
The Role of Oxidative Stress in Atherosclerosis

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
Global Burden of Cardiovascular Disease
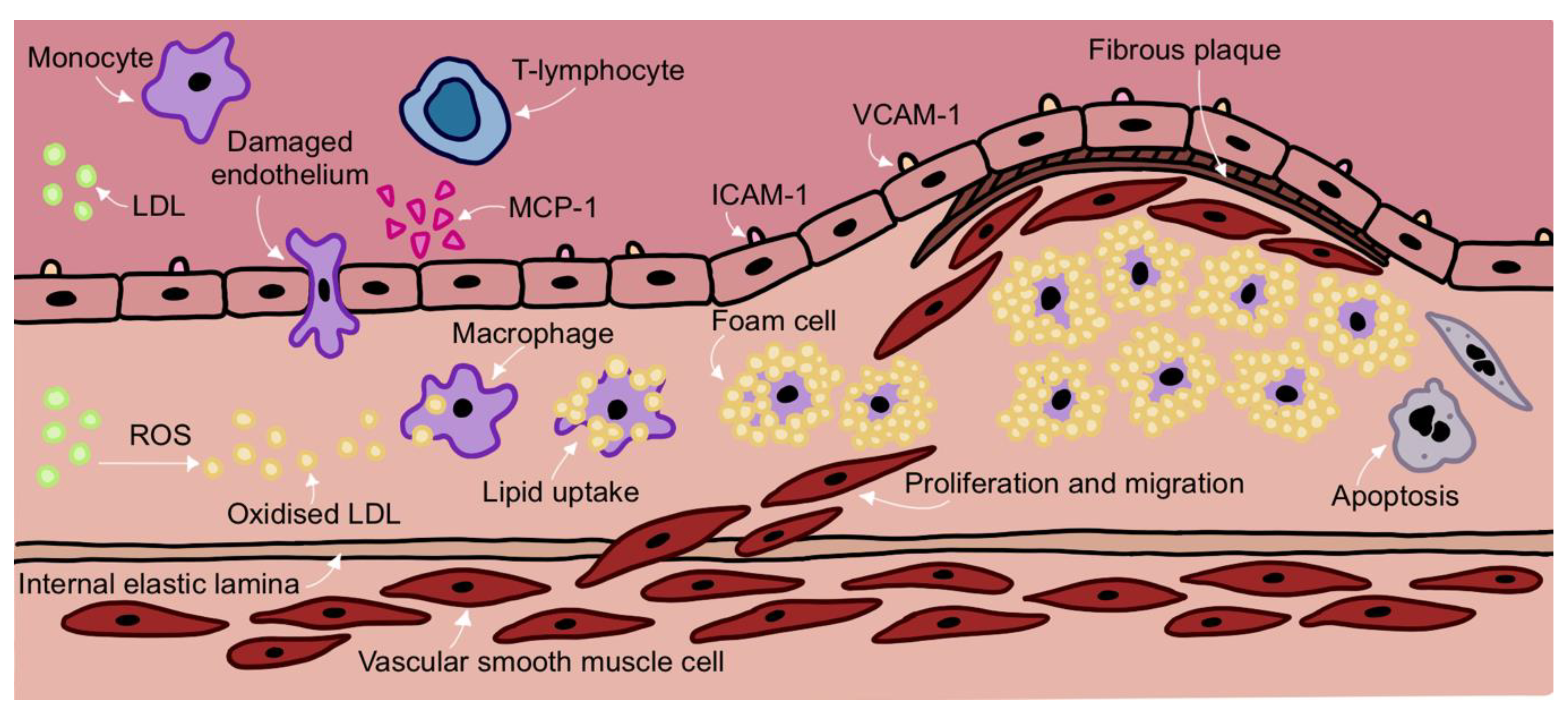
2. Development of Atherosclerosis

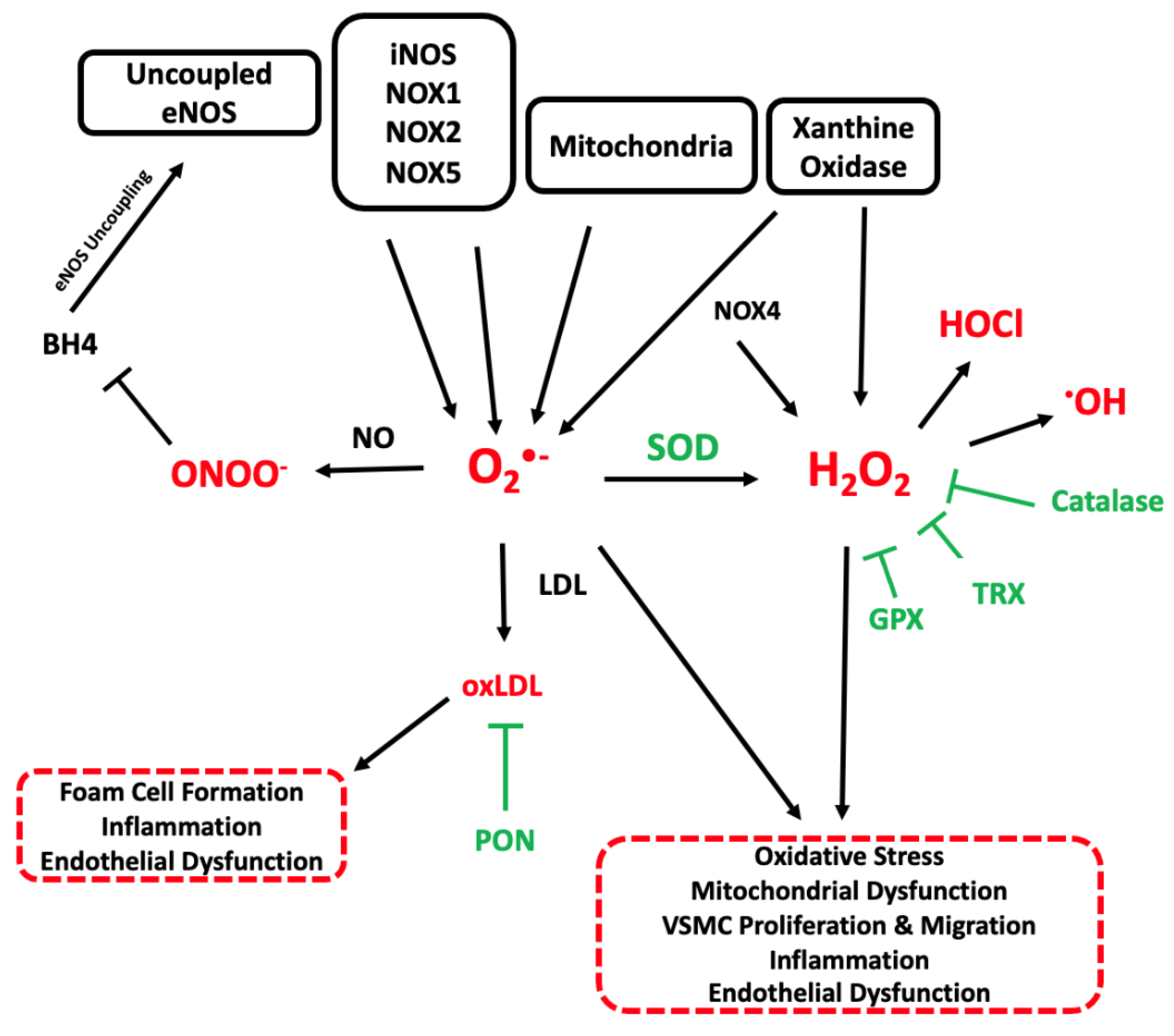
3. Overview of ROS
4. Sources of ROS in Atherosclerosis
4.1. Mitochondrial ROS
4.2. NADPH Oxidase
NADPH Oxidase in Atherosclerosis
4.3. Xanthine Oxidoreductase
4.4. Uncoupled Nitric Oxide Synthase
4.5. iNOS in Atherosclerosis
5. Antioxidant Systems in Atherosclerosis
