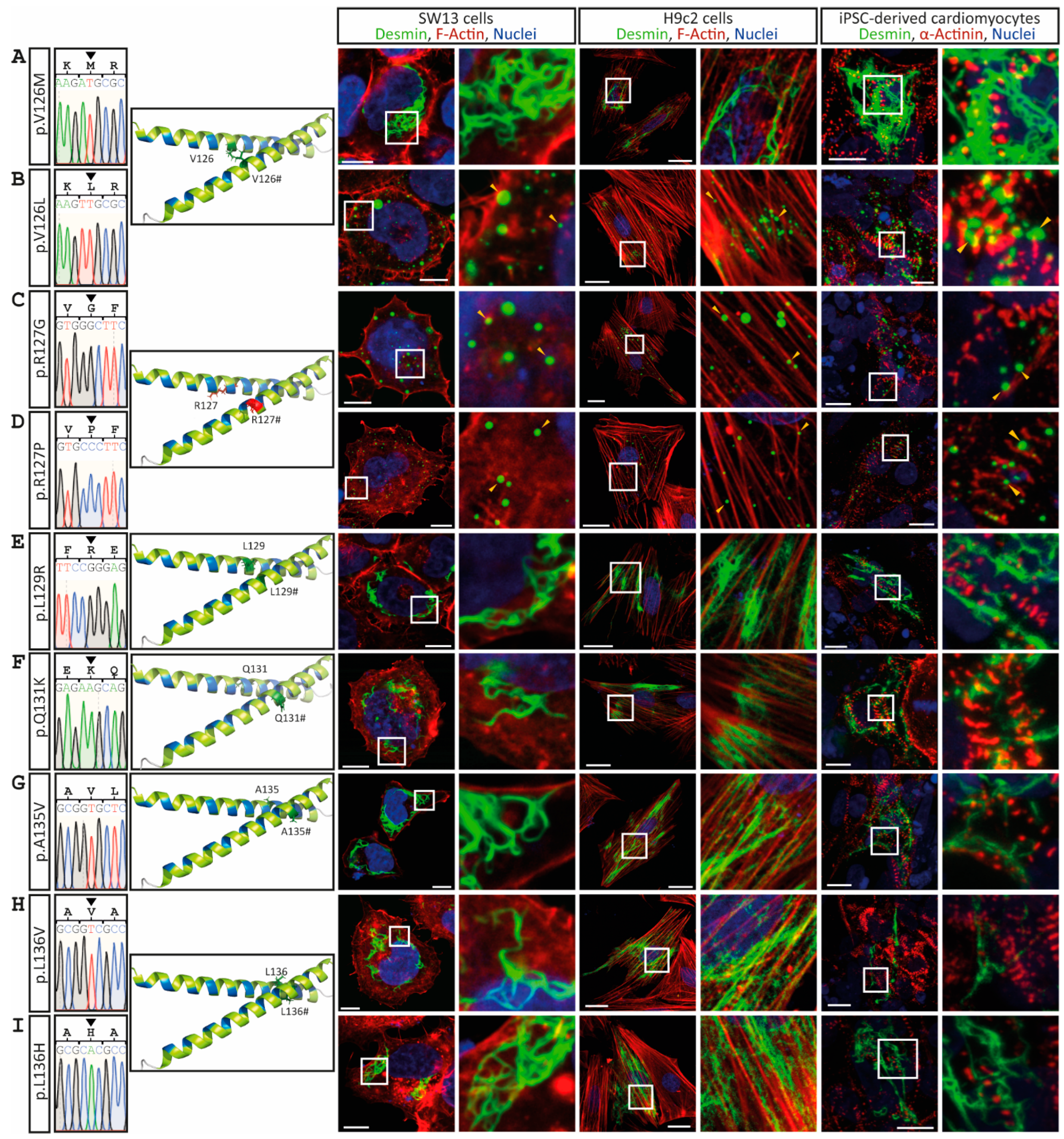
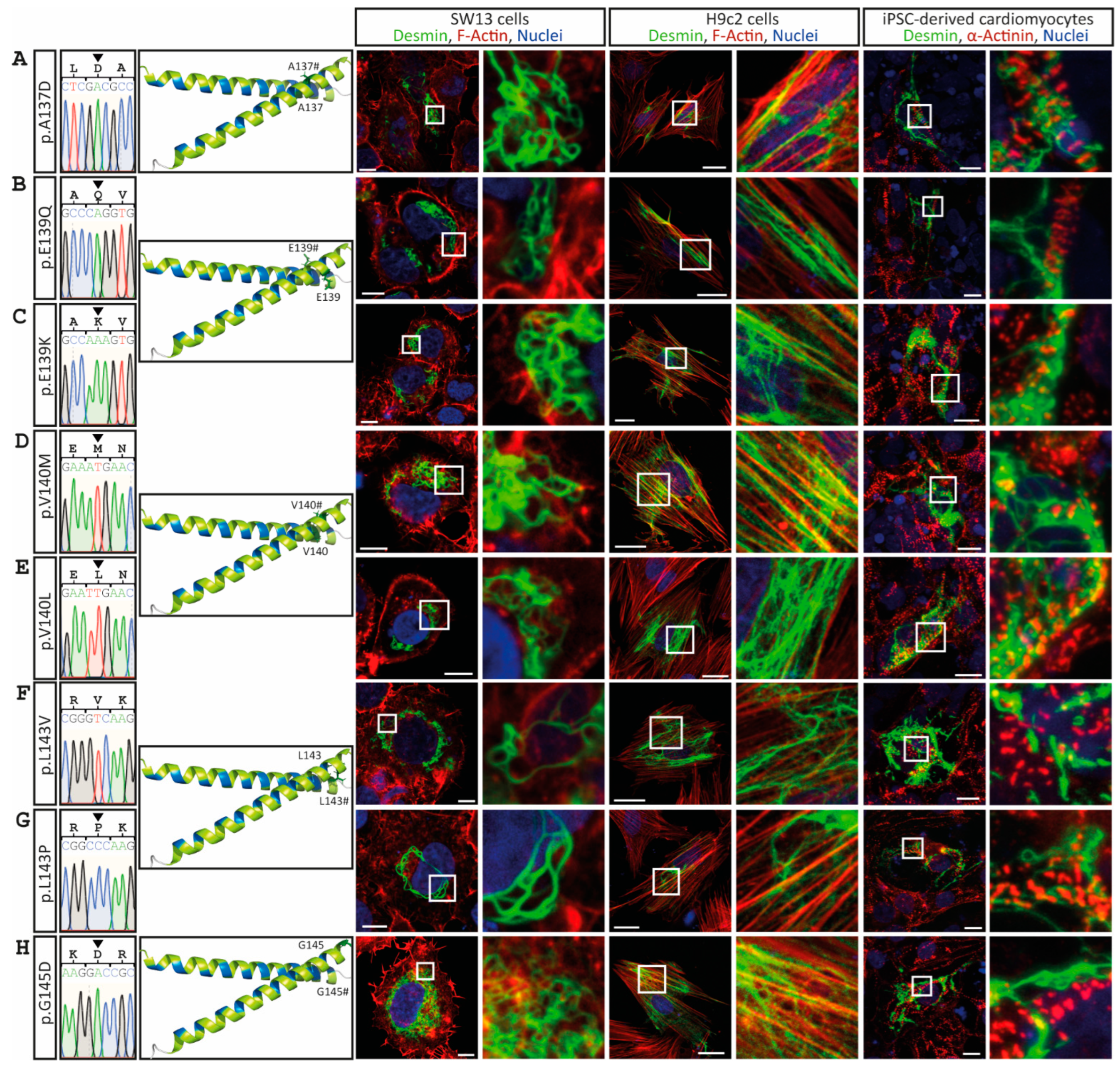
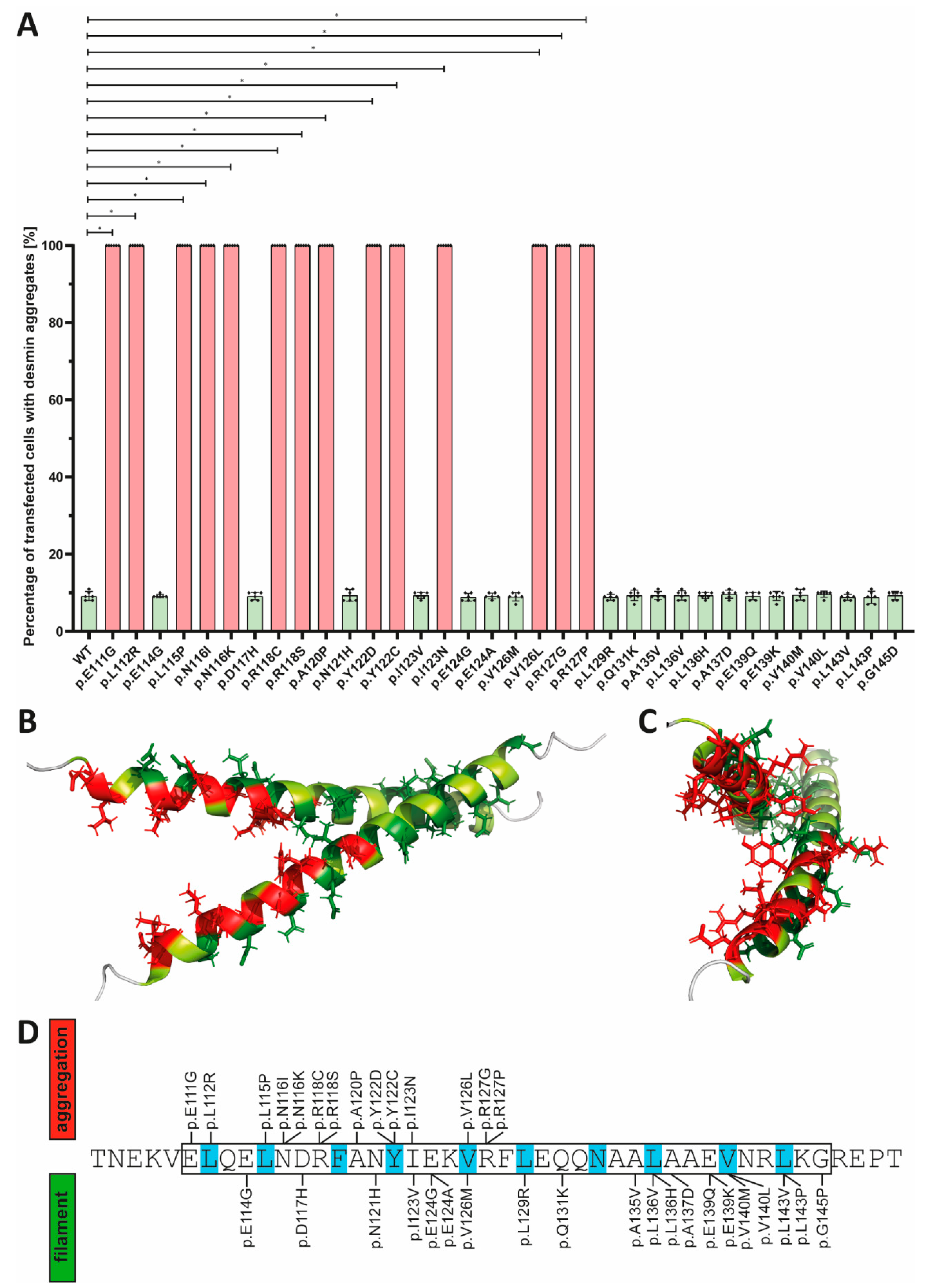
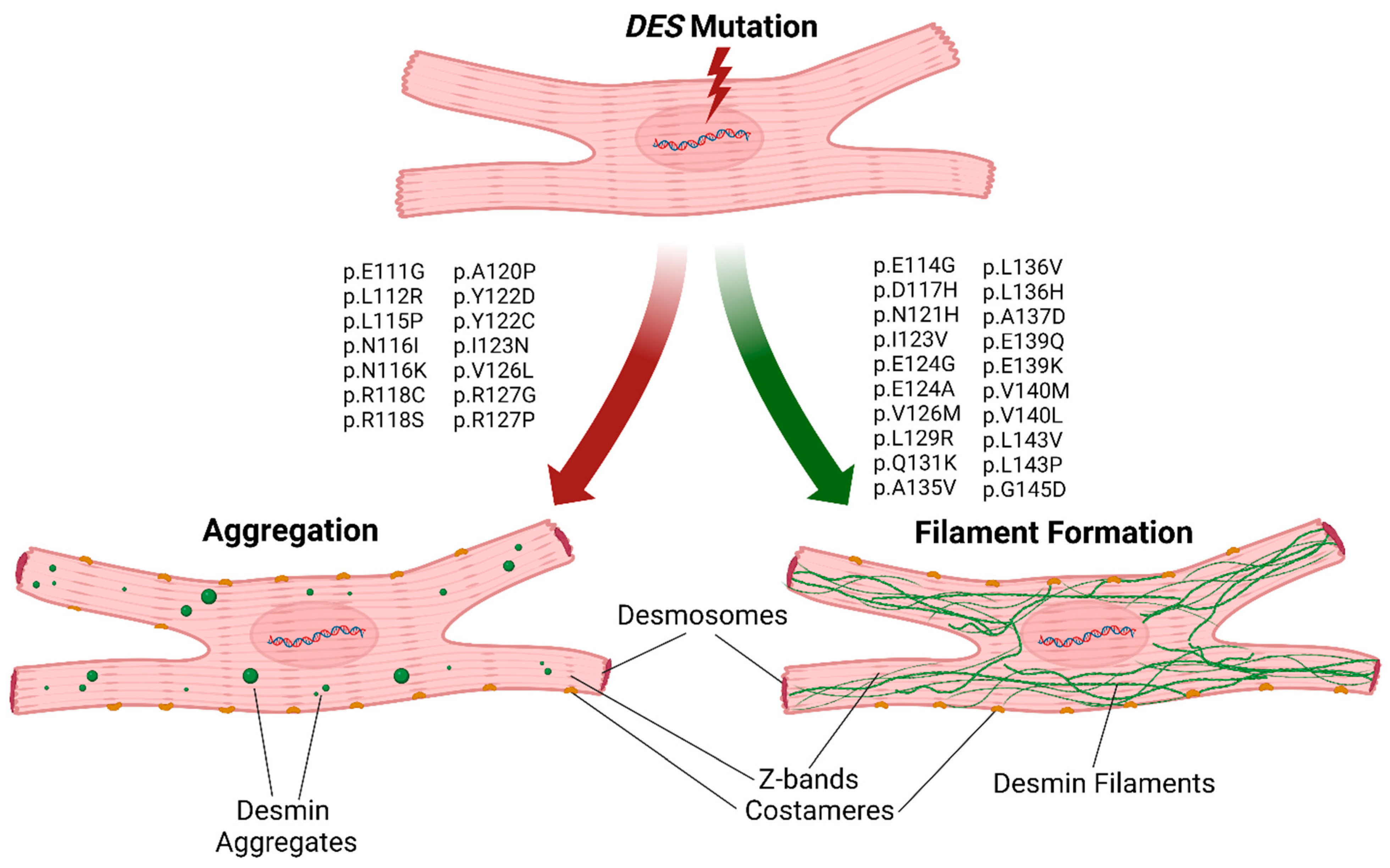
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′–3′) | Application |
|---|---|---|
| DES_E111G_for | CAACGAGAAGGTGGGGCTGCAGGAGCTCA | SDM |
| DES_E111G_rev | TGAGCTCCTGCAGCCCCACCTTCTCGTTG | SDM |
| DES_L112R_for | CGAGAAGGTGGAGCGGCAGGAGCTCAATG | SDM |
| DES_L112R_rev | CATTGAGCTCCTGCCGCTCCACCTTCTCG | SDM |
| DES_E114G_for | GTGGAGCTGCAGGGGCTCAATGACCGC | SDM |
| DES_E114G_rev | GCGGTCATTGAGCCCCTGCAGCTCCAC | SDM |
| DES_L115P_for | GGAGCTGCAGGAGCCCAATGACCGCTTCG | SDM |
| DES_L115P_rev | CGAAGCGGTCATTGGGCTCCTGCAGCTCC | SDM |
| DES_N116I_for | GTGGAGCTGCAGGAGCTCATTGACCGCTTC | SDM |
| DES_N116I_rev | GAAGCGGTCAATGAGCTCCTGCAGCTCCAC | SDM |
| DES_N116K_for | GGAGCTGCAGGAGCTCAAGGACCGCTTCG | SDM |
| DES_N116K_rev | CGAAGCGGTCCTTGAGCTCCTGCAGCTCC | SDM |
| DES_D117H_for | GGAGCTGCAGGAGCTCAATCACCGCTTCGC | SDM |
| DES_D117H_rev | GCGAAGCGGTGATTGAGCTCCTGCAGCTCC | SDM |
| DES_R118C_for | GCAGGAGCTCAATGACTGCTTCGCCAACTACAT | SDM |
| DES_R118C_rev | ATGTAGTTGGCGAAGCAGTCATTGAGCTCCTGC | SDM |
| DES_R118S_for | GCAGGAGCTCAATGACAGCTTCGCCAACTACAT | SDM |
| DES_R118S_rev | ATGTAGTTGGCGAAGCTGTCATTGAGCTCCTGC | SDM |
| DES_A120P_for | GCTCAATGACCGCTTCCCCAACTACATCGAGAA | SDM |
| DES_A120P_rev | TTCTCGATGTAGTTGGGGAAGCGGTCATTGAGC | SDM |
| DES_N121H_for | CAATGACCGCTTCGCCCACTACATCGAGAAGGT | SDM |
| DES_N121H_rev | ACCTTCTCGATGTAGTGGGCGAAGCGGTCATTG | SDM |
| DES_Y122D_for | GACCGCTTCGCCAACGACATCGAGAAGGTGC | SDM |
| DES_Y122D_rev | GCACCTTCTCGATGTCGTTGGCGAAGCGG | SDM |
| DES_Y122C_for | CCGCTTCGCCAACTGCATCGAGAAGGTGC | SDM |
| DES_Y122C_rev | GCACCTTCTCGATGCAGTTGGCGAAGCGG | SDM |
| DES_I123V_for | GCTTCGCCAACTACGTCGAGAAGGTGCGC | SDM |
| DES_I123V_rev | GCGCACCTTCTCGACGTAGTTGGCGAAGC | SDM |
| DES_I123N_for | ACCGCTTCGCCAACTACAACGAGAAGGTGC | SDM |
| DES_I123N_rev | GCACCTTCTCGTTGTAGTTGGCGAAGCGGT | SDM |
| DES_E124G_for | CGCCAACTACATCGGGAAGGTGCGCTTCC | SDM |
| DES_E124G_rev | GGAAGCGCACCTTCCCGATGTAGTTGGCG | SDM |
| DES_E124A_for | CGCCAACTACATCGCGAAGGTGCGCTTCC | SDM |
| DES_E124A_rev | GGAAGCGCACCTTCGCGATGTAGTTGGCG | SDM |
| DES_V126M_for | CAACTACATCGAGAAGATGCGCTTCCTGGAGCA | SDM |
| DES_V126M_rev | TGCTCCAGGAAGCGCATCTTCTCGATGTAGTTG | SDM |
