
The DARC Side of Inflamm-Aging: Duffy Antigen Receptor for Chemokines (DARC/ACKR1) as a Potential Biomarker of Aging, Immunosenescence, and Breast Oncogenesis among High-Risk Subpopulations

 , , , and
, , , and
Abstract
:1. Introduction

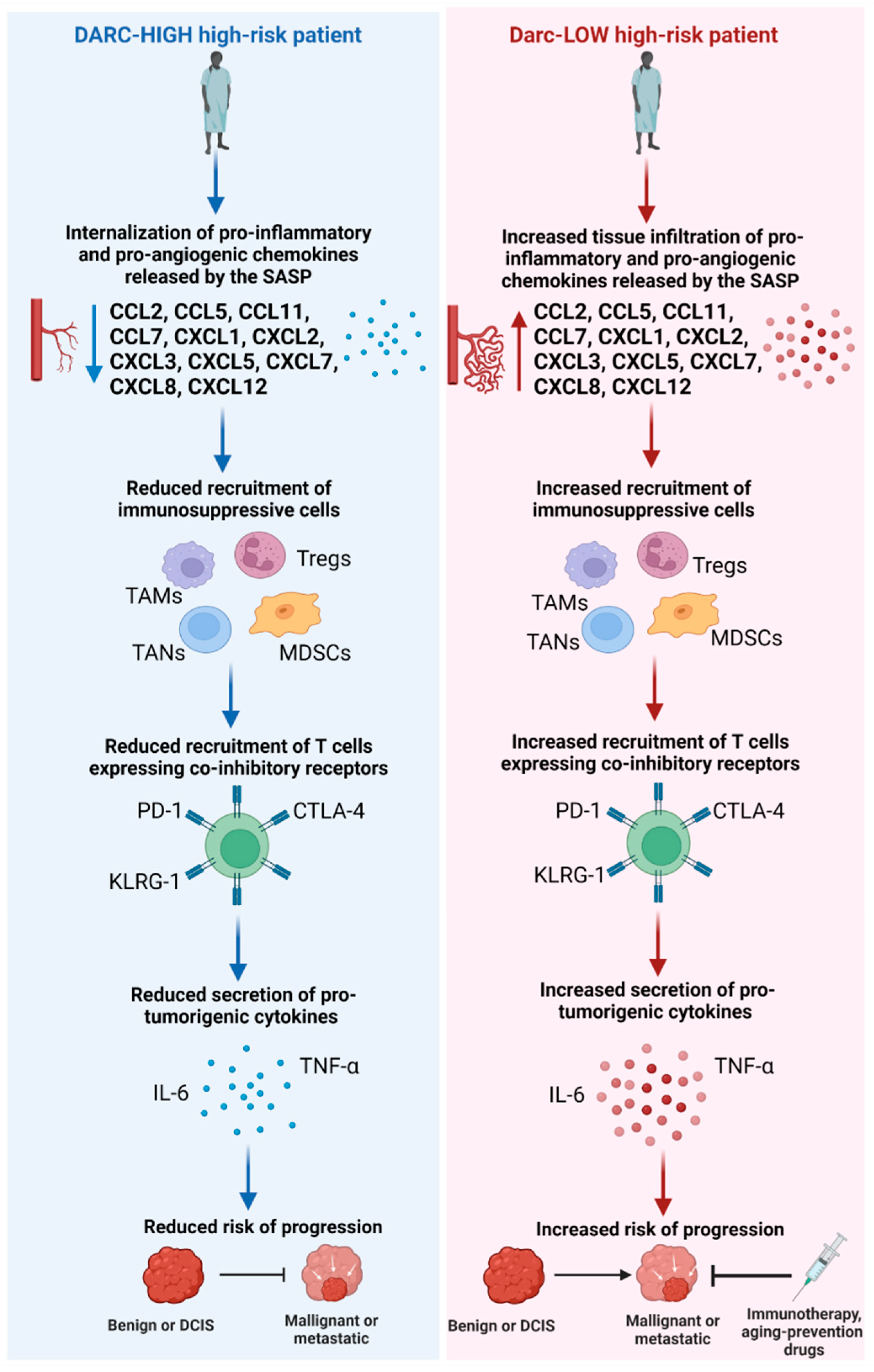{kind=link}
| Biomarker | Mechanism | Ref. |
|---|---|---|
| Immune cell markers | ||
| NK↑CD14+CD56dim/↓CD14+CD56bright | Accumulation of immature NK cells | [28,29] |
| NK↓NKP30, NKP46, DNAM-1 and ↑KIR, NKG2C | Decrease in activating receptors and increase in inhibitory receptors on NK cells | [29,30,31,32] |
| Monocyte↑CD14+(low)CD16+ and CD14++(high)CD16+/↓CD14+(low)CD16− | Increase in immature monocytes | [33] |
| Macrophage↓CD62L and TLR1/4/↑CD11b and TLR5 | Decrease in activating receptors and increase in inhibitory receptors on macrophages | [34] |
| Macrophage M1→M2 | Macrophage phenotype switch to proinflammatory | [35,36] |
| CD8+ naïve T cells | Decline in naïve T cells to reduce thymus T cell output | [37,38] |
| T cell↓CD27, CD28/↑KLRG-1, PD-1, CTLA-4, Tim-3, Tigit, CD57 | Downregulation of costimulatory molecules and upregulation of inhibitory molecules on T cells | [39,40] |
| B cell↓CD19+ | Downregulation in CD19+ B cells to impair B cell function | [41] |
| Cytokines | ||
| ↓IFN-γ, granzyme B, perforin | Reduction in dendritic cell and cytotoxic T cell activity | [42,43,44,45,46] |
| ↑IL-10, TGF-β, VEGF, indoleamine-2,3-dioxygenase | Increase in immunosuppressive cytokines | [8,47] |
| ↑IL-6, IL-1, TNF-α, CRP | Increase in proinflammatory cytokines | [8,48,49] |
| ↓IL-7 | Reduced activation of T cells | [11] |
| miRNAs | ||
| MiR-9, miRNA-17, miR-10a, miR-19a/b, miR-20, miRNA-21, miR-29a, miR-125a/b, miR-126, and miR-146a, miR-155, miR-181a/b, miR-187, miR-195, miR-199, miR-223, miR-517a/c, Let-7, Let-7i | Modulate signaling pathways implicated in inflamm-aging including NF-κB, mTOR, sirtuins, and TGF-β | [50,51] |
| Cell signaling pathways | ||
| NF-κB | A nuclear transcription factor that serves as the primary molecular switch for inflammatory pathways | [52] |
| mTOR | Activates NF-κB -mediated regulation of inflamm-aging | [53] |
| RIG-1 | Interacts with increased IL-6 and IL-8 levels in senescent cells and upregulates IL-6 expression | [54] |
| Notch | Induces senescence of endothelial cells | [55] |
| Sirtuin | Interacts with NF-κB to suppress its proinflammatory activity | [56] |
| TGF-β | Anti-inflammatory cytokine that deactivates macrophages to maintain immune homeostasis | [56] |
| Ras | Upregulates expression of proinflammatory cytokines | [57] |
2. Oncogenesis: Cellular Aging, Senescence, and the Senescence-Associated Secretory Phenotype
3. Inflamm-Aging: Illuminating Immunosenescence
3.1. Innate Immunosenescence
3.2. Adaptive Immunosenescence
3.3. Inflamm-Aging
4. A Light in the DARC: Gaining Back Time
4.1. Chemokines: Small Proteins, Big Roles
4.2. DARC: A Time Decoy
4.3. Shedding Light on Erythrocytic DARC
4.4. No Longer a DARC Secret: Endothelial DARC Mediates Chemokine Transcytosis
4.5. Shot in the DARC: How DARC Stops Circulating Tumor Cells in Their Tracks
5. Painting a DARC Picture: DARC, Immunosenescence, and Neoplastic Transformation