6. The Effects of ROS in Atherosclerosis
6.1. Lipid Oxidation
6.2. DNA Oxidation
6.3. Endothelial Dysfunction
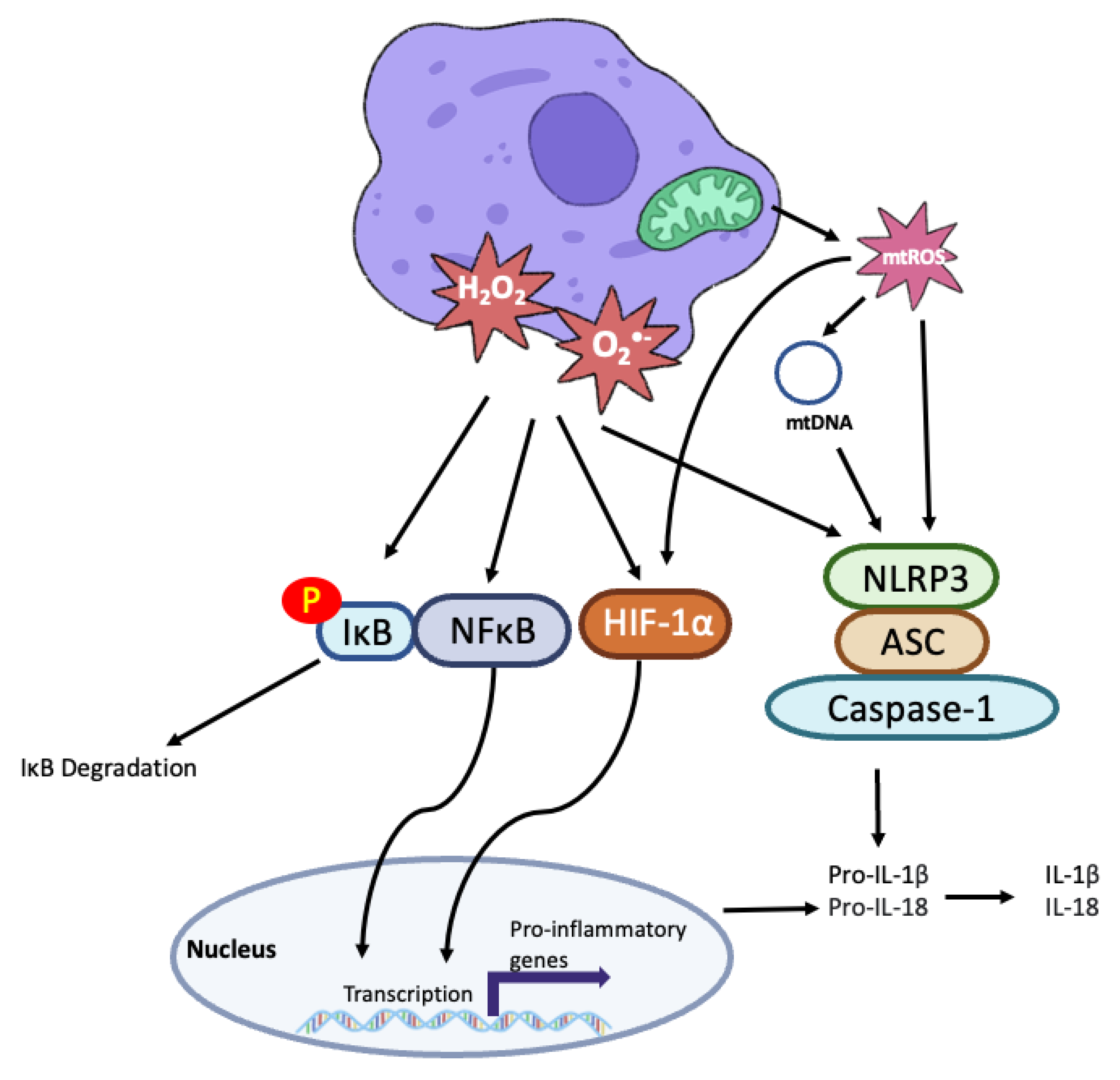
6.4. Inflammation

6.5. Fibrous Cap Stability
7. Antioxidant Therapies for Treating Atherosclerosis
8. Future Directions
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organisation, World Health Organisation Cardiovascular Disease Fact Sheet. 2021. Available online: https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 10 November 2022).
