
CAR Triggered Release of Type-1 Interferon Limits CAR T-Cell Activities by an Artificial Negative Autocrine Loop
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. CAR T-Cell Generation
2.3. Flow Cytometry
2.4. Cytokine Secretion
2.5. Bioactivity of IFNs
2.6. Cytotoxicity Assay
2.7. Repetitive Stimulation Assay
2.8. Statistical Analysis
3. Results
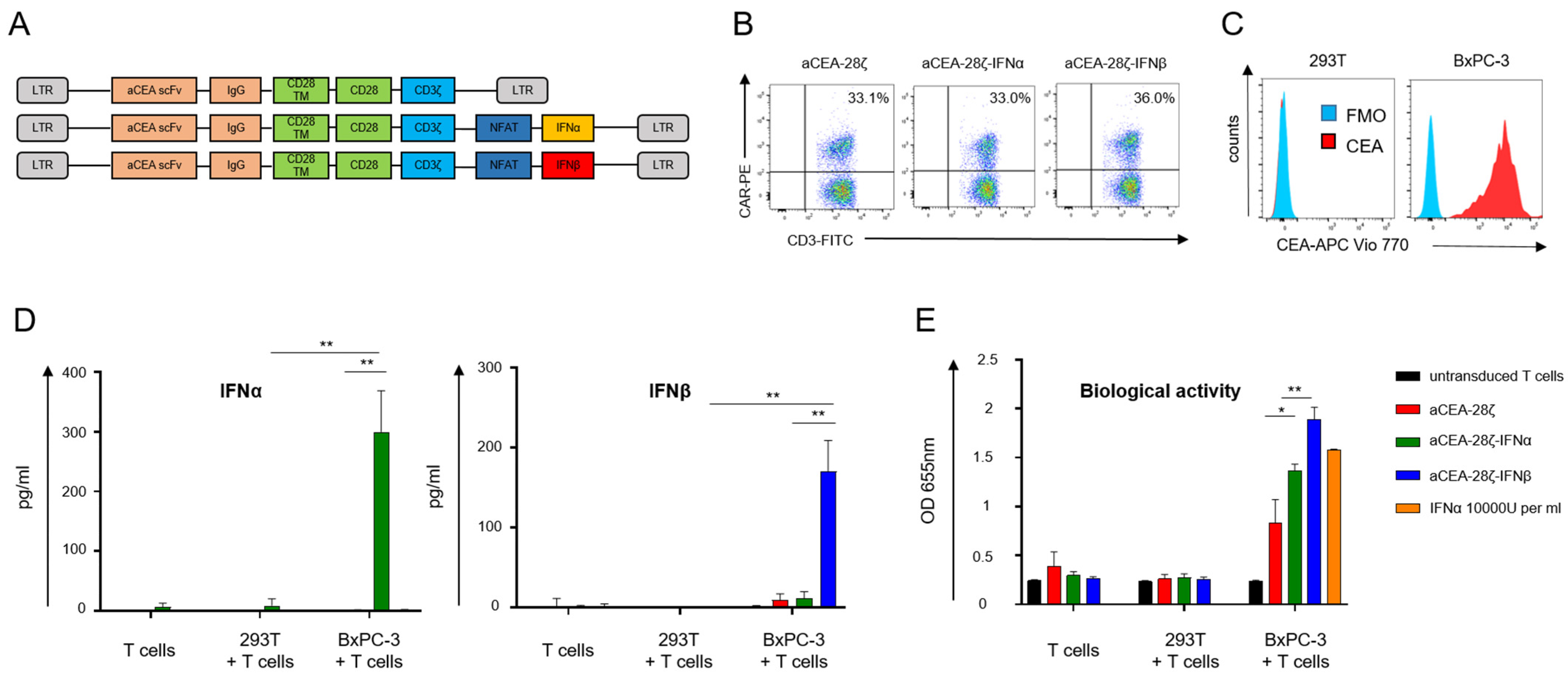
3.1. Development of CAR T-Cells with CAR Triggered Release of Bioactive IFN-α or IFN-β
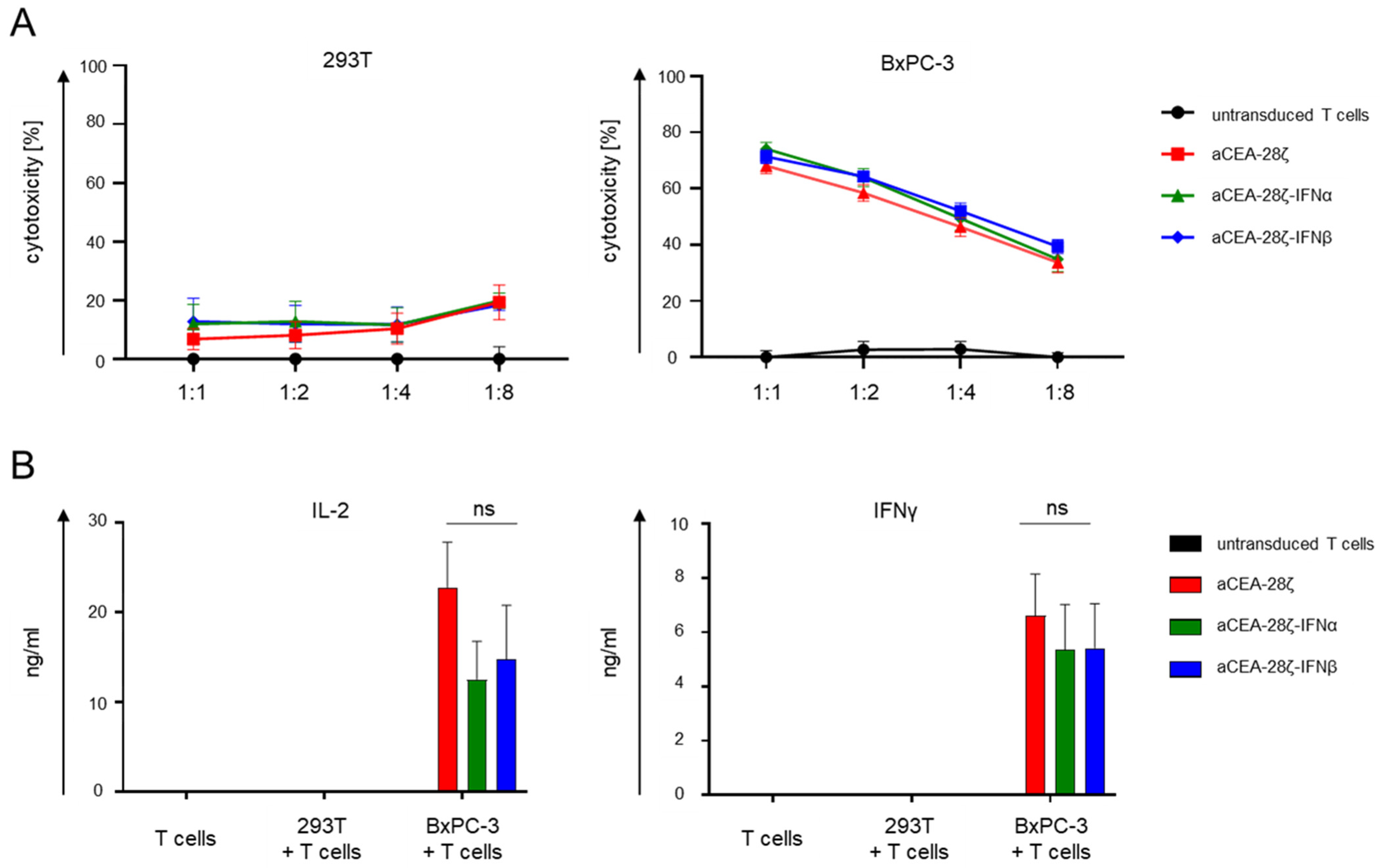
3.2. IFN-CAR T-Cells and Conventional CAR T-Cells Showed Similar Cytotoxicity and Cytokine Secretion
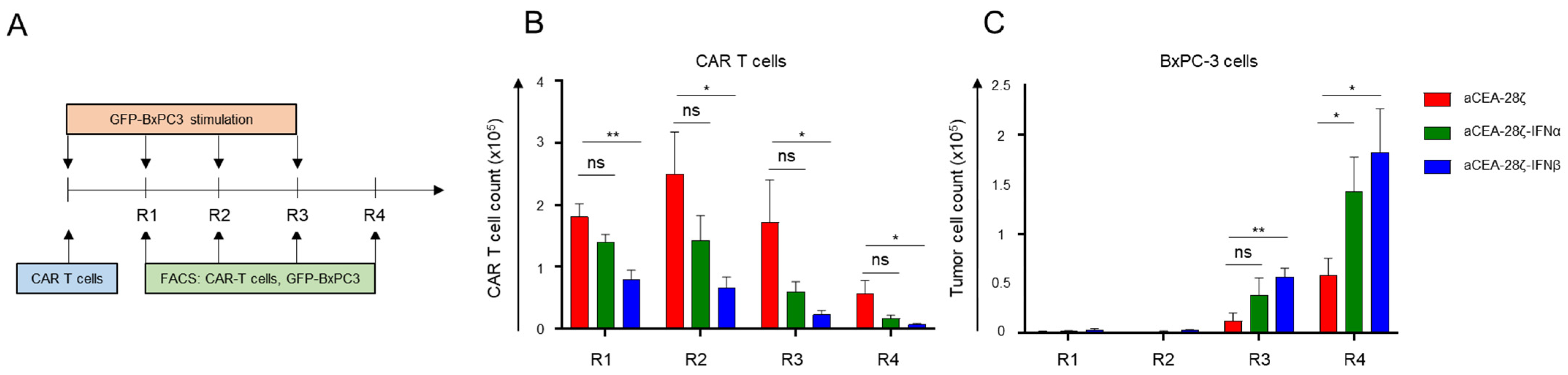
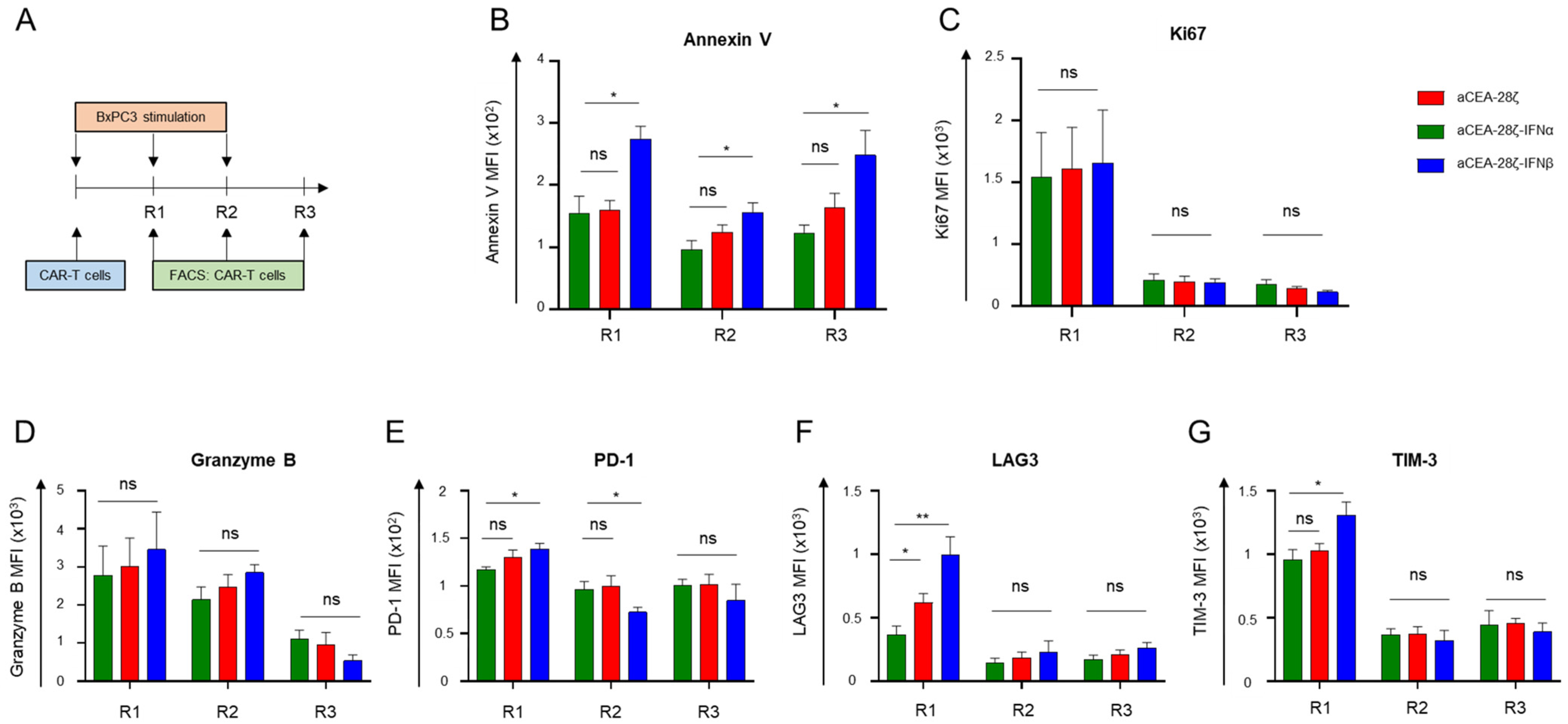
3.3. IFN-CAR T-Cells Exhibited Self-Limiting Activity upon CAR Signaling
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sadelain, M.; Rivière, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, A.; Abken, H. Treatment with Living Drugs: Pharmaceutical Aspects of CAR T Cells. Pharmacology 2022, 107, 446–463. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, A.; Abken, H. Advances and Challenges of CAR T Cells in Clinical Trials. Recent Results Cancer Res. 2020, 214, 93–128. [Google Scholar] [CrossRef]
- Castellarin, M.; Watanabe, K.; June, C.H.; Kloss, C.C.; Posey, A.D., Jr. Driving cars to the clinic for solid tumors. Gene Ther. 2018, 25, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert. Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, K.; Kuehle, J.; Dragon, A.C.; Galla, M.; Kloth, C.; Rudek, L.S.; Sandalcioglu, I.E.; Neyazi, B.; Moritz, T.; Meyer, J.; et al. Design and Characterization of an “All-in-One” Lentiviral Vector System Combining Constitutive Anti-G(D2) CAR Expression and Inducible Cytokines. Cancers (Basel) 2020, 12, 375. [Google