6. Turning Back the Clock on Time: Intervention Strategies for Employing DARC as a Biomarker of Breast Inflamm-Aging, Oncogenesis, and Immunotherapy Response among High-Risk Subpopulations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarmarkovich, M.; Farrel, A.; Sison, A., 3rd; di Marco, M.; Raman, P.; Parris, J.L.; Monos, D.; Lee, H.; Stevanovic, S.; Maris, J.M. Immunogenicity and Immune Silence in Human Cancer. Front. Immunol. 2020, 11, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, A.D.; Sivarapatna, A.; Gress, R.E. The aging immune system and its relationship with cancer. Aging Health 2011, 7, 707–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, A.; Bientinesi, E.; Monti, D. Immunosenescence and inflammaging in the aging process: Age-related diseases or longevity? Ageing Res. Rev. 2021, 71, 101422. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Fulop, T.; Larbi, A.; Witkowski, J.M.; Kotb, R.; Hirokawa, K.; Pawelec, G. Immunosenescence and cancer. Crit. Rev. Oncog. 2013, 18, 489–513. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Yue, Y.; Yu, W.; Zhang, Y. Immunosenescence: A key player in cancer development. J. Hematol. Oncol. 2020, 13, 151. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G. Immunosenescence comes of age. Symposium on Aging Research in Immunology: The Impact of Genomics. EMBO Rep. 2007, 8, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G. Age and immunity: What is “immunosenescence”? Exp. Gerontol. 2018, 105, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.B.; Walens, A.; Hire, R.; Mumin, K.; Brown, A.M.; Ford, D.; Howerth, E.W.; Monteil, M. Distinct Transcript Isoforms of the Atypical Chemokine Receptor 1 (ACKR1)/Duffy Antigen Receptor for Chemokines (DARC) Gene Are Expressed in Lymphoblasts and Altered Isoform Levels Are Associated with Genetic Ancestry and the Duffy-Null Allele. PLoS ONE 2015, 10, e0140098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meny, G.M. The Duffy blood group system: A review. Immunohematology 2010, 26, 51–56. [Google Scholar] [CrossRef]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Tournamille, C.; Colin, Y.; Cartron, J.P.; Le Van Kim, C. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat. Genet. 1995, 10, 224–228. [Google Scholar] [CrossRef]
- Lopez, G.H.; Morrison, J.; Condon, J.A.; Wilson, B.; Martin, J.R.; Liew, Y.W.; Flower, R.L.; Hyland, C.A. Duffy blood group phenotype-genotype correlations using high-resolution melting analysis PCR and microarray reveal complex cases including a new null FY*A allele: The role for sequencing in genotyping algorithms. Vox Sang. 2015, 109, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.H.; Mason, S.J.; Clyde, D.F.; McGinniss, M.H. The resistance factor to Plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N. Engl. J. Med. 1976, 295, 302–304. [Google Scholar] [CrossRef]
- Hodgson, J.A.; Pickrell, J.K.; Pearson, L.N.; Quillen, E.E.; Prista, A.; Rocha, J.; Soodyall, H.; Shriver, M.D.; Perry, G.H. Natural selection for the Duffy-null allele in the recently admixed people of Madagascar. Proc. Biol. Sci. 2014, 281, 20140930. [Google Scholar] [CrossRef] [Green Version]
- Howes, R.E.; Patil, A.P.; Piel, F.B.; Nyangiri, O.A.; Kabaria, C.W.; Gething, P.W.; Zimmerman, P.A.; Barnadas, C.; Beall, C.M.; Gebremedhin, A.; et al. The global distribution of the Duffy blood group. Nat. Commun. 2011, 2, 266. [Google Scholar] [CrossRef]