| DES_V126L_for | CAACTACATCGAGAAGTTGCGCTTCCTGGAGCA | SDM |
| DES_V126L_rev | TGCTCCAGGAAGCGCAACTTCTCGATGTAGTTG | SDM |
| DES_R127G_for | ACATCGAGAAGGTGGGCTTCCTGGAGCAG | SDM |
| DES_R127G_rev | CTGCTCCAGGAAGCCCACCTTCTCGATGT | SDM |
| DES_R127P_for | CATCGAGAAGGTGCCCTTCCTGGAGCAGC | SDM |
| DES_R127P_rev | GCTGCTCCAGGAAGGGCACCTTCTCGATG | SDM |
| DES_L129R_for | GAAGGTGCGCTTCCGGGAGCAGCAGAACG | SDM |
| DES_L129R_rev | CGTTCTGCTGCTCCCGGAAGCGCACCTTC | SDM |
| DES_Q131K_for | GCGCTTCCTGGAGAAGCAGAACGCGGC | SDM |
| DES_Q131K_rev | GCCGCGTTCTGCTTCTCCAGGAAGCGC | SDM |
| DES_A135V_for | GCAGAACGCGGTGCTCGCCGCCG | SDM |
| DES_A135V_rev | CGGCGGCGAGCACCGCGTTCTGC | SDM |
| DES_L136V_for | AGAACGCGGCGGTCGCCGCCGAA | SDM |
| DES_L136V_rev | TTCGGCGGCGACCGCCGCGTTCT | SDM |
| DES_L136H_for | GAACGCGGCGCACGCCGCCGAAG | SDM |
| DES_L136H_rev | CTTCGGCGGCGTGCGCCGCGTTC | SDM |
| DES_A137D_for | CGCGGCGCTCGACGCCGAAGTGA | SDM |
| DES_A137D_rev | TCACTTCGGCGTCGAGCGCCGCG | SDM |
| DES_E139Q_for | GCGCTCGCCGCCCAGGTGAACCGGCTC | SDM |
| DES_E139Q_rev | GAGCCGGTTCACCTGGGCGGCGAGCGC | SDM |
| DES_E139K_for | GCGCTCGCCGCCAAAGTGAACCGGC | SDM |
| DES_E139K_rev | GCCGGTTCACTTTGGCGGCGAGCGC | SDM |
| DES_V140M_for | CGCTCGCCGCCGAAATGAACCGGCTC | SDM |
| DES_V140M_rev | GAGCCGGTTCATTTCGGCGGCGAGCG | SDM |
| DES_V140L_for | CGCTCGCCGCCGAATTGAACCGGCTC | SDM |
| DES_V140L_rev | GAGCCGGTTCAATTCGGCGGCGAGCG | SDM |
| DES_L143V_for | GAAGTGAACCGGGTCAAGGGCCGCG | SDM |
| DES_L143V_rev | CGCGGCCCTTGACCCGGTTCACTTC | SDM |
| DES_L143P_for | GAAGTGAACCGGCCCAAGGGCCGCGAG | SDM |
| DES_L143P_rev | CTCGCGGCCCTTGGGCCGGTTCACTTC | SDM |
| DES_G145D_for | CCGGCTCAAGGACCGCGAGCCGA | SDM |
| DES_G145D_rev | TCGGCTCGCGGTCCTTGAGCCGG | SDM |
| CMV_for | CGCAAATGGGCGGTAGGCGTG | Sanger sequencing |
| EGFP_rev | CGTCGCCGTCCAGCTCGACCAG | Sanger sequencing |
References
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Mavroidis, M.; Psarras, S.; Capetanaki, Y.; Lattanzi, G. Skeletal and Cardiac Muscle Disorders Caused by Mutations in Genes Encoding Intermediate Filament Proteins. Int. J. Mol. Sci. 2021, 22, 4256. [Google Scholar] [CrossRef]