Scholar] [CrossRef]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, K.; Al-Obaidi, M.; Di Stasi, A. Generation of Suicide Gene-Modified Chimeric Antigen Receptor-Redirected T-Cells for Cancer Immunotherapy. Methods Mol. Biol. 2019, 1895, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Labanieh, L.; Majzner, R.G.; Klysz, D.; Sotillo, E.; Fisher, C.J.; Vilches-Moure, J.G.; Pacheco, K.Z.B.; Malipatlolla, M.; Xu, P.; Hui, J.H.; et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell 2022, 185, 1745–1763. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015, 350, aab4077. [Google Scholar] [CrossRef] [Green Version]
- Zajc, C.U.; Dobersberger, M.; Schaffner, I.; Mlynek, G.; Pühringer, D.; Salzer, B.; Djinović-Carugo, K.; Steinberger, P.; de Sousa Linhares, A.; Yang, N.J.; et al. A conformation-specific ON-switch for controlling CAR T cells with an orally available drug. Proc. Natl. Acad. Sci. USA 2020, 117, 14926–14935. [Google Scholar] [CrossRef]
- Bachmann, M. The UniCAR system: A modular CAR T cell approach to improve the safety of CAR T cells. Immunol. Lett. 2019, 211, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.; Fasslrinner, F.; Loureiro, L.R.; Koristka, S.; Feldmann, A.; Bachmann, M. Adaptor CAR Platforms-Next Generation of T Cell-Based Cancer Immunotherapy. Cancers 2020, 12, 1302. [Google Scholar] [CrossRef]
- Darowski, D.; Kobold, S.; Jost, C.; Klein, C. Combining the best of two worlds: Highly flexible chimeric antigen receptor adaptor molecules (CAR-adaptors) for the recruitment of chimeric antigen receptor T cells. MAbs 2019, 11, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Harrer, D.C.; Simon, B.; Fujii, S.-I.; Shimizu, K.; Uslu, U.; Schuler, G.; Gerer, K.F.; Hoyer, S.; Dörrie, J.; Schaft, N. RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: A safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer 2017, 17, 551. [Google Scholar] [CrossRef]