- Marczyk, M.; Qing, T.; O’Meara, T.; Yagahoobi, V.; Pelekanou, V.; Bai, Y.; Reisenbichler, E.; Cole, K.S.; Li, X.; Gunasekharan, V.; et al. Tumor immune microenvironment of self-identified African American and non-African American triple negative breast cancer. NPJ Breast Cancer 2022, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- O’Meara, T.; Safonov, A.; Casadevall, D.; Qing, T.; Silber, A.; Killelea, B.; Hatzis, C.; Pusztai, L. Immune microenvironment of triple-negative breast cancer in African-American and Caucasian women. Breast Cancer Res. Treat. 2019, 175, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.; Gong, Z.; Yao, S.; Bandera, E.V.; Zirpoli, G.; Hwang, H.; Roberts, M.; Ciupak, G.; Davis, W.; Sucheston, L.; et al. Cytokine and cytokine receptor genes of the adaptive immune response are differentially associated with breast cancer risk in American women of African and European ancestry. Int. J. Cancer 2014, 134, 1408–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdou, Y.; Attwood, K.; Cheng, T.D.; Yao, S.; Bandera, E.V.; Zirpoli, G.R.; Ondracek, R.P.; Stein, L.; Bshara, W.; Khoury, T.; et al. Racial differences in CD8(+) T cell infiltration in breast tumors from Black and White women. Breast Cancer Res. 2020, 22, 62. [Google Scholar] [CrossRef] [PubMed]
- Charan, M.; Verma, A.K.; Hussain, S.; Misri, S.; Mishra, S.; Majumder, S.; Ramaswamy, B.; Ahirwar, D.; Ganju, R.K. Molecular and Cellular Factors Associated with Racial Disparity in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 5936. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, B.D.; Martini, R.N.; Hire, R.; Brown, A.; Bennett, B.; Brown, I.; Howerth, E.W.; Egan, M.; Hodgson, J.; Yates, C.; et al. Atypical Chemokine Receptor 1 (DARC/ACKR1) in Breast Tumors Is Associated with Survival, Circulating Chemokines, Tumor-Infiltrating Immune Cells, and African Ancestry. Cancer Epidemiol. Biomark. Prev. 2019, 28, 690–700. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Cheng, T.D.; Elkhanany, A.; Yan, L.; Omilian, A.; Abrams, S.I.; Evans, S.; Hong, C.C.; Qi, Q.; Davis, W.; et al. Breast Tumor Microenvironment in Black Women: A Distinct Signature of CD8+ T-Cell Exhaustion. J. Natl. Cancer Inst. 2021, 113, 1036–1043. [Google Scholar] [CrossRef]
- Shehata, H.M.; Hoebe, K.; Chougnet, C.A. The aged nonhematopoietic environment impairs natural killer cell maturation and function. Aging Cell 2015, 14, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Manser, A.R.; Uhrberg, M. Age-related changes in natural killer cell repertoires: Impact on NK cell function and immune surveillance. Cancer Immunol. Immunother. 2016, 65, 417–426. [Google Scholar] [CrossRef]
- Tarazona, R.; Sanchez-Correa, B.; Casas-Aviles, I.; Campos, C.; Pera, A.; Morgado, S.; Lopez-Sejas, N.; Hassouneh, F.; Bergua, J.M.; Arcos, M.J.; et al. Immunosenescence: Limitations of natural killer cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2017, 66, 233–245. [Google Scholar] [CrossRef]
- Carlsten, M.; Bjorkstrom, N.K.; Norell, H.; Bryceson, Y.; van Hall, T.; Baumann, B.C.; Hanson, M.; Schedvins, K.; Kiessling, R.; Ljunggren, H.G.; et al. DNAX accessory molecule-1 mediated recognition of freshly isolated ovarian carcinoma by resting natural killer cells. Cancer Res. 2007, 67, 1317–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshmikanth, T.; Burke, S.; Ali, T.H.; Kimpfler, S.; Ursini, F.; Ruggeri, L.; Capanni, M.; Umansky, V.; Paschen, A.; Sucker, A.; et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J. Clin. Investig. 2009, 119, 1251–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyugen, J.; Agrawal, S.; Gollapudi, S.; Gupta, S. Impaired functions of peripheral blood monocyte subpopulations in aged humans. J. Clin. Immunol. 2010, 30, 806–813. [Google Scholar] [CrossRef] [Green Version]