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990-2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.K. Inflammation, Atherosclerosis, and Coronary Artery Disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim. Biophys. Acta—Bioenerg. 2006, 1757, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [Green Version]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorescu, D.; Weiss, D.; Lassègue, B.; Clempus, R.E.; Szöcs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [Green Version]
- Lassègue, B.; Griendling, K.K. NADPH Oxidases: Functions and Pathologies in the Vasculature. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef]
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2290–H2297. [Google Scholar] [CrossRef] [Green Version]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.P.; Di Marco, E.; Kennedy, K.; Chew, P.; Okabe, J.; El-Osta, A.; Calkin, A.C.; Biessen, E.A.L.; Touyz, R.M.; Cooper, M.E.; et al. Reactive Oxygen Species Can Provide Atheroprotection via NOX4-Dependent Inhibition of Inflammation and Vascular Remodeling. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, A.L.; Carrell, S.; Johnson, B.; Stanic, B.; Banfi, B.; Miller, F.J.J. Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis 2011, 216, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH Oxidase 1 Plays a Key Role in Diabetes Mellitus–Accelerated Atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef]
- Judkins, C.P.; Diep, H.; Broughton, B.R.S.; Mast, A.E.; Hooker, E.U.; Miller, A.A.; Selemidis, S.; Dusting, G.J.; Sobey, C.G.; Drummond, G.R. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE−/− mice. Am. J. Physiol. Circ. Physiol. 2009, 298, H24–H32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.; Ing, M.H.; Salazar, A.; Lassègue, B.; Griendling, K.; Navab, M.; Sevanian, A.; Hsiai, T.K. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: Implication for native LDL oxidation. Circ. Res. 2003, 93, 1225–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schürmann, C.; Rezende, F.; Kruse, C.; Yasar, Y.; Löwe, O.; Fork, C.; van de Sluis, B.; Bremer, R.; Weissmann, N.; Shah, A.M.; et al. The NADPH oxidase Nox4 has anti-atherosclerotic functions. Eur. Heart J. 2015, 36, 3447–3456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, A.; Frank, F.; Wolk, S.; Busch, A.; Klimova, A.; Sabarstinski, P.; Gerlach, M.; Egorov, D.; Kopaliani, I.; Weinert, S.; et al. NOX4 mRNA correlates with plaque stability in patients with carotid artery stenosis. Redox Biol. 2022, 57, 102473. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Cai, H.; Davis, M.E.; Ramasamy, S.; Harrison, D.G. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ. Res. 2000, 86, 347–354. [Google Scholar] [CrossRef]
- Ho, F.; Watson, A.M.D.; Elbatreek, M.H.; Kleikers, P.W.M.; Khan, W.; Sourris, K.C.; Dai, A.; Jha, J.; Schmidt, H.H.H.W.; Jandeleit-Dahm, K.A.M. Endothelial reactive oxygen-forming NADPH oxidase 5 is a possible player in diabetic aortic aneurysm but not atherosclerosis. Sci. Rep. 2022, 12, 11570. [Google Scholar] [CrossRef]
- Petheő, G.L.; Kerekes, A.; Mihálffy, M.; Donkó, Á.; Bodrogi, L.; Skoda, G.; Baráth, M.; Hoffmann, O.I.; Szeles, Z.; Balázs, B.; et al. Disruption of the NOX5 Gene Aggravates Atherosclerosis in Rabbits. Circ. Res. 2021, 128, 1320–1322. [Google Scholar] [CrossRef]
- Touyz, R.M.; Anagnostopoulou, A.; Rios, F.; Montezano, A.C.; Camargo, L.L. NOX5: Molecular biology and pathophysiology. Exp. Physiol. 2019, 104, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Battelli, M.G.; Polito, L.; Bolognesi, A. Xanthine oxidoreductase in atherosclerosis pathogenesis: Not only oxidative stress. Atherosclerosis 2014, 237, 562–567. [Google Scholar] [CrossRef]
- Ganji, M.; Nardi, V.; Prasad, M.; Jordan, K.L.; Bois, M.C.; Franchi, F.; Zhu, X.Y.; Tang, H.; Young, M.D.; Lerman, L.O.; et al. Carotid Plaques From Symptomatic Patients Are Characterized by Local Increase in Xanthine Oxidase Expression. Stroke 2021, 52, 2792–2801. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Tsukui, D.; Kono, H. Uric Acid in Inflammation and the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 12394. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, Y.; Dong, K.; Wang, A.; Yang, X.; Zhang, C.; Zhu, Y.; Wu, S.; Zhao, X. The Association between Serum Uric Acid Levels and the Prevalence of Vulnerable Atherosclerotic Carotid Plaque: A Cross-sectional Study. Sci. Rep. 2015, 5, 10003. [Google Scholar] [CrossRef] [Green Version]
- Hooper, D.C.; Spitsin, S.; Kean, R.B.; Champion, J.M.; Dickson, G.M.; Chaudhry, I.; Koprowski, H. Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. USA 1998, 95, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Kushiyama, A.; Okubo, H.; Sakoda, H.; Kikuchi, T.; Fujishiro, M.; Sato, H.; Kushiyama, S.; Iwashita, M.; Nishimura, F.; Fukushima, T.; et al. Xanthine Oxidoreductase Is Involved in Macrophage Foam Cell Formation and Atherosclerosis Development. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, J.; Busso, N.; Ives, A.; Matsui, C.; Tsujimoto, S.; Shirakura, T.; Tamura, M.; Kobayashi, T.; So, A.; Yamanaka, Y. Xanthine Oxidase Inhibition by Febuxostat Attenuates Experimental Atherosclerosis in Mice. Sci. Rep. 2014, 4, 4554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacIsaac, R.L.; Salatzki, J.; Higgins, P.; Walters, M.R.; Padmanabhan, S.; Dominiczak, A.F.; Touyz, R.M.; Dawson, J. Allopurinol and Cardiovascular Outcomes in Adults With Hypertension. Hypertens. 2016, 67, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.S.; Hawkey, C.J.; Ford, I.; Greenlaw, N.; Pigazzani, F.; Rogers, A.; Struthers, A.D.; Begg, A.G.; Wei, L.; Avery, A.J.; et al. Allopurinol versus usual care in UK patients with ischaemic heart disease (ALL-HEART): A multicentre, prospective, randomised, open-label, blinded-endpoint trial. Lancet 2022, 400, 1195–1205. [Google Scholar] [CrossRef]