- Krug, C.; Birkholz, K.; Paulus, A.; Schwenkert, M.; Schmidt, P.; Hoffmann, N.; Hombach, A.; Fey, G.; Abken, H.; Schuler, G.; et al. Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol. Immunother. 2015, 64, 1623–1635. [Google Scholar] [CrossRef]
- Aricò, E.; Castiello, L.; Capone, I.; Gabriele, L.; Belardelli, F. Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications. Cancers 2019, 11, 1943. [Google Scholar] [CrossRef] [Green Version]
- Borden, E.C. Interferons α and β in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cao, Y.; Markelc, B.; Kaeppler, J.; Vermeer, J.A.; Muschel, R.J. Type I IFN protects cancer cells from CD8+ T cell-mediated cytotoxicity after radiation. J. Clin. Investig. 2019, 129, 4224–4238. [Google Scholar] [CrossRef]
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 2015, 15, 231–242. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Murira, A.; Lamarre, A. Type-I Interferon Responses: From Friend to Foe in the Battle against Chronic Viral Infection. Front. Immunol. 2016, 7, 609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh, R.M.; Bahl, K.; Marshall, H.D.; Urban, S.L. Type 1 interferons and antiviral CD8 T-cell responses. PLoS Pathog. 2012, 8, e1002352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bon, A.; Durand, V.; Kamphuis, E.; Thompson, C.; Bulfone-Paus, S.; Rossmann, C.; Kalinke, U.; Tough, D.F. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J. Immunol. 2006, 176, 4682–4689. [Google Scholar] [CrossRef] [Green Version]
- Evgin, L.; Huff, A.L.; Wongthida, P.; Thompson, J.; Kottke, T.; Tonne, J.; Schuelke, M.; Ayasoufi, K.; Driscoll, C.B.; Shim, K.G.; et al. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat. Commun. 2020, 11, 3187. [Google Scholar] [CrossRef]
