

cAMP-Dependent Signaling and Ovarian Cancer

Abstract
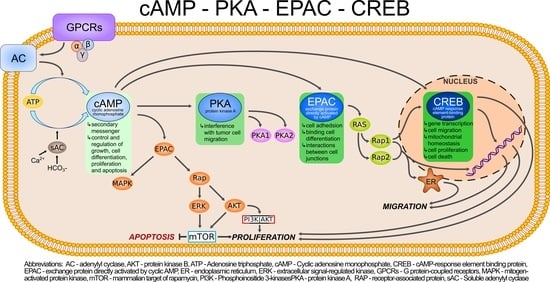
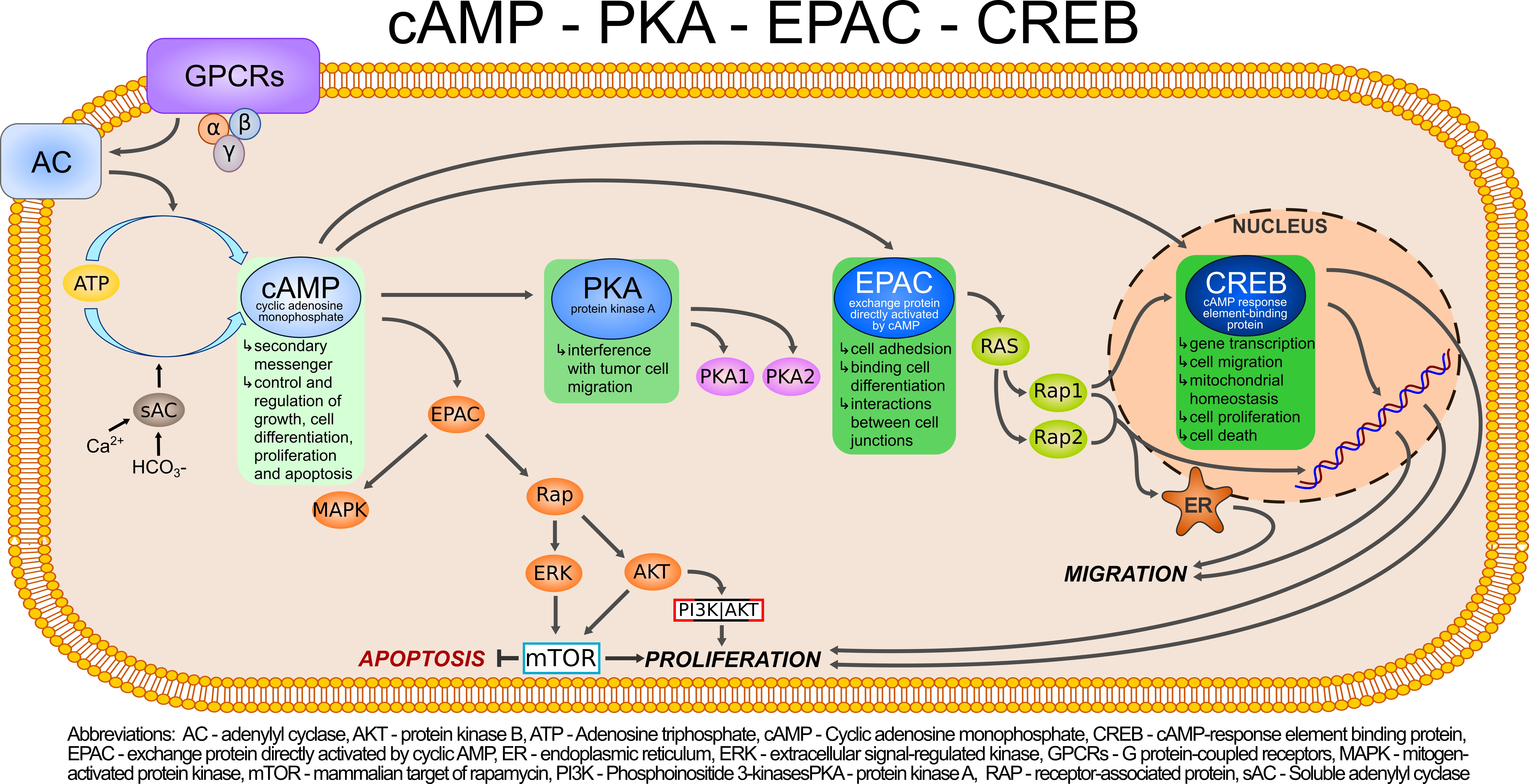
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. cAMP/PKA/CREB Cascade Signaling
Ovarian Intracellular cAMP and Stimulation of PKA Are Increased upon Gonadotropin Activity
3. The Bioinformatics Prediction in Ovarian Cancer
4. Signal Transduction Pathways as Potential Therapeutic Targets
5. Therapy
5.1. The Potential Role of PDE Inhibition in Ovarian Carcinoma Cells
5.2. AMP-PKA-pCREB, Leading to the Rapid Activity of PARP and DNA Repair
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Bai, X.; Dong, Q. Identification of novel candidate genes and small molecule drugs in ovarian cancer by bioinformatics strategy. Transl. Cancer Res. 2022, 11, 1630–1643. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Doubeni, C.A.; Doubeni, A.R.B.; Myers, A.E. Diagnosis and Management of Ovarian Cancer. Am. Fam. Physician 2016, 93, 937–944. [Google Scholar] [PubMed]
- Shanmughapriya, S.; Senthilkumar, G.; Vinodhini, K.; Das, B.C.; Vasanthi, N.; Natarajaseenivasan, K. Viral and bacterial aetiologies of epithelial ovarian cancer. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 2311–2317. [Google Scholar] [CrossRef] [PubMed]
- Mertens-Walker, I.; Bolitho, C.; Baxter, R.C.; Marsh, D.J. Gonadotropin-induced ovarian cancer cell migration and proliferation require extracellular signal-regulated kinase 1/2 activation regulated by calcium and protein kinase Cδ. Endocr Relat. Cancer 2010, 17, 335–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auersperg, N.; Wong, A.S.T.; Choi, K.C.; Kang, S.K.; Leung, P.C.K. Ovarian surface epithelium: Biology, endocrinology, and pathology. Endocr. Rev. 2001, 22, 255–288. [Google Scholar] [PubMed] [Green Version]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef] [Green Version]
- Karnezis, A.N.; Cho, K.R.; Gilks, C.B.; Pearce, C.L.; Huntsman, D.G. The disparate origins of ovarian cancers: Pathogenesis and prevention strategies. Nat. Rev. Cancer 2017, 17, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mei, Y.; Long, J.; Zhang, Y.; Hu, D.; Zhou, H. RIF1 promotes human epithelial ovarian cancer growth and progression via activating human telomerase reverse transcriptase expression. J. Exp. Clin. Cancer Res. 2018, 37, 182. [Google Scholar] [CrossRef]
- Prat, J.; D’Angelo, E.; Espinosa, I. Ovarian carcinomas: At least five different diseases with distinct histological features and molecular genetics. Hum. Pathol. 2018, 80, 11–27. [Google Scholar] [CrossRef] [PubMed]
- de Leo, A.; Santini, D.; Ceccarelli, C.; Santandrea, G.; Palicelli, A.; Acquaviva, G.; Chiarucci, F.; Rosini, F.; Ravegnini, G.; Pession, A.; et al. What is new on ovarian carcinoma: Integrated morphologic and molecular analysis following the new 2020 world health organization classification of female genital tumors. Diagnostics 2021, 11, 697. [Google Scholar] [CrossRef] [PubMed]
- Köbel, M.; Kang, E.Y. The Evolution of Ovarian Carcinoma Subclassification. Cancers 2022, 14, 416. [Google Scholar] [CrossRef] [PubMed]
- Murinaro, T.; Boruah, H.P.D.; Mintu, P. Diagnostic and prognostic biomarkers in ovarian cancer and the potential roles of cancer stem cells—an updated review. Exp. Cell Res. 2018, 326, 1–10. [Google Scholar]
- Gong, S.; Chen, Y.; Meng, F.; Zhang, Y.; Wu, H.; Wu, F. Roflumilast restores cAMP/PKA/CREB signaling axis for FtMt- mediated tumor inhibition of ovarian cancer. Oncotarget 2017, 8, 112341–112353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, W.; Ma, J.; Xiao, Y.; Wang, P.; Gu, X.; Xie, B.; Li, M. The Apoptotic Resistance of BRCA1-Deficient Ovarian Cancer Cells is Mediated by cAMP. Front. Cell Dev. Biol. 2022, 10, 889656. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, N.; Nagaraj, A.B.; Razi, A.; Singh, S.; Kamalakaran, S.; Banerjee, N.; Joseph, P.; Mankovich, A.; Mittal, P.; Difeo, A.; et al. InFlo: A novel systems biology framework identifies cAMP-CREB1 axis as a key modulator of platinum resistance in ovarian cancer. Oncogene 2017, 36, 2472–2482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubbay, O.; Rae, M.T.; McNeilly, A.S.; Donadeu, F.X.; Zeleznik, A.J.; Hillier, S.G. cAMP response element-binding (CREB) signalling and ovarian surface epithelial cell survival. J. Endocrinol. 