- Marahatha, R.; Basnet, S.; Bhattarai, B.R.; Budhathoki, P.; Aryal, B.; Adhikari, B.; Lamichhane, G.; Poudel, D.K.; Parajuli, N. Potential natural inhibitors of xanthine oxidase and HMG-CoA reductase in cholesterol regulation: In silico analysis. BMC Complement. Med. Ther. 2021, 21, 1. [Google Scholar] [CrossRef]
- Hong, F.-F.; Liang, X.-Y.; Liu, W.; Lv, S.; He, S.-J.; Kuang, H.-B.; Yang, S.-L. Roles of eNOS in atherosclerosis treatment. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2019, 68, 429–441. [Google Scholar] [CrossRef]
- Li, H.; Förstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharmacol. 2013, 13, 161–167. [Google Scholar] [CrossRef] [PubMed]
- van Haperen, R.; de Waard, M.; van Deel, E.; Mees, B.; Kutryk, M.; van Aken, T.; Hamming, J.; Grosveld, F.; Duncker, D.J.; de Crom, R. Reduction of blood pressure, plasma cholesterol, and atherosclerosis by elevated endothelial nitric oxide. J. Biol. Chem. 2002, 277, 48803–48807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozaki, M.; Kawashima, S.; Yamashita, T.; Hirase, T.; Namiki, M.; Inoue, N.; Hirata, K.; Yasui, H.; Sakurai, H.; Yoshida, Y.; et al. Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J. Clin. Investig. 2002, 110, 331–340. [Google Scholar] [CrossRef]
- Knowles, J.W.; Reddick, R.L.; Jennette, J.C.; Shesely, E.G.; Smithies, O.; Maeda, N. Enhanced atherosclerosis and kidney dysfunction in eNOS(-/-)Apoe(-/-) mice are ameliorated by enalapril treatment. J. Clin. Investig. 2000, 105, 451–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Baker, J.E.; Zhang, C.; Tweddell, J.S.; Su, J.; Pritchard, K.A.J. Chronic hypoxia increases endothelial nitric oxide synthase generation of nitric oxide by increasing heat shock protein 90 association and serine phosphorylation. Circ. Res. 2002, 91, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Zamora, R.; Vodovotz, Y.; Billiar, T.R. Inducible Nitric Oxide Synthase and Inflammatory Diseases. Mol. Med. 2000, 6, 347–373. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Roman, L.J.; Masters, B.S.; Zweier, J.L. Inducible nitric-oxide synthase generates superoxide from the reductase domain. J. Biol. Chem. 1998, 273, 22635–22639. [Google Scholar] [CrossRef] [Green Version]
- McNeill, E.; Crabtree, M.J.; Sahgal, N.; Patel, J.; Chuaiphichai, S.; Iqbal, A.J.; Hale, A.B.; Greaves, D.R.; Channon, K.M. Regulation of iNOS function and cellular redox state by macrophage Gch1 reveals specific requirements for tetrahydrobiopterin in NRF2 activation. Free Radic. Biol. Med. 2015, 79, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Sventek, P.; Larivière, R.; Thibault, G.; Schiffrin, E.L. Inducible nitric oxide synthase in vascular smooth muscle cells from prehypertensive spontaneously hypertensive rats. Am. J. Hypertens. 1996, 9, 867–877. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.; Yan, Y.; Zhang, R.; Xiong, H. Regulation of iNOS on Immune Cells and Its Role in Diseases. Int. J. Mol. Sci. 2018, 19, 3805. [Google Scholar] [CrossRef]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Depre, C.; Havaux, X.; Renkin, J.; Vanoverschelde, J.L.J.; Wijns, W. Expression of inducible nitric oxide synthase in human coronary atherosclerotic plaque. Cardiovasc. Res. 1999, 41, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Detmers, P.A.; Hernandez, M.; Mudgett, J.; Hassing, H.; Burton, C.; Mundt, S.; Chun, S.; Fletcher, D.; Card, D.J.; Lisnock, J.; et al. Deficiency in inducible nitric oxide synthase results in reduced atherosclerosis in apolipoprotein E-deficient mice. J. Immunol. 2000, 165, 3430–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhlencordt, P.J.; Chen, J.; Han, F.; Astern, J.; Huang, P.L. Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein E-knockout mice. Circulation 2001, 103, 3099–3104. [Google Scholar] [CrossRef]
- Lubrano, V.; Balzan, S. Enzymatic antioxidant system in vascular inflammation and coronary artery disease. World J. Exp. Med. 2015, 5, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Sodhi, S.; Mahajan, V. Correlation of antioxidants with lipid peroxidation and lipid profile in patients suffering from coronary artery disease. Expert Opin. Ther. Targets 2009, 13, 889–894. [Google Scholar] [CrossRef]
- Ninić, A.; Bogavac-Stanojević, N.; Sopić, M.; Munjas, J.; Kotur-Stevuljević, J.; Miljković, M.; Gojković, T.; Kalimanovska-Oštrić, D.; Spasojević-Kalimanovska, V. Superoxide Dismutase Isoenzymes Gene Expression in Peripheral Blood Mononuclear Cells in Patients with Coronary Artery Disease. J. Med. Biochem. 2019, 38, 284–291. [Google Scholar] [CrossRef]