- Golumba-Nagy, V.; Kuehle, J.; Abken, H. Genetic Modification of T Cells with Chimeric Antigen Receptors: A Laboratory Manual. Hum. Gene Ther. Methods 2017, 28, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J. Immunol. 2001, 167, 6123–6131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hombach, A.A.; Rappl, G.; Abken, H. Blocking CD30 on T Cells by a Dual Specific CAR for CD30 and Colon Cancer Antigens Improves the CAR T Cell Response against CD30(-) Tumors. Mol. Ther. 2019, 27, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Medrano, R.F.V.; Hunger, A.; Mendonça, S.A.; Barbuto, J.A.M.; Strauss, B.E. Immunomodulatory and antitumor effects of type I interferons and their application in cancer therapy. Oncotarget 2017, 8, 71249–71284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piehler, J.; Thomas, C.; Garcia, K.C.; Schreiber, G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol. Rev. 2012, 250, 317–334. [Google Scholar] [CrossRef] [Green Version]
- Filipi, M.; Jack, S. Interferons in the Treatment of Multiple Sclerosis: A Clinical Efficacy, Safety, and Tolerability Update. Int. J. MS Care 2020, 22, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Vitale, G.; van Eijck, C.H.; van Koetsveld Ing, P.M.; Erdmann, J.I.; Speel, E.J.M.; van der Wansem Ing, K.; Mooij, D.M.; Colao, A.; Lombardi, G.; Croze, E.; et al. Type I interferons in the treatment of pancreatic cancer: Mechanisms of action and role of related receptors. Ann. Surg. 2007, 246, 259–268. [Google Scholar] [CrossRef]