- Hazeldine, J.; Lord, J.M. Innate immunesenescence: Underlying mechanisms and clinical relevance. Biogerontology 2015, 16, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Gayoso, I.; Sanchez-Correa, B.; Campos, C.; Alonso, C.; Pera, A.; Casado, J.G.; Morgado, S.; Tarazona, R.; Solana, R. Immunosenescence of human natural killer cells. J. Innate. Immun. 2011, 3, 337–343. [Google Scholar] [CrossRef]
- van Beek, A.A.; Van den Bossche, J.; Mastroberardino, P.G.; de Winther, M.P.J.; Leenen, P.J.M. Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends Immunol. 2019, 40, 113–127. [Google Scholar] [CrossRef]
- Thomas, R.; Wang, W.; Su, D.M. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun. Ageing 2020, 17, 2. [Google Scholar] [CrossRef] [Green Version]
- Palmer, D.B. The effect of age on thymic function. Front. Immunol. 2013, 4, 316. [Google Scholar] [CrossRef] [Green Version]
- Henson, S.M.; Riddell, N.E.; Akbar, A.N. Properties of end-stage human T cells defined by CD45RA re-expression. Curr. Opin. Immunol. 2012, 24, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Huff, W.X.; Kwon, J.H.; Henriquez, M.; Fetcko, K.; Dey, M. The Evolving Role of CD8(+)CD28(−) Immunosenescent T Cells in Cancer Immunology. Int. J. Mol. Sci. 2019, 20, 2810. [Google Scholar] [CrossRef]
- Kogut, I.; Scholz, J.L.; Cancro, M.P.; Cambier, J.C. B cell maintenance and function in aging. Semin. Immunol. 2012, 24, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Agrawal, S.; Gupta, S. Role of Dendritic Cells in Inflammation and Loss of Tolerance in the Elderly. Front. Immunol. 2017, 8, 896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, A.; Gupta, S. Impact of aging on dendritic cell functions in humans. Ageing Res. Rev. 2011, 10, 336–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, J.K.; Mamotte, C.D.S.; Jackaman, C.; Nelson, D.J. Modulation of dendritic cell and T cell cross-talk during aging: The potential role of checkpoint inhibitory molecules. Ageing Res. Rev. 2017, 38, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Yang, O.O.; Lin, H.; Dagarag, M.; Ng, H.L.; Effros, R.B.; Uittenbogaart, C.H. Decreased perforin and granzyme B expression in senescent HIV-1-specific cytotoxic T lymphocytes. Virology 2005, 332, 16–19. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): Impact on cancer and aging. J. Mol. Med. 2019, 97, 1049–1064. [Google Scholar] [CrossRef] [Green Version]
- Callender, L.A.; Carroll, E.C.; Beal, R.W.J.; Chambers, E.S.; Nourshargh, S.; Akbar, A.N.; Henson, S.M. Human CD8(+) EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell 2018, 17, e12675. [Google Scholar] [CrossRef]
- Pangrazzi, L.; Weinberger, B. T cells, aging and senescence. Exp. Gerontol. 2020, 134, 110887. [Google Scholar] [CrossRef]
- Giuliani, A.; Gaetani, S.; Sorgentoni, G.; Agarbati, S.; Laggetta, M.; Matacchione, G.; Gobbi, M.; Rossi, T.; Galeazzi, R.; Piccinini, G.; et al. Circulating Inflamma-miRs as Potential Biomarkers of Cognitive Impairment in Patients Affected by Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 647015. [Google Scholar] [CrossRef]