2006, 191, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, S.Y.; Park, J.J.; Shin, E.Y.; Kim, E.G. PAK4 signaling in health and disease: Defining the PAK4–CREB axis. Exp. Mol. Med. 2019, 51, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fajardo, A.M.; Piazza, G.A.; Tinsley, H.N. The role of cyclic nucleotide signaling pathways in cancer: Targets for prevention and treatment. Cancers 2014, 6, 436–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taussig, R.; Gilman, A.G. Mammalian Membrane-bound Adenylyl Cyclases. J. Biol. Chem. 1995, 270, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Hunzicker-Dunn, M.E.; Lopez-Biladeau, B.; Law, N.C.; Fiedler, S.E.; Carr, D.W.; Maizels, E.T. PKA and GAB2 play central roles in the FSH signaling pathway to PI3K and AKT in ovarian granulosa cells. Proc. Natl. Acad. Sci. USA 2012, 109, E2979–E2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheadle, C.; Nesterova, M.; Watkins, T.; Barnes, K.C.; Hall, J.C.; Rosen, A.; Becker, K.G.; Cho-Chung, Y.S. Regulatory subunits of PKA define an axis of cellular proliferation/differentiation in ovarian cancer cells. BMC Med. Genom. 2008, 1, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Beavo, J.; Brunton, L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–717. [Google Scholar] [CrossRef]
- Robichaux, W.G.; Cheng, X.X. Intracellular cAMP sensor Epac: Physiology, pathophysiology, and therapeutics development. Physiol. Rev. 2018, 98, 919–1053. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.B.; Agarwal, S.R.; Harvey, R.D.; Ostrom, R.S. cAMP Signaling Compartmentation: Adenylyl Cyclases as Anchors of Dynamic Signaling Complexes. Mol. Pharm. 2018, 93, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Vigone, G.; Shuhaibar, L.C.; Egbert, J.R.; Uliasz, T.F.; Movsesian, M.A.; Jaffe, L.A. Multiple cAMP Phosphodiesterases Act Together to Prevent Premature Oocyte Meiosis and Ovulation. Endocrinology 2018, 159, 2142–2152. [Google Scholar] [CrossRef] [Green Version]
- Carr, D.W.; DeManno, D.A.; Atwood, A.; Hunzicker-Dunn, M.; Scott, J.D. Follicle-stimulating hormone regulation of A-kinase anchoring proteins in granulosa cells. J. Biol. Chem. 1993, 268, 20729–20732. [Google Scholar] [CrossRef]
- Parker, T.; Wang, K.W.; Manning, D.; Dart, C. Soluble adenylyl cyclase links Ca2+ entry to Ca2+/cAMP-response element binding protein (CREB) activation in vascular smooth muscle. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP Produced inside Mitochondria Regulates Oxidative Phosphorylation. Cell Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zippin, J.H.; Farrell, J.; Huron, D.; Kamenetsky, M.; Hess, K.C.; Fischman, D.A.; Levin, L.R.; Buck, J. Bicarbonate-responsive “soluble” adenylyl cyclase defines a nuclear cAMP microdomain. J. Cell Biol. 2004, 164, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, K.F.; LaVigne, J.E.; Brust, T.F.; Seifert, R.; Dessauer, C.W.; Watts, V.J.; Ostrom, R.S. Physiological roles of mammalian transmembrane adenylyl cyclase isoforms. Physiol. Rev. 2022, 102, 815–857. [Google Scholar] [CrossRef] [PubMed]
- Hanoune, J.; Defer, N. Regulation and role of adenylyl cyclase isoforms. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 145–174. [Google Scholar] [CrossRef]
- Ferreira, J.; Levin, L.R.; Buck, J. Strategies to safely target widely expressed soluble adenylyl cyclase for contraception. Front. Pharmacol. 2022, 13, 953903. [Google Scholar] [CrossRef]
- Chang, J.-C.; Oude-Elferink, R.P.J. Role of the bicarbonate-responsive soluble adenylyl cyclase in pH sensing and metabolic regulation. Front. Physiol. 2014, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wiggins, S.v.; Steegborn, C.; Levin, L.R.; Buck, J. Pharmacological modulation of the CO2/HCO3−/pH-, calcium-, and ATP-sensing soluble adenylyl cyclase. Pharmacol. Ther. 2018, 190, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, H.C. Amplification of FSH signalling by CFTR and nuclear soluble adenylyl cyclase in the ovary. Clin. Exp. Pharm. Physiol. 2017, 44, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilanowska, A.; Ziółkowska, A. Role of phosphodiesterase in the biology and pathology of diabetes. Int. J. Mol. Sci. 2020, 21, 8244. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, M.F.; Faucz, F.R.; Bimpaki, E.; Horvath, A.; Levy, I.; de Alexandre, R.B.; Ahmad, F.; Manganiello, V.; Stratakis, C.A. Clinical and molecular genetics of thephosphodiesterases (PDEs). Endocr. Rev. 2014, 35, 195–233. [Google Scholar] [CrossRef]
- Lugnier, C.; Meyer, A.; Talha, S.; Geny, B. Pharmacology & Therapeutics Cyclic nucleotide phosphodiesterases: New targets in the metabolic syndrome? A. General structure of PDE isozyme monomer N-terminal regulatory domain Conserved core catalytic site (270 aa) C-terminal domain. Pharm. Ther. 2020, 208, 107475. [Google Scholar]
- Menon, K.M.J.; Menon, B. Structure, Function and Regulation of Gonadotropin Receptors- A Perspective. Mol. Cell Endocrinol. 2012, 356, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Conti, M. Specificity of the Cyclic Adenosine 3’,5’-Monophosphate Signalin Granulosa Cell Function. Biol. Reprod. 2002, 67, 1653–1661. [Google Scholar] [CrossRef]
- Petersen, T.S.; Kristensen, S.G.; Jeppesen, J.V.; Grøndahl, M.L.; Wissing, M.L.; Macklone, K.T.; Andersen, C.Y. Distribution and function of 3′, 5′ -Cyclic-AMP phosphodiesterases in the human ovary. Mol. Cell Endocrinol. 2015, 403, 10–20. [Google Scholar] [CrossRef] [PubMed]