- Brodehl, A.; Dieding, M.; Biere, N.; Unger, A.; Klauke, B.; Walhorn, V.; Gummert, J.; Schulz, U.; Linke, W.A.; Gerull, B.; et al. Functional characterization of the novel DES mutation p.L136P associated with dilated cardiomyopathy reveals a dominant filament assembly defect. J. Mol. Cell. Cardiol. 2016, 91, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Xing, Y.L.; Li, H.W. Heterozygous desmin gene (DES) mutation contributes to familial dilated cardiomyopathy. J. Int. Med. Res. 2021, 49, 03000605211006598. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.; Dittmann, S.; Brodehl, A.; Unger, A.; Stallmeyer, B.; Paul, M.; Seebohm, G.; Kayser, A.; Peischard, S.; Linke, W.A.; et al. Functional characterization of novel alpha-helical rod domain desmin (DES) pathogenic variants associated with dilated cardiomyopathy, atrioventricular block and a risk for sudden cardiac death. Int. J. Cardiol. 2021, 329, 167–174. [Google Scholar] [CrossRef]
- Protonotarios, A.; Brodehl, A.; Asimaki, A.; Jager, J.; Quinn, E.; Stanasiuk, C.; Ratnavadivel, S.; Futema, M.; Akhtar, M.M.; Gossios, T.D.; et al. The Novel Desmin Variant p.Leu115Ile Is Associated With a Unique Form of Biventricular Arrhythmogenic Cardiomyopathy. Can. J. Cardiol. 2021, 37, 857–866. [Google Scholar] [CrossRef]
- Bermudez-Jimenez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.A.; Alvarez, M.; Lopez-Fernandez, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef]
- Oka, H.; Nakau, K.; Imanishi, R.; Furukawa, T.; Tanabe, Y.; Hirono, K.; Hata, Y.; Nishida, N.; Azuma, H. A Case Report of a Rare Heterozygous Variant in the Desmin Gene Associated With Hypertrophic Cardiomyopathy and Complete Atrioventricular Block. CJC Open 2021, 3, 1195–1198. [Google Scholar] [CrossRef]
- Harada, H.; Hayashi, T.; Nishi, H.; Kusaba, K.; Koga, Y.; Koga, Y.; Nonaka, I.; Kimura, A. Phenotypic expression of a novel desmin gene mutation: Hypertrophic cardiomyopathy followed by systemic myopathy. J. Hum. Genet. 2018, 63, 249–254. [Google Scholar] [CrossRef]
- Brodehl, A.; Gerull, B. Genetic Insights into Primary Restrictive Cardiomyopathy. J. Clin. Med. 2022, 11, 2094. [Google Scholar] [CrossRef]
- Brodehl, A.; Hain, C.; Flottmann, F.; Ratnavadivel, S.; Gaertner, A.; Klauke, B.; Kalinowski, J.; Korperich, H.; Gummert, J.; Paluszkiewicz, L.; et al. The Desmin Mutation DES-c.735G>C Causes Severe Restrictive Cardiomyopathy by Inducing In-Frame Skipping of Exon-3. Biomedicines 2021, 9, 1400. [Google Scholar] [CrossRef]