- Olivieri, F.; Rippo, M.R.; Monsurro, V.; Salvioli, S.; Capri, M.; Procopio, A.D.; Franceschi, C. MicroRNAs linking inflamm-aging, cellular senescence and cancer. Ageing Res. Rev. 2013, 12, 1056–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Stanfel, M.N.; Shamieh, L.S.; Kaeberlein, M.; Kennedy, B.K. The TOR pathway comes of age. Biochim. Biophys. Acta 2009, 1790, 1067–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Wu, S.; Ren, H.; Gu, J. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat. Cell Biol. 2011, 13, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Charreau, B. Impact of notch signaling on inflammatory responses in cardiovascular disorders. Int. J. Mol. Sci. 2013, 14, 6863–6888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, R.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An Update on Inflamm-Aging: Mechanisms, Prevention, and Treatment. J. Immunol. Res. 2016, 2016, 8426874. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Yoshida, T.; Tateno, K.; Miyauchi, H.; Zou, Y.; Toko, H.; Komuro, I. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation 2003, 108, 2264–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, T. Educational initiatives in geriatric oncology—Who, why, and how? J. Geriatr. Oncol. 2016, 7, 390–396. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Aleskandarany, M.A.; Rakha, E.A.; Ahmed, M.A.; Powe, D.G.; Ellis, I.O.; Green, A.R. Clinicopathologic and molecular significance of phospho-Akt expression in early invasive breast cancer. Breast Cancer Res. Treat. 2011, 127, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Garbe, J.C.; Bhattacharya, S.; Merchant, B.; Bassett, E.; Swisshelm, K.; Feiler, H.S.; Wyrobek, A.J.; Stampfer, M.R. Molecular distinctions between stasis and telomere attrition senescence barriers shown by long-term culture of normal human mammary epithelial cells. Cancer Res. 2009, 69, 7557–7568. [Google Scholar] [CrossRef] [Green Version]
- Garbe, J.C.; Holst, C.R.; Bassett, E.; Tlsty, T.; Stampfer, M.R. Inactivation of p53 function in cultured human mammary epithelial cells turns the telomere-length dependent senescence barrier from agonescence into crisis. Cell Cycle 2007, 6, 1927–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbe, J.C.; Vrba, L.; Sputova, K.; Fuchs, L.; Novak, P.; Brothman, A.R.; Jackson, M.; Chin, K.; LaBarge, M.A.; Watts, G.; et al. Immortalization of normal human mammary epithelial cells in two steps by direct targeting of senescence barriers does not require gross genomic alterations. Cell Cycle 2014, 13, 3423–3435. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Regulski, M.J. Cellular Senescence: What, Why, and How. Wounds 2017, 29, 168–174. [Google Scholar] [PubMed]
- West, M.D.; Pereira-Smith, O.M.; Smith, J.R. Replicative senescence of human skin fibroblasts correlates with a loss of regulation and overexpression of collagenase activity. Exp. Cell Res. 1989, 184, 138–147. [Google Scholar] [CrossRef]
- Millis, A.J.; Hoyle, M.; McCue, H.M.; Martini, H. Differential expression of metalloproteinase and tissue inhibitor of metalloproteinase genes in aged human fibroblasts. Exp. Cell Res. 1992, 201, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Millis, A.J. Differential regulation of collagenase and stromelysin mRNA in late passage cultures of human fibroblasts. Exp. Cell Res. 1996, 222, 150–156. [Google Scholar] [CrossRef]