- di Benedetto, G.; Gerbino, A.; Lefkimmiatis, K. Shaping mitochondrial dynamics: The role of cAMP signalling. Biochem. Biophys. Res. Commun. 2018, 500, 65–74. [Google Scholar] [CrossRef]
- Langeberg, L.K.; Scott, J.D. Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 2015, 16, 232–244. [Google Scholar] [CrossRef] [Green Version]
- Torres-Quesada, O.; Mayrhofer, J.E.; Stefan, E. The many faces of compartmentalized PKA signalosomes. Cell Signal. 2017, 37, 1–11. [Google Scholar] [CrossRef]
- Zaccolo, M.; Zerio, A.; Lobo, M.J. Subcellular Organization of the cAMP Signaling Pathway. Pharm. Rev. 2021, 73, 278–309. [Google Scholar] [CrossRef]
- Greenwald, E.C.; Saucerman, J.J. Bigger, better, faster: Principles and models of AKAP signaling. J. Cardiovasc. Pharmacol. 2011, 58, 462–469. [Google Scholar] [CrossRef]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap. Science 1998, 282, 2275–2279. [Google Scholar]
- Kumar, N.; Prasad, P.; Jash, E.; Saini, M.; Husain, A.; Goldman, A.; Sehrawat, S. Insights into exchange factor directly activated by cAMP (EPAC) as potential target for cancer treatment. Mol. Cell Biochem. 2018, 447, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex roles of cAMP–PKA–CREB signaling in cancer. Exp. Hematol. Oncol. 2020, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. From Ras to Rap and Back, a Journey of 35 Years. Cold Spring Harb Perspect Med. 2018, 8, a031468. [Google Scholar] [CrossRef] [Green Version]
- Rehmann, H.; Das, J.; Knipscheer, P.; Wittinghofer, A.; Bos, J.L. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature 2006, 439, 625–628. [Google Scholar] [CrossRef]
- Rehmann, H.; Arias-Palomo, E.; Hadders, M.A.; Schwede, F.; Llorca, O.; Bos, J.L. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature 2008, 455, 124–127. [Google Scholar] [CrossRef]
- Bose, C.K. Follicle Stimulating Hormone Receptor in Ovarian Surface Epithelium and Epithelial Ovarian Cancer. Oncol Res. 2008, 17, 231–238. [Google Scholar] [CrossRef]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Gao, M.; Ma, Y.; Bast, R.C.; Yue, J.; Lu, L.; Liu, Y.; Sun, Y.; Fang, Z.; Zhang, L.; Wang, X.; et al. Epac1 knockdown inhibits the proliferation of ovarian cancer cells by inactivating AKT / Cyclin D1 / CDK4 pathway in vitro and in vivo. Med. Oncol. 2016, 33, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Dekker, F.J.; Maarsingh, H. Exchange protein directly activated by cAMP (epac): A multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol. Rev. 2013, 65, 670–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wayne, C.M.; Fan, H.-Y.; Cheng, X.; Richards, J.S. Follicle-Stimulating Hormone Induces Multiple Signaling Cascades: Evidence that Activation of Rous Sarcoma Oncogene, RAS, and the Epidermal Growth Factor Receptor Are Critical for Granulosa Cell Differentiation. Mol. Endocrinol. 2007, 21, 1940–1957. [Google Scholar] [CrossRef] [Green Version]
- Ponsioen, B.; Gloerich, M.; Ritsma, L.; Rehmann, H.; Bos, J.L.; Jalink, K. Direct Spatial Control of Epac1 by Cyclic AMP. Mol. Cell Biol. 2009, 29, 2521–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloerich, M.; Ponsioen, B.; Vliem, M.J.; Zhang, Z.; Zhao, J.; Kooistra, M.R.; Price, L.S.; Ritsma, L.; Zwartkruis, F.J.; Rehmann, H.; et al. Spatial Regulation of Cyclic AMP-Epac1 Signaling in Cell Adhesion by ERM Proteins. Mol. Cell Biol. 2010, 30, 5421–5431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montminy, M.R.; Bilezikjian, L.M. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature 1987, 328, 175–178. [Google Scholar] [CrossRef]
- Lonze, B.E.; Ginty, D.D. Function and Regulation of CREB Family Transcription Factors in the Nervous System CREB and its close relatives are now widely accepted. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, V.O.; Dobroff, A.S.; Zigler, M.; Villares, G.J.; Braeuer, R.R.; Wang, H.; Huang, L.; Bar-Eli, M. CREB inhibits AP-2α expression to regulate the malignant phenotype of melanoma. PLoS ONE 2010, 5, e12452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannessen, M.; Pedersen, M.; Moens, D.U. What turns CREB on? Cell Signal. 2004, 16, 1211–1227. [Google Scholar] [CrossRef] [PubMed]
- Shaywitz, A.J.; Greenberg, M.E. CREB: A Stimulus-Induced Transcription Factor Activated by A Diverse Array of Extracellular Signals. Annu. Rev. Biochem. 1999, 68, 821–861. [Google Scholar] [CrossRef]
- Wang, F.; Marshall, C.B.; Ikura, M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: Structural and functional versatility in target recognition. Cell. Mol. Life Sci. 2013, 70, 3989–4008. [Google Scholar] [CrossRef]
- Bhartiya, D.; Singh, J. FSH-FSHR3-stem cells in ovary surface epithelium: Basis for adult ovarian biology, failure, aging, and cancer. Reproduction 2015, 149, R35–R48. [Google Scholar] [CrossRef] [Green Version]