- Ballinger, S.W.; Patterson, C.; Knight-Lozano, C.A.; Burow, D.L.; Conklin, C.A.; Hu, Z.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G.C.; et al. Mitochondrial integrity and function in atherogenesis. Circulation 2002, 106, 544–549. [Google Scholar] [CrossRef]
- Fukai, T.; Siegfried, M.R.; Ushio-Fukai, M.; Cheng, Y.; Kojda, G.; Harrison, D.G. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J. Clin. Investig. 2000, 105, 1631–1639. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Folz, R.J.; Landmesser, U.; Harrison, D.G. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc. Res. 2002, 55, 239–249. [Google Scholar] [CrossRef]
- Sentman, M.L.; Brännström, T.; Westerlund, S.; Laukkanen, M.O.; Ylä-Herttuala, S.; Basu, S.; Marklund, S.L. Extracellular superoxide dismutase deficiency and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1477–1482. [Google Scholar] [CrossRef] [Green Version]
- Rosenblat, M.; Volkova, N.; Coleman, R.; Aviram, M. Anti-oxidant and anti-atherogenic properties of liposomal glutathione: Studies in vitro, and in the atherosclerotic apolipoprotein E-deficient mice. Atherosclerosis 2007, 195, e61–e68. [Google Scholar] [CrossRef]
- Torzewski, M.; Ochsenhirt, V.; Kleschyov, A.L.; Oelze, M.; Daiber, A.; Li, H.; Rossmann, H.; Tsimikas, S.; Reifenberg, K.; Cheng, F.; et al. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 850–857. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Torzewski, M.; Degreif, A.; Rossmann, H.; Canisius, A.; Lackner, K.J. Impact of glutathione peroxidase-1 deficiency on macrophage foam cell formation and proliferation: Implications for atherogenesis. PLoS ONE 2013, 8, e72063. [Google Scholar] [CrossRef] [Green Version]
- Meilhac, O.; Zhou, M.; Santanam, N.; Parthasarathy, S. Lipid peroxides induce expression of catalase in cultured vascular cells. J. Lipid Res. 2000, 41, 1205–1213. [Google Scholar] [CrossRef]
- Yang, H.; Roberts, L.J.; Shi, M.J.; Zhou, L.C.; Ballard, B.R.; Richardson, A.; Guo, Z.M. Retardation of Atherosclerosis by Overexpression of Catalase or Both Cu/Zn-Superoxide Dismutase and Catalase in Mice Lacking Apolipoprotein E. Circ. Res. 2004, 95, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Mackness, B.; Mackness, M. Anti-Inflammatory Properties of Paraoxonase-1. In Atherosclerosis BT—Paraoxonases in Inflammation, Infection, and Toxicology; Reddy, S.T., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 143–151. [Google Scholar]
- Tward, A.; Xia, Y.-R.; Wang, X.-P.; Shi, Y.-S.; Park, C.; Castellani, L.W.; Lusis, A.J.; Shih, D.M. Decreased Atherosclerotic Lesion Formation in Human Serum Paraoxonase Transgenic Mice. Circulation 2002, 106, 484–490. [Google Scholar] [CrossRef] [Green Version]
- Ikhlef, S.; Berrougui, H.; Kamtchueng Simo, O.; Zerif, E.; Khalil, A. Human paraoxonase 1 overexpression in mice stimulates HDL cholesterol efflux and reverse cholesterol transport. PLoS ONE 2017, 12, e0173385. [Google Scholar] [CrossRef] [Green Version]
- Devarajan, A.; Bourquard, N.; Hama, S.; Navab, M.; Grijalva, V.R.; Morvardi, S.; Clarke, C.F.; Vergnes, L.; Reue, K.; Teiber, J.F.; et al. Paraoxonase 2 deficiency alters mitochondrial function and exacerbates the development of atherosclerosis. Antioxid. Redox Signal. 2011, 14, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Marsillach, J.; Becker, J.O.; Vaisar, T.; Hahn, B.H.; Brunzell, J.D.; Furlong, C.E.; de Boer, I.H.; McMahon, M.A.; Hoofnagle, A.N. Paraoxonase-3 is depleted from the high-density lipoproteins of autoimmune disease patients with subclinical atherosclerosis. J. Proteome Res. 2015, 14, 2046–2054. [Google Scholar] [CrossRef]
- Patwari, P.; Higgins, L.J.; Chutkow, W.A.; Yoshioka, J.; Lee, R.T. The Interaction of Thioredoxin with Txnip: Evidence for Formation of a Mixed Disulfide by Disulfide Exchange. J. Biol. Chem. 2006, 281, 21884–21891. [Google Scholar] [CrossRef] [Green Version]
- Furman, C.; Rundlöf, A.-K.; Larigauderie, G.; Jaye, M.; Bricca, G.; Copin, C.; Kandoussi, A.M.; Fruchart, J.-C.; Arnér, E.S.J.; Rouis, M. Thioredoxin reductase 1 is upregulated in atherosclerotic plaques: Specific induction of the promoter in human macrophages by oxidized low-density lipoproteins. Free Radic. Biol. Med. 2004, 37, 71–85. [Google Scholar] [CrossRef]
- Jekell, A.; Hossain, A.; Alehagen, U.; Dahlström, U.; Rosén, A. Elevated circulating levels of thioredoxin and stress in chronic heart failure. Eur. J. Heart Fail. 2004, 6, 883–890. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ji, N.; Gong, X.; Ni, S.; Xu, L.; Zhang, H. Thioredoxin-1 attenuates atherosclerosis development through inhibiting NLRP3 inflammasome. Endocrine 2020, 70, 65–70. [Google Scholar] [CrossRef]
- Rousset, S.; Alves-Guerra, M.-C.; Mozo, J.; Miroux, B.; Cassard-Doulcier, A.-M.; Bouillaud, F.; Ricquier, D. The Biology of Mitochondrial Uncoupling Proteins. Diabetes 2004, 53, S130–S135. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Della-Morte, D.; Wang, L.; Cabral, D.; Beecham, A.; McClendon, M.S.; Luca, C.C.; Blanton, S.H.; Sacco, R.L.; Rundek, T. Association of the sirtuin and mitochondrial uncoupling protein genes with carotid plaque. PLoS ONE 2011, 6, e27157. [Google Scholar] [CrossRef] [Green Version]
- Blanc, J.; Alves-Guerra, M.C.; Esposito, B.; Rousset, S.; Gourdy, P.; Ricquier, D.; Tedgui, A.; Miroux, B.; Mallat, Z. Protective role of uncoupling protein 2 in atherosclerosis. Circulation 2003, 107, 388–390. [Google Scholar] [CrossRef] [Green Version]