- Parrinello, S.; Coppe, J.P.; Krtolica, A.; Campisi, J. Stromal-epithelial interactions in aging and cancer: Senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci. 2005, 118, 485–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Hornsby, P.J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007, 67, 3117–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasi, F.; Carmeliet, P. uPAR: A versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Rasoamanantena, P.; Thweatt, R.; Labat-Robert, J.; Goldstein, S. Altered regulation of fibronectin gene expression in Werner syndrome fibroblasts. Exp. Cell Res. 1994, 213, 121–127. [Google Scholar] [CrossRef]
- Kumazaki, T.; Kobayashi, M.; Mitsui, Y. Enhanced expression of fibronectin during in vivo cellular aging of human vascular endothelial cells and skin fibroblasts. Exp. Cell Res. 1993, 205, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Sato, I.; Morita, I.; Kaji, K.; Ikeda, M.; Nagao, M.; Murota, S. Reduction of nitric oxide producing activity associated with in vitro aging in cultured human umbilical vein endothelial cell. Biochem. Biophys. Res. Commun. 1993, 195, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macip, S.; Igarashi, M.; Fang, L.; Chen, A.; Pan, Z.Q.; Lee, S.W.; Aaronson, S.A. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002, 21, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.G.; Zhang, J.; Block, E.R.; Patel, J.M. Senescence-enhanced oxidative stress is associated with deficiency of mitochondrial cytochrome c oxidase in vascular endothelial cells. Mech Ageing Dev. 2003, 124, 911–919. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Finkel, T.; Serrano, M.; Blasco, M.A. The common biology of cancer and ageing. Nature 2007, 448, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, L.P.; Teixeira, V.R.; Alencar-Silva, T.; Simonassi-Paiva, B.; Pereira, R.W.; Pogue, R.; Carvalho, J.L. Hallmarks of aging and immunosenescence: Connecting the dots. Cytokine Growth Factor Rev. 2021, 59, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Locati, M.; Vecchi, A.; Sozzani, S.; Allavena, P. Decoy receptors: A strategy to regulate inflammatory cytokines and chemokines. Trends Immunol. 2001, 22, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Richmond, A. Constitutive IkappaB kinase activity correlates with nuclear factor-kappaB activation in human melanoma cells. Cancer Res. 2001, 61, 4901–4909. [Google Scholar] [PubMed]
- Richmond, A.; Lawson, D.H.; Nixon, D.W.; Stevens, J.S.; Chawla, R.K. Extraction of a melanoma growth-stimulatory activity from culture medium conditioned by the Hs0294 human melanoma cell line. Cancer Res. 1983, 43, 2106–2112. [Google Scholar] [PubMed]
- Richmond, A.; Lawson, D.H.; Nixon, D.W.; Chawla, R.K. Characterization of autostimulatory and transforming growth factors from human melanoma cells. Cancer Res. 1985, 45, 6390–6394. [Google Scholar] [PubMed]
- Fimmel, S.; Devermann, L.; Herrmann, A.; Zouboulis, C. GRO-alpha: A potential marker for cancer and aging silenced by RNA interference. Ann. N. Y. Acad. Sci. 2007, 1119, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintas-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Myeloid-derived suppressor cells (MDSC): An important partner in cellular/tissue senescence. Biogerontology 2018, 19, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Di Mitri, D.; Toso, A.; Chen, J.J.; Sarti, M.; Pinton, S.; Jost, T.R.; D’Antuono, R.; Montani, E.; Garcia-Escudero, R.; Guccini, I.; et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 2014, 515, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Su, D.M.; Aw, D.; Palmer, D.B. Immunosenescence: A product of the environment? Curr. Opin. Immunol. 