- Kulikova, O.; Brodehl, A.; Kiseleva, A.; Myasnikov, R.; Meshkov, A.; Stanasiuk, C.; Gartner, A.; Divashuk, M.; Sotnikova, E.; Koretskiy, S.; et al. The Desmin (DES) Mutation p.A337P Is Associated with Left-Ventricular Non-Compaction Cardiomyopathy. Genes 2021, 12, 121. [Google Scholar] [CrossRef]
- Marakhonov, A.V.; Brodehl, A.; Myasnikov, R.P.; Sparber, P.A.; Kiseleva, A.V.; Kulikova, O.V.; Meshkov, A.N.; Zharikova, A.A.; Koretsky, S.N.; Kharlap, M.S.; et al. Noncompaction cardiomyopathy is caused by a novel in-frame desmin (DES) deletion mutation within the 1A coiled-coil rod segment leading to a severe filament assembly defect. Hum. Mutat. 2019, 40, 734–741. [Google Scholar] [CrossRef]
- Bergman, J.E.; Veenstra-Knol, H.E.; van Essen, A.J.; van Ravenswaaij, C.M.; den Dunnen, W.F.; van den Wijngaard, A.; van Tintelen, J.P. Two related Dutch families with a clinically variable presentation of cardioskeletal myopathy caused by a novel S13F mutation in the desmin gene. Eur. J. Med. Genet. 2007, 50, 355–366. [Google Scholar] [CrossRef]
- Goldfarb, L.G.; Park, K.Y.; Cervenakova, L.; Gorokhova, S.; Lee, H.S.; Vasconcelos, O.; Nagle, J.W.; Semino-Mora, C.; Sivakumar, K.; Dalakas, M.C. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat. Genet. 1998, 19, 402–403. [Google Scholar] [CrossRef]
- Munoz-Marmol, A.M.; Strasser, G.; Isamat, M.; Coulombe, P.A.; Yang, Y.; Roca, X.; Vela, E.; Mate, J.L.; Coll, J.; Fernandez-Figueras, M.T.; et al. A dysfunctional desmin mutation in a patient with severe generalized myopathy. Proc. Natl. Acad. Sci. USA 1998, 95, 11312–11317. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.L.; Lilienbaum, A.; Butler-Browne, G.; Paulin, D. Human desmin-coding gene: Complete nucleotide sequence, characterization and regulation of expression during myogenesis and development. Gene 1989, 78, 243–254. [Google Scholar] [CrossRef]
- Cetin, N.; Balci-Hayta, B.; Gundesli, H.; Korkusuz, P.; Purali, N.; Talim, B.; Tan, E.; Selcen, D.; Erdem-Ozdamar, S.; Dincer, P. A novel desmin mutation leading to autosomal recessive limb-girdle muscular dystrophy: Distinct histopathological outcomes compared with desminopathies. J. Med. Genet. 2013, 50, 437–443. [Google Scholar] [CrossRef]
- McLaughlin, H.M.; Kelly, M.A.; Hawley, P.P.; Darras, B.T.; Funke, B.; Picker, J. Compound heterozygosity of predicted loss-of-function DES variants in a family with recessive desminopathy. BMC Med. Genet. 2013, 14, 78. [Google Scholar] [CrossRef] [Green Version]