- Casarini, L.; Santi, D.; Brigante, G.; Simoni, M. Two hormones for one receptor: Evolution, biochemistry, actions, and pathophysiology of LH and hCG. Endocr. Rev. 2018, 39, 549–592. [Google Scholar] [CrossRef]
- Casarini, L.; Lispi, M.; Longobardi, S.; Milosa, F.; la Marca, A.; Tagliasacchi, D.; Pignatti, E.; Simoni, M. LH and hCG Action on the Same Receptor Results in Quantitatively and Qualitatively Different Intracellular Signalling. PLoS ONE 2012, 7, e46682. [Google Scholar] [CrossRef] [Green Version]
- Riccetti, L.; Sperduti, S.; Lazzaretti, C.; Casarini, L.; Simoni, M. The cAMP/PKA pathway: Steroidogenesis of the antral follicular stage. Minerva Ginecol. 2018, 70, 516–524. [Google Scholar] [CrossRef]
- Gershon, E.; Dekel, N. Newly identified regulators of ovarian folliculogenesis and ovulation. Int. J. Mol. Sci. 2020, 21, 4565. [Google Scholar] [CrossRef]
- Chauvin, S.; Cohen-Tannoudji, J.; Guigon, C.J. Estradiol Signaling at the Heart of Folliculogenesis: Its Potential Deregulation in Human Ovarian Pathologies. Int. J. Mol. Sci. 2022, 23, 512. [Google Scholar] [CrossRef]
- Richards, J.S.; Pangas, S.A. Review series The ovary: Basic biology and clinical implications. J. Clin. Investig. 2010, 120, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Casarini, L.; Crépieux, P. Molecular mechanisms of action of FSH. Front. Endocrinol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chin, E.C.; Harris, T.E.; Abayasekara, D.R.E. Changes in cAMP-dependent protein kinase (PKA) and progesterone secretion in luteinizing human granulosa cells. J. Endocrinol. 2004, 183, 39–50. [Google Scholar] [CrossRef]
- Hurley, J.H. Structure, Mechanism, and Regulation of Mammalian Adenylyl Cyclase. J. Biol. Chem. 1999, 274, 7599–7602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steegborn, C. Biochimica et Biophysica Acta Structure, mechanism, and regulation of soluble adenylyl cyclases—similarities and differences to transmembrane adenylyl cyclases ☆. BBA Mol. Basis Dis. 2014, 1842, 2535–2547. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.M.F. Compartmentalization of adenylate cyclase and cAMP signalling. Biochem. Soc. Trans. 2005, 6, 1319–1322. [Google Scholar] [CrossRef] [Green Version]
- Flynn, M.P.; Maizels, E.T.; Karlsson, A.B.; Mcavoy, T.; Ahn, J.; Nairn, A.C.; Hunzicker-Dunn, M. Luteinizing Hormone Receptor Activation in Ovarian Granulosa Cells Promotes Protein Kinase A-Dependent Dephosphorylation of Microtubule- Associated Protein 2D. Mol. Endocrinol. 2008, 22, 1695–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.; Park-Sarge, O.-K.; Mayo, K.E. Gonadotropins Induce Rapid Phosphorylation of the 3’,5’-Cyclic Adenosine Monophosphate Response Element Binding in Ovarian Granulosa Cells. Endocrinology 1996, 137, 3234–3245. [Google Scholar] [CrossRef] [PubMed]
- Casarini, L.; Moriondo, V.; Marino, M.; Adversi, F.; Capodanno, F.; Grisolia, C.; la Marca, A.; la Sala, G.B.; Simoni, M. FSHR polymorphism p.N680S mediates different responses to FSH in vitro. Mol. Cell Endocrinol. 2014, 393, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Cottom, J.; Salvador, L.M.; Maizels, E.T.; Reierstad, S.; Park, Y.; Carr, D.W.; Davare, M.A.; Hell, J.W.; Palmer, S.S.; Dent, P.; et al. Follicle-stimulating Hormone Activates Extracellular Signal-regulated Kinase but Not Extracellular Signal-regulated Kinase Kinase through a 100-kDa Phosphotyrosine Phosphatase. J. Biol. Chem. 2003, 278, 7167–7179. [Google Scholar] [CrossRef] [PubMed]
- Salvador, L.M.; Park, Y.; Cottom, J.; Maizels, E.T.; Jones, J.C.R.; Schillace, R.V.; Carr, D.W.; Cheung, P.; Allis, C.D.; Jameson, J.L.; et al. Follicle-stimulating Hormone Stimulates Protein Kinase A-mediated Histone H3 Phosphorylation and Acetylation Leading to Select Gene Activation in Ovarian Granulosa Cells. J. Biol. Chem. 2001, 276, 40146–40155. [Google Scholar] [CrossRef] [Green Version]
- DeManno, D.A.; Cottom, J.E.; Kline, M.P.; Peters, C.A.; Maizels, E.T.; Hunzicker-Dunn, M. Follicle-Stimulating Hormone Promotes Histone H3 Phosphorylation on Serine. Mol. Endocrinol. 1999, 13, 91–105. [Google Scholar] [CrossRef]
- Hunzicker-Dunn, M.; Maizels, E.T. FSH signaling pathways in immature granulosa cells that regulate target gene expression: Branching out from protein kinase A. Cell Signal. 2006, 18, 1351–1359. [Google Scholar] [CrossRef] [Green Version]
- Puri, P.; Little-Ihrig, L.; Chandran, U.; Law, N.C.; Hunzicker-Dunn, M.; Zeleznik, A.J. Protein Kinase A: A Master Kinase of Granulosa Cell Differentiation. Sci. Rep. 2016, 6, 28132. [Google Scholar] [CrossRef] [Green Version]
- Kayampilly, P.P.; Menon, K.M.J. Follicle-Stimulating Hormone Inhibits Adenosine 5′-Monophosphate-Activated Protein Kinase Activation and Promotes Cell Proliferation of Primary Granulosa Cells in Culture through an Akt-Dependent Pathway. Endocrinology 2009, 150, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Godin, P.; Tsoi, M.F.; Morin, M.; Gévry, N.; Boerboom, D. The granulosa cell response to luteinizing hormone is partly mediated by YAP1-dependent induction of amphiregulin. Cell Commun. Signal. 2022, 20, 72. [Google Scholar] [CrossRef]