- Cominacini, L.; Mozzini, C.; Garbin, U.; Pasini, A.; Stranieri, C.; Solani, E.; Vallerio, P.; Tinelli, I.A.; Fratta Pasini, A. Endoplasmic reticulum stress and Nrf2 signaling in cardiovascular diseases. Free Radic. Biol. Med. 2015, 88, 233–242. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.-I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1862–H1870. [Google Scholar] [CrossRef]
- Zakkar, M.; Van der Heiden, K.; Luong, L.A.; Chaudhury, H.; Cuhlmann, S.; Hamdulay, S.S.; Krams, R.; Edirisinghe, I.; Rahman, I.; Carlsen, H.; et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1851–1857. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.R.; Gupte, A.A.; Ji, R.; Ramirez, M.R.; Minze, L.J.; Liu, J.Z.; Arredondo, M.; Ren, Y.; Deng, T.; Wang, J.; et al. Myeloid deletion of nuclear factor erythroid 2-related factor 2 increases atherosclerosis and liver injury. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2839–2846. [Google Scholar] [CrossRef] [Green Version]
- Barajas, B.; Che, N.; Yin, F.; Rowshanrad, A.; Orozco, L.D.; Gong, K.W.; Wang, X.; Castellani, L.W.; Reue, K.; Lusis, A.J.; et al. NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Freigang, S.; Ampenberger, F.; Spohn, G.; Heer, S.; Shamshiev, A.T.; Kisielow, J.; Hersberger, M.; Yamamoto, M.; Bachmann, M.F.; Kopf, M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 2011, 41, 2040–2051. [Google Scholar] [CrossRef]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef]
- Cyrus, T.; Praticò, D.; Zhao, L.; Witztum, J.L.; Rader, D.J.; Rokach, J.; FitzGerald, G.A.; Funk, C.D. Absence of 12/15-Lipoxygenase Expression Decreases Lipid Peroxidation and Atherogenesis in Apolipoprotein E–Deficient Mice. Circulation 2001, 103, 2277–2282. [Google Scholar] [CrossRef] [Green Version]
- Shih, D.M.; Xia, Y.R.; Wang, X.P.; Miller, E.; Castellani, L.W.; Subbanagounder, G.; Cheroutre, H.; Faull, K.F.; Berliner, J.A.; Witztum, J.L.; et al. Combined serum paraoxonase knockout/apolipoprotein E knockout mice exhibit increased lipoprotein oxidation and atherosclerosis. J. Biol. Chem. 2000, 275, 17527–17535. [Google Scholar] [CrossRef] [Green Version]
- Petrosillo, G.; Ruggiero, F.M.; Paradies, G. Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FASEB J. 2003, 17, 2202–2208. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Ye, C.; McCain, K.; Greenberg, M.L. The Role of Cardiolipin in Cardiovascular Health. Biomed Res. Int. 2015, 2015, 891707. [Google Scholar] [CrossRef]
- Wan, M.; Hua, X.; Su, J.; Thiagarajan, D.; Frostegård, A.G.; Haeggström, J.Z.; Frostegård, J. Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by Annexin A5. Atherosclerosis 2014, 235, 592–598. [Google Scholar] [CrossRef]
- Mercer, J.R.; Cheng, K.-K.; Figg, N.; Gorenne, I.; Mahmoudi, M.; Griffin, J.; Vidal-Puig, A.; Logan, A.; Murphy, M.P.; Bennett, M. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ. Res. 2010, 107, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Botto, N.; Rizza, A.; Colombo, M.G.; Mazzone, A.M.; Manfredi, S.; Masetti, S.; Clerico, A.; Biagini, A.; Andreassi, M.G. Evidence for DNA damage in patients with coronary artery disease. Mutat. Res. 2001, 493, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Knaapen, M.W.M.; De Meyer, G.R.Y.; Herman, A.G.; Kockx, M.M. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 2002, 106, 927–932. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Gray, K.; Figg, N.; Finigan, A.; Starks, L.; Bennett, M. Defective Base Excision Repair of Oxidative DNA Damage in Vascular Smooth Muscle Cells Promotes Atherosclerosis. Circulation 2018, 138, 1446–1462. [Google Scholar] [CrossRef]
- Yu, E.; Calvert, P.A.; Mercer, J.R.; Harrison, J.; Baker, L.; Figg, N.L.; Kumar, S.; Wang, J.C.; Hurst, L.A.; Obaid, D.R.; et al. Mitochondrial DNA Damage Can Promote Atherosclerosis Independently of Reactive Oxygen Species Through Effects on Smooth Muscle Cells and Monocytes and Correlates With Higher-Risk Plaques in Humans. Circulation 2013, 128, 702–712. [Google Scholar] [CrossRef] [Green Version]
- Gimbrone, M.A.J.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Ponnuswamy, P.; Schröttle, A.; Ostermeier, E.; Grüner, S.; Huang, P.L.; Ertl, G.; Hoffmann, U.; Nieswandt, B.; Kuhlencordt, P.J. eNOS Protects from Atherosclerosis Despite Relevant Superoxide Production by the Enzyme in apoE−/− Mice. PLoS ONE 2012, 7, e30193. [Google Scholar] [CrossRef] [Green Version]
- Shafique, E.; Torina, A.; Reichert, K.; Colantuono, B.; Nur, N.; Zeeshan, K.; Ravichandran, V.; Liu, Y.; Feng, J.; Zeeshan, K.; et al. Mitochondrial redox plays a critical role in the paradoxical effects of NAPDH oxidase-derived ROS on coronary endothelium. Cardiovasc. Res. 2017, 113, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.D.; Zimmerman, G.A.; Prescott, S.M.; McEver, R.P.; McIntyre, T.M. Oxygen radicals induce human endothelial cells to express GMP-140 and bind neutrophils. J. Cell Biol. 1991, 112, 749–759. [Google Scholar] [CrossRef]
- Zhang, H.; Park, Y.; Wu, J.; Chen, X.P.; Lee, S.; Yang, J.; Dellsperger, K.C.; Zhang, C. Role of TNF-alpha in vascular dysfunction. Clin. Sci. 2009, 116, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Corda, S.; Laplace, C.; Vicaut, E.; Duranteau, J. Rapid Reactive Oxygen Species Production by Mitochondria in Endothelial Cells Exposed to Tumor Necrosis Factor- α Is Mediated by Ceramide. Am. J. Respir. Cell Mol. Biol. 2001, 24, 762–768. [Google Scholar] [CrossRef]