- Agnetti, G.; Herrmann, H.; Cohen, S. New roles for desmin in the maintenance of muscle homeostasis. FEBS J. 2022, 289, 2755–2770. [Google Scholar] [CrossRef]
- Granger, B.L.; Lazarides, E. The existence of an insoluble Z disc scaffold in chicken skeletal muscle. Cell 1978, 15, 1253–1268. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M. Costameres: The Achilles’ heel of Herculean muscle. J. Biol. Chem. 2003, 278, 13591–13594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.J.; Park-Snyder, S.; Pascoe, L.T.; Green, K.J.; Weis, W.I. Structures of two intermediate filament-binding fragments of desmoplakin reveal a unique repeat motif structure. Nat. Struct. Biol. 2002, 9, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Lowery, J.; Kuczmarski, E.R.; Herrmann, H.; Goldman, R.D. Intermediate Filaments Play a Pivotal Role in Regulating Cell Architecture and Function. J. Biol. Chem. 2015, 290, 17145–17153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldfarb, L.G.; Dalakas, M.C. Tragedy in a heartbeat: Malfunctioning desmin causes skeletal and cardiac muscle disease. J. Clin. Investig. 2009, 119, 1806–1813. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Aebi, U. Intermediate Filaments: Structure and Assembly. Cold Spring Harb. Perspect. Biol. 2016, 8, a018242. [Google Scholar] [CrossRef] [Green Version]
- Vermeire, P.J.; Stalmans, G.; Lilina, A.V.; Fiala, J.; Novak, P.; Herrmann, H.; Strelkov, S.V. Molecular Interactions Driving Intermediate Filament Assembly. Cells 2021, 10, 2457. [Google Scholar] [CrossRef]
- Chernyatina, A.A.; Nicolet, S.; Aebi, U.; Herrmann, H.; Strelkov, S.V. Atomic structure of the vimentin central alpha-helical domain and its implications for intermediate filament assembly. Proc. Natl. Acad. Sci. USA 2012, 109, 13620–13625. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Strelkov, S.V.; Feja, B.; Rogers, K.R.; Brettel, M.; Lustig, A.; Haner, M.; Parry, D.A.; Steinert, P.M.; Burkhard, P.; et al. The intermediate filament protein consensus motif of helix 2B: Its atomic structure and contribution to assembly. J. Mol. Biol. 2000, 298, 817–832. [Google Scholar] [CrossRef]
- Herrmann, H.; Haner, M.; Brettel, M.; Ku, N.O.; Aebi, U. Characterization of distinct early assembly units of different intermediate filament proteins. J. Mol. Biol. 1999, 286, 1403–1420. [Google Scholar] [CrossRef]