- Heuser, C.; Guhlke, S.; Matthies, A.; Bender, H.; Barth, S.; Diehl, V.; Abken, H.; Hombach, A. Anti-CD30-scFv-Fc-IL-2 antibody-cytokine fusion protein that induces resting NK cells to highly efficient cytolysis of Hodgkin’s lymphoma derived tumour cells. Int. J. Cancer 2004, 110, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Di Franco, S.; Turdo, A.; Todaro, M.; Stassi, G. Role of Type I and II Interferons in Colorectal Cancer and Melanoma. Front. Immunol. 2017, 8, 878. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.; Mu, W.; Zhao, Y.; Jia, X.; Liu, J.; Wei, Q.; Tan, T.; Zhou, J. The rational development of CD5-targeting biepitopic CARs with fully human heavy-chain-only antigen recognition domains. Mol. Ther. 2021, 29, 2707–2722. [Google Scholar] [CrossRef]
- Mackensen, A.; Müller, F.; Mougiakakos, D.; Böltz, S.; Wilhelm, A.; Aigner, M.; Völkl, S.; Simon, D.; Kleyer, A.; Munoz, L.; et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 2022, 28, 2124–2132. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrer, D.C.; Schenkel, C.; Bezler, V.; Kaljanac, M.; Hartley, J.; Barden, M.; Pan, H.; Holzinger, A.; Herr, W.; Abken, H. CAR Triggered Release of Type-1 Interferon Limits CAR T-Cell Activities by an Artificial Negative Autocrine Loop. Cells 2022, 11, 3839. https://doi.org/10.3390/cells11233839
Harrer DC, Schenkel C, Bezler V, Kaljanac M, Hartley J, Barden M, Pan H, Holzinger A, Herr W, Abken H. CAR Triggered Release of Type-1 Interferon Limits CAR T-Cell Activities by an Artificial Negative Autocrine Loop. Cells. 2022; 11(23):3839. https://doi.org/10.3390/cells11233839
Chicago/Turabian StyleHarrer, Dennis Christoph, Charlotte Schenkel, Valerie Bezler, Marcell Kaljanac, Jordan Hartley, Markus Barden, Hong Pan, Astrid Holzinger, Wolfgang Herr, and Hinrich Abken. 2022. "CAR Triggered Release of Type-1 Interferon Limits CAR T-Cell Activities by an Artificial Negative Autocrine Loop" Cells 11, no. 23: 3839. https://doi.org/10.3390/cells11233839