- Li, J.-Y.; Li, C.-J.; Lin, L.-T.; Tsui, K.-H. Multi-Omics Analysis Identifying Key Biomarkers in Ovarian Cancer. Cancer Control 2020, 27, 1–10. [Google Scholar] [CrossRef]
- Kroeger, P.T., Jr.; Drapkin, R. Pathogenesis and heterogeneity of ovarian cancer. Curr. Opin. Obs. Gynecol. 2017, 29, 26. [Google Scholar] [CrossRef] [Green Version]
- Konecny, G.E.; Winterhoff, B.; Wang, C. Gene-expression signatures in ovarian cancer: Promise and challenges for patient stratification. Gynecol. Oncol. 2016, 141, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Januchowski, R.; Sterzyńska, K.; Zawierucha, P.; Ruciński, M.; Świerczewska, M.; Partyka, M.; Bednarek-Rajewska, K.; Brazert, M.; Nowicki, M.; Zabel, M.; et al. Microarray-based detection and expression analysis of new genes associated with drug resistance in ovarian cancer cell lines. Oncotarget 2017, 8, 49944–49958. [Google Scholar] [CrossRef]
- Kazmierczak, D.; Jopek, K.; Sterzynska, K.; Ginter-Matuszewska, B.; Nowicki, M.; Rucinski, M.; Januchowski, R. The significance of MicroRNAs expression in regulation of extracellular matrix and other drug resistant genes in drug resistant ovarian cancer cell lines. Int. J. Mol. Sci. 2020, 21, 2619. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Layton, O.; Hong, L. Identification of genes and pathways involved in ovarian epithelial cancer by bioinformatics analysis. J. Cancer 2018, 9, 3016–3022. [Google Scholar] [CrossRef]
- Huang, H.; Xu, S.; Chen, A.; Li, F.; Wu, J.; Tu, X.; Hu, K. Identification of a 5-Gene-Based Scoring System by WGCNA and LASSO to Predict Prognosis for Rectal Cancer Patients. Anal. Cell. Pathol. 2021, 2021, 1–17. [Google Scholar] [CrossRef]
- Yin, X.; Wang, P.; Yang, T.; Li, G.; Teng, X.; Huang, W.; Yu, H. Identification of key modules and genes associated with breast cancer prognosis using WGCNA and ceRNA network analysis. Aging 2021, 13, 2519–2538. [Google Scholar] [CrossRef]
- Nomiri, S.; Karami, H.; Baradaran, B.; Javadrashid, D.; Derakhshani, A.; Nourbakhsh, N.S.; Shadbad, M.A.; Solimando, A.G.; Tabrizi, N.J.; Brunetti, O.; et al. Exploiting systems biology to investigate the gene modules and drugs in ovarian cancer: A hypothesis based on the weighted gene co-expression network analysis. Biomed. Pharmacother. 2022, 146, 112537. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Wang, C.; Gu, Y.; Zhang, K.; Xie, K.; Zhu, M.; Dai, N.; Jiang, Y.; Guo, X.; Liu, M.; Dai, J.; et al. Systematic identification of genes with a cancer-testis expression pattern in 19 cancer types. Nat. Commun. 2016, 7, 10499. [Google Scholar] [CrossRef] [Green Version]
- Xie, K.; Fu, C.; Wang, S.; Xu, H.; Liu, S.; Shao, Y.; Gong, Z.; Wu, X.; Xu, B.; Han, J.; et al. Cancer-testis antigens in ovarian cancer: Implication for biomarkers and therapeutic targets. J. Ovarian Res. 2019, 12, 1–13. [Google Scholar] [CrossRef]
- Agarwal, S.; Saini, S.; Parashar, D.; Verma, A.; Sinha, A.; Jagadish, N.; Batra, A.; Suri, S.; Gupta, A.; Ansari, A.S.; et al. The novel cancer-testis antigen A-kinase anchor protein 4 (AKAP4) is a potential target for immunotherapy of ovarian serous carcinoma. Oncoimmunology 2013, 2, e24270. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, K.; Ono, T.; Matsushita, H.; Shimono, M.; Noguchi, Y.; Mizutani, Y.; Kodama, J.; Kudo, T.; Nakayama, E. A-kinase anchoring protein 3 messenger RNA expression in ovarian cancer and its implication on prognosis. Int. J. Cancer 2004, 108, 86–90. [Google Scholar] [CrossRef]
- Li, C.J.; Lin, L.T.; Chu, P.Y.; Chiang, A.J.; Tsai, H.W.; Chiu, Y.H.; Huang, M.S.; Wen, Z.H.; Tsui, K.H. Identification of novel biomarkers and candidate drug in ovarian cancer. J. Pers Med. 2021, 11, 316. [Google Scholar] [CrossRef]
- Chan, D.W.; Lam, W.-Y.; Chen, F.; Yung, M.M.H.; Chan, Y.S.; Chan, W.S.; He, F.; Liu, S.S.; Chan, K.K.L.; Li, B.; et al. Genome-wide DNA methylome analysis identifies methylation signatures associated with survival and drug resistance of ovarian cancers. Clin. Epigenetics 2021, 13, 142. [Google Scholar] [CrossRef]
- Liu, G.F.; Ruan, G.Y.; Huang, M.M.; Chen, L.L.; Sun, P.M. Genome-wide DNA copy number profiling and bioinformatics analysis of ovarian cancer reveals key genes and pathways associated with distinct invasive/migratory capabilities. Aging 2020, 12, 178–192. [Google Scholar] [CrossRef]
- Calura, E.; Ciciani, M.; Sambugaro, A.; Paracchini, L.; Benvenuto, G.; Milite, S.; Martini, P.; Beltrame, L.; Zane, F.; Fruscio, R.; et al. Transcriptional Characterization of Stage I Epithelial Ovarian Cancer: A Multicentric Study. Cells 2019, 8, 1554. [Google Scholar] [CrossRef] [Green Version]