- Winheim, S.; Hieb, A.R.; Silbermann, M.; Surmann, E.M.; Wedig, T.; Herrmann, H.; Langowski, J.; Mucke, N. Deconstructing the late phase of vimentin assembly by total internal reflection fluorescence microscopy (TIRFM). PLoS ONE 2011, 6, e19202. [Google Scholar] [CrossRef] [Green Version]
- Colakoglu, G.; Brown, A. Intermediate filaments exchange subunits along their length and elongate by end-to-end annealing. J. Cell. Biol. 2009, 185, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Bar, H.; Mucke, N.; Kostareva, A.; Sjoberg, G.; Aebi, U.; Herrmann, H. Severe muscle disease-causing desmin mutations interfere with in vitro filament assembly at distinct stages. Proc. Natl. Acad. Sci. USA 2005, 102, 15099–15104. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Hedde, P.N.; Dieding, M.; Fatima, A.; Walhorn, V.; Gayda, S.; Saric, T.; Klauke, B.; Gummert, J.; Anselmetti, D.; et al. Dual color photoactivation localization microscopy of cardiomyopathy-associated desmin mutants. J. Biol. Chem. 2012, 287, 16047–16057. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Pour Hakimi, S.A.; Stanasiuk, C.; Ratnavadivel, S.; Hendig, D.; Gaertner, A.; Gerull, B.; Gummert, J.; Paluszkiewicz, L.; Milting, H. Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes 2019, 10, 918. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Dieding, M.; Klauke, B.; Dec, E.; Madaan, S.; Huang, T.; Gargus, J.; Fatima, A.; Saric, T.; Cakar, H.; et al. The novel desmin mutant p.A120D impairs filament formation, prevents intercalated disk localization, and causes sudden cardiac death. Circ. Cardiovasc. Genet. 2013, 6, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Vernengo, L.; Chourbagi, O.; Panuncio, A.; Lilienbaum, A.; Batonnet-Pichon, S.; Bruston, F.; Rodrigues-Lima, F.; Mesa, R.; Pizzarossa, C.; Demay, L.; et al. Desmin myopathy with severe cardiomyopathy in a Uruguayan family due to a codon deletion in a new location within the desmin 1A rod domain. Neuromuscul. Disord. 2010, 20, 178–187. [Google Scholar] [CrossRef]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedberg, K.K.; Chen, L.B. Absence of intermediate filaments in a human adrenal cortex carcinoma-derived cell line. Exp. Cell Res. 1986, 163, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Hescheler, J.; Meyer, R.; Plant, S.; Krautwurst, D.; Rosenthal, W.; Schultz, G. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ. Res. 1991, 69, 1476–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodehl, A.; Weiss, J.; Debus, J.D.; Stanasiuk, C.; Klauke, B.; Deutsch, M.A.; Fox, H.; Bax, J.; Ebbinghaus, H.; Gartner, A.; et al. A homozygous DSC2 deletion associated with arrhythmogenic cardiomyopathy is caused by uniparental isodisomy. J. Mol. Cell. Cardiol. 2020, 141, 17–29. [Google Scholar] [CrossRef]
- Debus, J.D.; Milting, H.; Brodehl, A.; Kassner, A.; Anselmetti, D.; Gummert, J.; Gaertner-Rommel, A. In vitro analysis of arrhythmogenic cardiomyopathy associated desmoglein-2 (DSG2) mutations reveals diverse glycosylation patterns. J. Mol. Cell. Cardiol. 2019, 129, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Burridge, P.W.; Holmstrom, A.; Wu, J.C. Chemically Defined Culture and Cardiomyocyte Differentiation of Human Pluripotent Stem Cells. Curr. Protoc. Hum. Genet. 2015, 87, 21–23. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hsu, L.A.; Ko, Y.S.; Yeh, Y.H.; Chang, C.J.; Chan, Y.H.; Kuo, C.T.; Tsai, H.Y.; Chang, G.J. A Novel DES L115F Mutation Identified by Whole Exome Sequencing is Associated with Inherited Cardiac Conduction Disease. Int. J. Mol. Sci. 2019, 20, 6227. [Google Scholar] [CrossRef] [Green Version]
- Bar, H.; Goudeau, B.; Walde, S.; Casteras-Simon, M.; Mucke, N.; Shatunov, A.; Goldberg, Y.P.; Clarke, C.; Holton, J.L.; Eymard, B.; et al. Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum. Mutat. 2007, 28, 374–386. [Google Scholar] [CrossRef]
- Bar, H.; Kostareva, A.; Sjoberg, G.; Sejersen, T.; Katus, H.A.; Herrmann, H. Forced expression of desmin and desmin mutants in cultured cells: Impact of myopathic missense mutations in the central coiled-coil domain on network formation. Exp. Cell Res. 2006, 312, 1554–1565. [Google Scholar] [CrossRef]
- Kreplak, L.; Bar, H. Severe myopathy mutations modify the nanomechanics of desmin intermediate filaments. J. Mol. Biol. 2009, 385, 1043–1051. [Google Scholar] [CrossRef]