2013, 25, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Ecker, B.L.; Kaur, A.; Douglass, S.M.; Webster, M.R.; Almeida, F.V.; Marino, G.E.; Sinnamon, A.J.; Neuwirth, M.G.; Alicea, G.M.; Ndoye, A.; et al. Age-Related Changes in HAPLN1 Increase Lymphatic Permeability and Affect Routes of Melanoma Metastasis. Cancer Discov. 2019, 9, 82–95. [Google Scholar] [CrossRef] [Green Version]
- Kaur, A.; Webster, M.R.; Marchbank, K.; Behera, R.; Ndoye, A.; Kugel, C.H., 3rd; Dang, V.M.; Appleton, J.; O’Connell, M.P.; Cheng, P.; et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 2016, 532, 250–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accardi, G.; Caruso, C. Immune-inflammatory responses in the elderly: An update. Immun. Ageing 2018, 15, 11. [Google Scholar] [CrossRef]
- Yang, L.; Li, A.; Lei, Q.; Zhang, Y. Tumor-intrinsic signaling pathways: Key roles in the regulation of the immunosuppressive tumor microenvironment. J. Hematol. Oncol. 2019, 12, 125. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Campos, C.; Pera, A.; Bergua, J.M.; Arcos, M.J.; Banas, H.; Casado, J.G.; Morgado, S.; Duran, E.; Solana, R.; et al. Natural killer cell immunosenescence in acute myeloid leukaemia patients: New targets for immunotherapeutic strategies? Cancer Immunol. Immunother. 2016, 65, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Solana, R.; Campos, C.; Pera, A.; Tarazona, R. Shaping of NK cell subsets by aging. Curr. Opin. Immunol. 2014, 29, 56–61. [Google Scholar] [CrossRef]
- Ginhoux, F.; Schultze, J.L.; Murray, P.J.; Ochando, J.; Biswas, S.K. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat. Immunol. 2016, 17, 34–40. [Google Scholar] [CrossRef]
- Jackaman, C.; Tomay, F.; Duong, L.; Abdol Razak, N.B.; Pixley, F.J.; Metharom, P.; Nelson, D.J. Aging and cancer: The role of macrophages and neutrophils. Ageing Res. Rev. 2017, 36, 105–116. [Google Scholar] [CrossRef]
- Plackett, T.P.; Boehmer, E.D.; Faunce, D.E.; Kovacs, E.J. Aging and innate immune cells. J. Leukoc. Biol. 2004, 76, 291–299. [Google Scholar] [CrossRef]
- Wenisch, C.; Patruta, S.; Daxbock, F.; Krause, R.; Horl, W. Effect of age on human neutrophil function. J. Leukoc. Biol. 2000, 67, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Shaheen, E.; Drake, R.R.; Chen, N.; Gravenstein, S.; Deng, Y. Aging is associated with a numerical and functional decline in plasmacytoid dendritic cells, whereas myeloid dendritic cells are relatively unaltered in human peripheral blood. Hum. Immunol. 2009, 70, 777–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacca, E.R.; Crespo, M.I.; Acland, R.P.; Roselli, E.; Nunez, N.G.; Maccioni, M.; Maletto, B.A.; Pistoresi-Palencia, M.C.; Moron, G. Aging Impairs the Ability of Conventional Dendritic Cells to Cross-Prime CD8+ T Cells upon Stimulation with a TLR7 Ligand. PLoS ONE 2015, 10, e0140672. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Albergante, L.; Blackburn, C.C.; Newman, T.J. Thymic involution and rising disease incidence with age. Proc. Natl. Acad. Sci. USA 2018, 115, 1883–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, J.S.; Boursalian, T.E.; Turk, G.L.; Fink, P.J. Thymic output in aged mice. Proc. Natl. Acad. Sci. USA 2006, 103, 8447–8452. [Google Scholar] [CrossRef] [Green Version]