- Cheaib, B.; Auguste, A.; Leary, A. The PI3K/Akt/mTOR pathway in ovarian cancer: Therapeutic opportunities and challenges. Chin. J. Cancer 2015, 34, 4–16. [Google Scholar] [CrossRef] [Green Version]
- Ducie, J.; Dao, F.; Considine, M.; Olvera, N.; Shaw, P.A.; Kurman, R.J.; Shih, I.M.; Soslow, R.A.; Cope, L.; Levine, D.A. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat. Commun. 2017, 8, 990. [Google Scholar] [CrossRef] [Green Version]
- Vang, R.; Shih, I.M.; Kurman, R.J. Fallopian tube precursors of ovarian low- and high-grade serous neoplasms. Histopathology 2013, 62, 44–58. [Google Scholar] [CrossRef]
- van der Ploeg, P.; Uittenboogaard, A.; Bosch, S.L.; van Diest, P.J.; Wesseling-Rozendaal, Y.J.W.; van de Stolpe, A.; Lambrechts, S.; Bekkers, R.L.M.; Piek, J.M.J. Signal transduction pathway activity in high-grade serous carcinoma, its precursors and Fallopian tube epithelium. Gynecol. Oncol. 2022, 165, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Hou, J.; He, Z.; Liu, T.; Chen, D.; Wang, B.; Wen, Q.; Zheng, X. Evolution of Molecular Targeted Cancer Therapy: Mechanisms of Drug Resistance and Novel Opportunities Identified by CRISPR-Cas9 Screening. Front. Oncol. 2022, 12, 755053. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Li, A.; Yi, M.; Yu, S.; Zhang, M.; Wu, K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J. Hematol. Oncol. 2019, 12, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [Green Version]
- Drozdz, M.M.; Doane, A.S.; Alkallas, R.; Desman, G.; Bareja, R.; Reilly, M.; Bang, J.; Yusupova, M.; You, J.; Eraslan, Z.; et al. A nuclear cAMP microdomain suppresses tumor growth by Hippo pathway inactivation. Cell Rep. 2022, 40, 111412. [Google Scholar] [CrossRef]
- Ramos-Espiritu, L.; Diaz, A.; Nardin, C.; Saviola, A.J.; Shaw, F.; Plitt, T.; Yang, X.; Wolchok, J.; Pirog, E.C.; Desman, G.; et al. The metabolic/pH sensor soluble adenylyl cyclase is a tumor suppressor protein. Oncotarget 2016, 7, 45597–45607. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.; Ladilov, Y. Regulation of mitochondrial homeostasis by sac-derived camp pool: Basic and translational aspects. Cells 2021, 10, 473. [Google Scholar] [CrossRef]
- Creed, S.J.; Le, C.P.; Hassan, M.; Pon, C.K.; Albold, S.; Chan, K.T.; Berginski, M.E.; Huang, Z.; Bear, J.E.; Lane, J.R.; et al. β2-adrenoceptor signaling regulates invadopodia formation to enhance tumor cell invasion. Breast Cancer Res. 2015, 17, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, Y.; He, Z.; Yin, K.; Li, B.; Zhang, L.; Xu, Z. Chronic stress promotes gastric cancer progression and metastasis: An essential role for ADRB. Cell Death Dis. 2019, 10, 788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; Liu, T.; Shasaltaneh, M.D.; Wang, X.; Imani, S.; Wen, Q.L. Targeting Adenylate Cyclase Family: New Concept of Targeted Cancer Therapy. Front. Oncol. 2022, 12, 829212. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T.; Matsuda, Y.; Haniu, H. Cyclic phosphatidic acid stimulates cAMP production and inhibits growth in human colon cancer cells. PLoS ONE 2013, 8, e81139. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.; Rheinstein, P.H. The ADRB1 (Adrenoceptor Beta 1) and ADRB2 Genes Significantly Co-express with Commonly Mutated Genes in Prostate Cancer. Discov. Med. 2020, 30, 163–171. [Google Scholar] [PubMed]
- Wong, W.; Scott, J.D. AKAP signalling complexes: Focal points in space and time. Nat. Rev. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef]
- Howe, A.K.; Baldor, L.C.; Hogan, B.P. Spatial regulation of the cAMP-dependent protein kinase during chemotactic cell migration. Proc. Natl. Acad. Sci. USA 2005, 102, 14320–14325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.J.; Kain, K.H.; Tkachenko, E.; Goldfinger, L.E.; Gutierrez, E.; Allen, M.D.; Groisman, A.; Zhang, J.; Ginsberg, M.H.; Schwarzbauer, J.E. Integrin-mediated Protein Kinase A Activation at the Leading Edge of Migrating Cells. Mol. Biol. Cell 2008, 19, 4930–4941. [Google Scholar] [CrossRef] [Green Version]
- Paulucci-Holthauzen, A.A.; Vergara, L.A.; Bellot, L.J.; Canton, D.; Scott, J.D.; O’Connor, K.L. Spatial distribution of protein kinase A activity during cell migration is mediated by A-kinase anchoring protein AKAP Lbc. J. Biol. Chem. 2009, 284, 5956–5967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, A.J.; Campbell, S.L.; Howe, A.K. Protein kinase a activity and anchoring are required for ovarian cancer cell migration and invasion. PLoS ONE 2011, 6, e26552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDaid, H.M.; Cairns, M.T.; Atkinson, R.J.; Mcaleer, S.; Harkin, D.P.; Gilmore, P.; Johnston, P.G. Increased expression of the RIα subunit of the cAMP-dependent protein kinase A is associated with advanced stage ovarian cancer. Br. J. Cancer 1999, 79, 933–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadish, N.; Parashar, D.; Gupta, N.; Agarwal, S.; Sharma, A.; Fatima, R.; Suri, V.; Kumar, R.; Gupta, A.; Lohiya, N.K.; et al. A novel cancer testis antigen target A-kinase anchor protein (AKAP4) for the early diagnosis and immunotherapy of colon cancer. Oncoimmunology 2016, 5, e1078965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Jagadish, N.; Suri, A. Role of A-Kinase anchor protein (AKAP4) in growth and survival of ovarian cancer cells. Oncotarget 2017, 8, 53124–53136. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, A.J.; Svec, K.V.; Williams, T.F.; Howe, A.K. Protein kinase A activity is regulated by actomyosin contractility during cell migration and is required for durotaxis. Mol. Biol. Cell 2020, 31, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Enserink, J.M.; Kuiperij, H.B.; de Rooij, J.; Price, L.S.; Schwede, F.; Bos, J.L. Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon stimulation of the β2-adrenergic receptor. J. Cell Biol. 2003, 160, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.; Brock, E.J.; Ji, K.; Mattingly, R.R. Seminars in Cancer Biology Ras and Rap1: A tale of two GTPases. Semin Cancer Biol. 2019, 54, 29–39. [Google Scholar] [CrossRef]
- Lyle, K.S.; Raaijmakers, J.H.; Bruinsma, W.; Bos, J.L.; de Rooij, J. cAMP-induced Epac-Rap activation inhibits epithelial cell migration by modulating focal adhesion and leading edge dynamics. Cell Signal. 2008, 20, 1104–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magliozzi, R.; Low, T.Y.; Weijts, B.G.M.W.; Cheng, T.; Spanjaard, E.; Mohammed, S.; van Veen, A.; Ovaa, H.; de Rooij, J.; Zwartkruis, F.J.T.; et al. Control of Epithelial Cell Migration and Invasion by the IKKb-and CK1a -Mediated Degradation of RAPGEF. Dev. Cell 2013, 27, 574–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.; Yeh, Y.; Chuang, C.; Yang, S.Y.; Chang, J.; Sun, S.; Wang, Y.; Chao, K.; Wang, L. Glucocorticoids mediate induction of microRNA-708 to suppress ovarian cancer metastasis through targeting Rap1B. Nat. Commun. 2015, 6, 5917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onodera, Y.; Nam, J.; Bissell, M.J. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J. Clin. Investig. 2014, 124, 367–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Chen, C.L.; Poon, S.L.; Wang, H.S.; Leung, P.C.K. Gonadotropin-stimulated epidermal growth factor receptor expression in human ovarian surface epithelial cells: Involvement of cyclic AMP-dependent exchange protein activated by cAMP pathway. Endocr. Relat. Cancer 2009, 16, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Silva, M.A.; Li, H.; Zhu, L.; Li, P.; Li, X.; Wang, X.; Gao, J.; Wang, P.; Zhang, Z. Long noncoding RNA DQ786243 interacts with miR-506 and promotes progression of ovarian cancer through targeting cAMP responsive element binding protein. J. Cell Biochem. 2018, 119, 9764–9780. [Google Scholar] [CrossRef] [PubMed]
- Linnerth, N.M.; Greenaway, J.B.; Petrik, J.J.; Moorehead, R.A. cAMP response element-binding protein is expressed at high levels in human ovarian adenocarcinoma and regulates ovarian tumor cell proliferation. Int. J. Gynecol. Cancer 2008, 18, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Deng, Y.; Liao, Y.; Li, X.; Liu, J.U.N.; Yao, S. CREB5 promotes tumor cell invasion and correlates with poor prognosis in epithelial ovarian cancer. Oncol. Lett. 2017, 14, 8156–8161. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.B.; Alghamdi, A.A.A.; Islam, S.U.; Lee, J.-S.; Lee, Y.-S. cAMP Signaling in Cancer: A PKA-CREB and EPAC-Centric Approach. Cells 2022, 11, 2020. [Google Scholar] [CrossRef] [PubMed]
- Steven, A.; Seliger, B. Control of CREB expression in tumors: From molecular mechanisms and signal transduction pathways to therapeutic target. Oncotarget 2016, 7, 35454–35465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirotkin, A.V.; Benčo, A.; Mlynček, M.; Harrath, A.H.; Alwasel, S.; Kotwica, J. The involvement of the phosphorylatable and nonphosphorylatable transcription factor CREB-1 in the control of human ovarian cell functions. Comptes Rendus Biol. 2019, 342, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Lu, Y.; Hu, Y. A role of CREB in BRCA1 constitutive promoter activity and aromatase basal expression. Int. J. Biomed. Sci. 2008, 4, 260–265. [Google Scholar]
- Sapio, L.; Salzillo, A.; Ragone, A.; Illiano, M.; Spina, A.; Naviglio, S. Targeting CREB in Cancer Therapy: A Key Candidate or One of Many? An Update. Cancers 2020, 12, 3166. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, S.-T.; Zhang, Z.-J.; Zhou, Q.; Peng, B.-G. Original Article CREB5 promotes cell proliferation and correlates with poor prognosis in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2018, 11, 4908–4916. [Google Scholar]
- Wang, S.; Qiu, J.; Liu, L.; Su, C.; Qi, L.; Huang, C.; Chen, X.; Zhang, Y.; Ye, Y.; Ding, Y.; et al. CREB5 promotes invasiveness and metastasis in colorectal cancer by directly activating MET. J. Exp. Clin. Cancer Res. 2020, 39, 168. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Wang, Y.; Ao, Y.; Sun, X. CREB1 induced lncRNA HAS2-AS1 promotes epithelial ovarian cancer proliferation and invasion via the miR-466/RUNX2 axis. Biomed. Pharmacother. 2019, 115, 108891. [Google Scholar] [CrossRef]