- Prust, M.; Wang, J.; Morizono, H.; Messing, A.; Brenner, M.; Gordon, E.; Hartka, T.; Sokohl, A.; Schiffmann, R.; Gordish-Dressman, H.; et al. GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology 2011, 77, 1287–1294. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Johnson, A.B.; Salomons, G.; Goldman, J.E.; Naidu, S.; Quinlan, R.; Cree, B.; Ruyle, S.Z.; Banwell, B.; D’Hooghe, M.; et al. Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann. Neurol. 2005, 57, 310–326. [Google Scholar] [CrossRef]
- Brenner, M.; Johnson, A.B.; Boespflug-Tanguy, O.; Rodriguez, D.; Goldman, J.E.; Messing, A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat. Genet. 2001, 27, 117–120. [Google Scholar] [CrossRef]
- Gorospe, J.R.; Naidu, S.; Johnson, A.B.; Puri, V.; Raymond, G.V.; Jenkins, S.D.; Pedersen, R.C.; Lewis, D.; Knowles, P.; Fernandez, R.; et al. Molecular findings in symptomatic and pre-symptomatic Alexander disease patients. Neurology 2002, 58, 1494–1500. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.; Yang, H.; Yuan, Y.; Bonnemann, C.; Chang, X.; Wang, S.; Wu, Y.; Wu, X.; Xiong, H. Phenotype-Genotype Analysis of Chinese Patients with Early-Onset LMNA-Related Muscular Dystrophy. PLoS ONE 2015, 10, e0129699. [Google Scholar] [CrossRef]
- Benedetti, S.; Menditto, I.; Degano, M.; Rodolico, C.; Merlini, L.; D’Amico, A.; Palmucci, L.; Berardinelli, A.; Pegoraro, E.; Trevisan, C.P.; et al. Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology 2007, 69, 1285–1292. [Google Scholar] [CrossRef]
- Prigogine, C.; Richard, P.; Van den Bergh, P.; Groswasser, J.; Deconinck, N. Novel LMNA mutation presenting as severe congenital muscular dystrophy. Pediatric Neurol. 2010, 43, 283–286. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brodehl, A.; Holler, S.; Gummert, J.; Milting, H. The N-Terminal Part of the 1A Domain of Desmin Is a Hot Spot Region for Putative Pathogenic DES Mutations Affecting Filament Assembly. Cells 2022, 11, 3906. https://doi.org/10.3390/cells11233906
Brodehl A, Holler S, Gummert J, Milting H. The N-Terminal Part of the 1A Domain of Desmin Is a Hot Spot Region for Putative Pathogenic DES Mutations Affecting Filament Assembly. Cells. 2022; 11(23):3906. https://doi.org/10.3390/cells11233906
Chicago/Turabian StyleBrodehl, Andreas, Stephanie Holler, Jan Gummert, and Hendrik Milting. 2022. "The N-Terminal Part of the 1A Domain of Desmin Is a Hot Spot Region for Putative Pathogenic DES Mutations Affecting Filament Assembly" Cells 11, no. 23: 3906. https://doi.org/10.3390/cells11233906