- Li, D.; Mehta, J.L. Upregulation of Endothelial Receptor for Oxidized LDL (LOX-1) by Oxidized LDL and Implications in Apoptosis of Human Coronary Artery Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1116–1122. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Abid, M.D.N.; Xiong, Y.; Chen, Q.; Chen, J. ox-LDL downregulates eNOS activity via LOX-1-mediated endoplasmic reticulum stress. Int. J. Mol. Med. 2013, 32, 1442–1450. [Google Scholar] [CrossRef] [Green Version]
- Akhmedov, A.; Rozenberg, I.; Paneni, F.; Camici, G.G.; Shi, Y.; Doerries, C.; Sledzinska, A.; Mocharla, P.; Breitenstein, A.; Lohmann, C.; et al. Endothelial overexpression of LOX-1 increases plaque formation and promotes atherosclerosis in vivo. Eur. Heart J. 2014, 35, 2839–2848. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Canty, T.G.J.; Boyle, E.M.J.; Farr, A.; Morgan, E.N.; Verrier, E.D.; Pohlman, T.H. Oxidative stress induces NF-kappaB nuclear translocation without degradation of IkappaBalpha. Circulation 1999, 100, II361–II364. [Google Scholar] [CrossRef]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: Evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Jung, H.Y.; Park, E.Y.; Kim, J.; Lee, W.J.; Bae, Y.S. Cutting edge: Direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J. Immunol. 2004, 173, 3589–3593. [Google Scholar] [CrossRef] [Green Version]
- Mohan, S.; Koyoma, K.; Thangasamy, A.; Nakano, H.; Glickman, R.D.; Mohan, N. Low shear stress preferentially enhances IKK activity through selective sources of ROS for persistent activation of NF-kappaB in endothelial cells. Am. J. Physiol. Cell Physiol. 2007, 292, C362–C371. [Google Scholar] [CrossRef]
- Sorescu, G.P.; Song, H.; Tressel, S.L.; Hwang, J.; Dikalov, S.; Smith, D.A.; Boyd, N.L.; Platt, M.O.; Lassègue, B.; Griendling, K.K.; et al. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase. Circ. Res. 2004, 95, 773–779. [Google Scholar] [CrossRef] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.; O’Neill, L.A.J. Succinate: A metabolic signal in inflammation. Trends Cell Biol. 2014, 24, 313–320. [Google Scholar] [CrossRef] [Green Version]
- Ives, A.; Nomura, J.; Martinon, F.; Roger, T.; LeRoy, D.; Miner, J.N.; Simon, G.; Busso, N.; So, A. Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat. Commun. 2015, 6, 6555. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef]
- Bevilacqua, M.P.; Pober, J.S.; Wheeler, M.E.; Cotran, R.S.; Gimbrone, M.A.J. Interleukin-1 activation of vascular endothelium. Effects on procoagulant activity and leukocyte adhesion. Am. J. Pathol. 1985, 121, 394–403. [Google Scholar]
- Libby, P.; Warner, S.J.; Friedman, G.B. Interleukin 1: A mitogen for human vascular smooth muscle cells that induces the release of growth-inhibitory prostanoids. J. Clin. Investig. 1988, 81, 487–498. [Google Scholar] [CrossRef]
- Loppnow, H.; Libby, P. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin 6. J. Clin. Investig. 1990, 85, 731–738. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Virmani, R.; Burke, A.P.; Farb, A.; Kolodgie, F.D. Pathology of the vulnerable plaque. J. Am. Coll. Cardiol. 2006, 47, C13–C18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenglet, S.; Mach, F.; Montecucco, F. Role of matrix metalloproteinase-8 in atherosclerosis. Mediators Inflamm. 2013, 2013, 659282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, S.H.; Cho, C.-H.; Kim, H.O.; Jo, Y.H.; Yoon, K.-S.; Lee, J.H.; Park, J.-C.; Park, K.C.; Ahn, T.-B.; Chung, K.C.; et al. Plaque rupture is a determinant of vascular events in carotid artery atherosclerotic disease: Involvement of matrix metalloproteinases 2 and 9. J. Clin. Neurol. 2011, 7, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newby, A.C. Metalloproteinases promote plaque rupture and myocardial infarction: A persuasive concept waiting for clinical translation. Matrix Biol. 2015, 44–46, 157–166. [Google Scholar] [CrossRef]
- Gough, P.J.; Gomez, I.G.; Wille, P.T.; Raines, E.W. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J. Clin. Investig. 2006, 116, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.L.; George, S.J.; Newby, A.C.; Jackson, C.L. Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc. Natl. Acad. Sci. USA 2005, 102, 15575–15580. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Mironova, M.; Lopes-Virella, M.F. Oxidized LDL Stimulates Matrix Metalloproteinase-1 Expression in Human Vascular Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2640–2647. [Google Scholar] [CrossRef] [Green Version]
- Valentin, F.; Bueb, J.-L.; Kieffer, P.; Tschirhart, E.; Atkinson, J. Oxidative stress activates MMP-2 in cultured human coronary smooth muscle cells. Fundam. Clin. Pharmacol. 2005, 19, 661–667. [Google Scholar] [CrossRef]
- Xu, X.P.; Meisel, S.R.; Ong, J.M.; Kaul, S.; Cercek, B.; Rajavashisth, T.B.; Sharifi, B.; Shah, P.K. Oxidized low-density lipoprotein regulates matrix metalloproteinase-9 and its tissue inhibitor in human monocyte-derived macrophages. Circulation 1999, 99, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K.K.; Melendez, J.A. Mitochondrial redox control of matrix metalloproteinases. Free Radic. Biol. Med. 2004, 37, 768–784. [Google Scholar] [CrossRef] [PubMed]