- Kandettu, A.; Adiga, D.; Devi, V.; Suresh, P.S.; Chakrabarty, S.; Radhakrishnan, R.; Kabekkodu, S.P. Deregulated miRNA clusters in ovarian cancer: Imperative implications in personalized medicine. Genes Dis. 2022, 9, 1443–1465. [Google Scholar] [CrossRef]
- Chen, B.; Zhao, Q.; Guan, L.; Lv, H.; Bie, L.; Huang, J.; Chen, X.B. Long non-coding RNA NNT-AS1 sponges miR-424/E2F1 to promote the tumorigenesis and cell cycle progression of gastric cancer. J. Cell Mol. Med. 2018, 22, 4751–4759. [Google Scholar] [CrossRef] [Green Version]
- Braga, E.A.; Fridman, M.V.; Moscovtsev, A.A.; Filippova, E.A.; Dmitriev, A.A.; Kushlinskii, N.E. LncRNAs in ovarian cancer progression, metastasis, and main pathways: ceRNA and alternative mechanisms. Int. J. Mol. Sci. 2020, 21, 8855. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.F.; Zheng, X.; Fan, X.; Lin, A. fu Role of cytoplasmic lncRNAs in regulating cancer signaling pathways. J. Zhejiang Univ. Sci. B Biomed. Biotechnol. 2019, 20, 1–8. [Google Scholar]
- Shan, T.; Fan, J.; Zhao, Q.; Deng, K.; Xia, J. Upregulation of long non-coding RNA DQ786243 promotes the progression of gastric cancer. Mol. Med. Rep. 2017, 16, 3761–3768. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Liu, C.; Wu, M. New insights into long noncoding RNAs and their roles in glioma. Mol. Cancer 2018, 17, 61. [Google Scholar] [CrossRef]
- Skiriute, D.; Stakaitis, R.; Steponaitis, G.; Tamasauskas, A.; Vaitkiene, P. The role of CASC2 and miR-21 interplay in glioma malignancy and patient outcome. Int. J. Mol. Sci. 2020, 21, 7962. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Chen, Y.; Meng, F.; Zhang, Y.; Li, C.; Zhang, G.; Huan, W.; Wu, F. Roflumilast enhances cisplatin-sensitivity and reverses cisplatin-resistance of ovarian cancer cells via cAMP/PKA/CREB-FtMt signalling axis. Cell Prolif. 2018, 51, e12474. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Nam, H.J. PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them. Int. J. Mol. Sci. 2022, 23, 8412. [Google Scholar] [CrossRef] [PubMed]
- Franz, A.; Coscia, F.; Shen, C.; Charaoui, L.; Mann, M.; Sander, C. Molecular response to PARP1 inhibition in ovarian cancer cells as determined by mass spectrometry based proteomics. J. Ovarian Res. 2021, 14, 140. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Coleman, R.L.; González-Martín, A.; Moore, K.N.; Colombo, N.; Ray-Coquard, I.; Pignata, S. The forefront of ovarian cancer therapy: Update on PARP inhibitors. Ann. Oncol. 2020, 31, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Tew, W.P.; Lacchetti, C.; Ellis, A.; Maxian, K.; Banerjee, S.; Bookman, M.; Jones, M.B.; Lee, J.M.; Lheureux, S.; Liu, J.F.; et al. PARP Inhibitors in the Management of Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 3468–3493. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Qi, G.; Han, F.; Lu, W.; Peng, J.; Li, R.; Yan, S.; Yuan, C.; Kong, B. HMGB3 promotes PARP inhibitor resistance through interacting with PARP1 in ovarian cancer. Cell Death Dis. 2022, 13, 263. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: The First Synthetic Lethal Targeted Therapy. Science 2017, 355, 1152–1158. [Google Scholar]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [Green Version]
- Franzese, E.; Centonze, S.; Diana, A.; Carlino, F.; Guerrera, L.P.; di Napoli, M.; de Vita, F.; Pignata, S.; Ciardiello, F.; Orditura, M. PARP inhibitors in ovarian cancer. Cancer Treat. Rev. 2019, 73, 1–9. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.Y.; Wu, N.; Chen, Y.C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef]
- Nishio, K.; Morikage, T.; Kubota, N.; Ohmori, T.; Takeda, Y.; Fujiwara, Y.; Miki, K.; Abe, K.; Saijo, N. Alteration of type II regulatory subunit of cAMP-dependent protein kinase in human cisplatin-resistant cells as a basis of collateral sensitivity to 8-chloro-cAMP. Jpn. J. Cancer Res. 1992, 83, 754–760. [Google Scholar] [CrossRef]
- Pulliam, N.; Fang, F.; Ozes1, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA-mutations. Clin. Cancer Res. 2018, 24, 3163–3175. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kilanowska, A.; Ziółkowska, A.; Stasiak, P.; Gibas-Dorna, M. cAMP-Dependent Signaling and Ovarian Cancer. Cells 2022, 11, 3835. https://doi.org/10.3390/cells11233835
Kilanowska A, Ziółkowska A, Stasiak P, Gibas-Dorna M. cAMP-Dependent Signaling and Ovarian Cancer. Cells. 2022; 11(23):3835. https://doi.org/10.3390/cells11233835
Chicago/Turabian StyleKilanowska, Agnieszka, Agnieszka Ziółkowska, Piotr Stasiak, and Magdalena Gibas-Dorna. 2022. "cAMP-Dependent Signaling and Ovarian Cancer" Cells 11, no. 23: 3835. https://doi.org/10.3390/cells11233835