- Zalba, G.; Fortuño, A.; Orbe, J.; San José, G.; Moreno, M.U.; Belzunce, M.; Rodríguez, J.A.; Beloqui, O.; Páramo, J.A.; Díez, J. Phagocytic NADPH Oxidase-Dependent Superoxide Production Stimulates Matrix Metalloproteinase-9. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 587–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjelakovic, G.; Nikolova, D.; Gluud, C. Antioxidant Supplements to Prevent Mortality. JAMA 2013, 310, 1178–1179. [Google Scholar] [CrossRef]
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vascul. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Cook, N.R.; Lee, I.-M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.; Sharma, S.; Xu, S.; Tewari, D.; Fang, J. Curcumin as a Natural Remedy for Atherosclerosis: A Pharmacological Review. Molecules 2021, 26, 36. [Google Scholar] [CrossRef]
- Siasos, G.; Tousoulis, D.; Tsigkou, V.; Kokkou, E.; Oikonomou, E.; Vavuranakis, M.; Basdra, E.K.; Papavassiliou, A.G.; Stefanadis, C. Flavonoids in atherosclerosis: An overview of their mechanisms of action. Curr. Med. Chem. 2013, 20, 2641–2660. [Google Scholar] [CrossRef]
- Dalgaard, F.; Bondonno, N.P.; Murray, K.; Bondonno, C.P.; Lewis, J.R.; Croft, K.D.; Kyrø, C.; Gislason, G.; Scalbert, A.; Cassidy, A.; et al. Associations between habitual flavonoid intake and hospital admissions for atherosclerotic cardiovascular disease: A prospective cohort study. Lancet. Planet. Health 2019, 3, e450–e459. [Google Scholar] [CrossRef] [Green Version]
- Peterson, J.J.; Dwyer, J.T.; Jacques, P.F.; McCullough, M.L. Associations between flavonoids and cardiovascular disease incidence or mortality in European and US populations. Nutr. Rev. 2012, 70, 491–508. [Google Scholar] [CrossRef] [Green Version]
- Sesso, H.D.; Manson, J.E.; Aragaki, A.K.; Rist, P.M.; Johnson, L.G.; Friedenberg, G.; Copeland, T.; Clar, A.; Mora, S.; Moorthy, M.V.; et al. Effect of cocoa flavanol supplementation for the prevention of cardiovascular disease events: The COcoa Supplement and Multivitamin Outcomes Study (COSMOS) randomized clinical trial. Am. J. Clin. Nutr. 2022, 115, 1490–1500. [Google Scholar] [CrossRef]
- Agarwal, B.; Campen, M.J.; Channell, M.M.; Wherry, S.J.; Varamini, B.; Davis, J.G.; Baur, J.A.; Smoliga, J.M. Resveratrol for primary prevention of atherosclerosis: Clinical trial evidence for improved gene expression in vascular endothelium. Int. J. Cardiol. 2013, 166, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Santana, T.M.; Ogawa, L.Y.; Rogero, M.M.; Barroso, L.P.; Alves de Castro, I. Effect of resveratrol supplementation on biomarkers associated with atherosclerosis in humans. Complement. Ther. Clin. Pract. 2022, 46, 101491. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; McMurray, J.J.V.; Klug, E.; Small, R.; Schumi, J.; Choi, J.; Cooper, J.; Scott, R.; Lewis, E.F.; L’Allier, P.L.; et al. Effects of succinobucol (AGI-1067) after an acute coronary syndrome: A randomised, double-blind, placebo-controlled trial. Lancet 2008, 371, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Taguchi, I.; Teragawa, H.; Ishizaka, N.; Kanzaki, Y.; Tomiyama, H.; Sata, M.; Sezai, A.; Eguchi, K.; Kato, T.; et al. Febuxostat does not delay progression of carotid atherosclerosis in patients with asymptomatic hyperuricemia: A randomized, controlled trial. PLoS Med. 2020, 17, e1003095. [Google Scholar] [CrossRef] [Green Version]
- White, H.D.; Held, C.; Stewart, R.; Tarka, E.; Brown, R.; Davies, R.Y.; Budaj, A.; Harrington, R.A.; Steg, P.G.; Ardissino, D.; et al. Darapladib for preventing ischemic events in stable coronary heart disease. N. Engl. J. Med. 2014, 370, 1702–1711. [Google Scholar] [CrossRef] [Green Version]
- O’Donoghue, M.L.; Braunwald, E.; White, H.D.; Lukas, M.A.; Tarka, E.; Steg, P.G.; Hochman, J.S.; Bode, C.; Maggioni, A.P.; Im, K.; et al. Effect of darapladib on major coronary events after an acute coronary syndrome: The SOLID-TIMI 52 randomized clinical trial. JAMA 2014, 312, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- Martin-Ventura, J.L.; Rodrigues-Diez, R.; Martinez-Lopez, D.; Salaices, M.; Blanco-Colio, L.M.; Briones, A.M. Oxidative Stress in Human Atherothrombosis: Sources, Markers and Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 2315. [Google Scholar] [CrossRef] [Green Version]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Chegodaev, Y.S.; Wu, W.-K.; Orekhov, A.N. Oxidative Stress and Antioxidants in Atherosclerosis Development and Treatment. Biology 2020, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- López-Alarcón, C.; Denicola, A. Evaluating the antioxidant capacity of natural products: A review on chemical and cellular-based assays. Anal. Chim. Acta 2013, 763, 1–10. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batty, M.; Bennett, M.R.; Yu, E. The Role of Oxidative Stress in Atherosclerosis. Cells 2022, 11, 3843. https://doi.org/10.3390/cells11233843
Batty M, Bennett MR, Yu E. The Role of Oxidative Stress in Atherosclerosis. Cells. 2022; 11(23):3843. https://doi.org/10.3390/cells11233843
Chicago/Turabian StyleBatty, Matthew, Martin R. Bennett, and Emma Yu. 2022. "The Role of Oxidative Stress in Atherosclerosis" Cells 11, no. 23: 3843. https://doi.org/10.3390/cells11233843