- Rezzani, R.; Nardo, L.; Favero, G.; Peroni, M.; Rodella, L.F. Thymus and aging: Morphological, radiological, and functional overview. Age 2014, 36, 313–351. [Google Scholar] [CrossRef] [Green Version]
- Chinn, I.K.; Blackburn, C.C.; Manley, N.R.; Sempowski, G.D. Changes in primary lymphoid organs with aging. Semin. Immunol. 2012, 24, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Pulko, V.; Davies, J.S.; Martinez, C.; Lanteri, M.C.; Busch, M.P.; Diamond, M.S.; Knox, K.; Bush, E.C.; Sims, P.A.; Sinari, S.; et al. Human memory T cells with a naive phenotype accumulate with aging and respond to persistent viruses. Nat. Immunol. 2016, 17, 966–975. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Huang, X.; Hsueh, E.C.; Zhang, Q.; Ma, C.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Human regulatory T cells induce T-lymphocyte senescence. Blood 2012, 120, 2021–2031. [Google Scholar] [CrossRef]
- Ye, J.; Ma, C.; Hsueh, E.C.; Eickhoff, C.S.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Tumor-derived gammadelta regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J. Immunol. 2013, 190, 2403–2414. [Google Scholar] [CrossRef]
- Liu, X.; Mo, W.; Ye, J.; Li, L.; Zhang, Y.; Hsueh, E.C.; Hoft, D.F.; Peng, G. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat. Commun. 2018, 9, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Yang, L.; Yue, D.; Cao, L.; Li, L.; Wang, D.; Ping, Y.; Shen, Z.; Zheng, Y.; Wang, L.; et al. Macrophage-derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 2019, 452, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, F.; Yu, M.; Cavanagh, M.M.; Hutter Saunders, J.; Qi, Q.; Ye, Z.; Le Saux, S.; Sultan, W.; Turgano, E.; Dekker, C.L.; et al. Expression of CD39 on Activated T Cells Impairs their Survival in Older Individuals. Cell Rep. 2016, 14, 1218–1231. [Google Scholar] [CrossRef] [Green Version]
- Shimatani, K.; Nakashima, Y.; Hattori, M.; Hamazaki, Y.; Minato, N. PD-1+ memory phenotype CD4+ T cells expressing C/EBPalpha underlie T cell immunodepression in senescence and leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 15807–15812. [Google Scholar] [CrossRef] [Green Version]
- Warren, L.A.; Rossi, D.J. Stem cells and aging in the hematopoietic system. Mech. Ageing Dev. 2009, 130, 46–53. [Google Scholar] [CrossRef]
- Hashimoto, K.; Kouno, T.; Ikawa, T.; Hayatsu, N.; Miyajima, Y.; Yabukami, H.; Terooatea, T.; Sasaki, T.; Suzuki, T.; Valentine, M.; et al. Single-cell transcriptomics reveals expansion of cytotoxic CD4 T cells in supercentenarians. Proc. Natl. Acad. Sci. USA 2019, 116, 24242–24251. [Google Scholar] [CrossRef] [Green Version]
- Pinti, M.; Appay, V.; Campisi, J.; Frasca, D.; Fulop, T.; Sauce, D.; Larbi, A.; Weinberger, B.; Cossarizza, A. Aging of the immune system: Focus on inflammation and vaccination. Eur. J. Immunol. 2016, 46, 2286–2301. [Google Scholar] [CrossRef] [Green Version]
- Bulati, M.; Caruso, C.; Colonna-Romano, G. From lymphopoiesis to plasma cells differentiation, the age-related modifications of B cell compartment are influenced by “inflamm-ageing”. Ageing Res. Rev. 2017, 36, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Gibson, K.L.; Wu, Y.C.; Barnett, Y.; Duggan, O.; Vaughan, R.; Kondeatis, E.; Nilsson, B.O.; Wikby, A.; Kipling, D.; Dunn-Walters, D.K. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell 2009, 8, 18–25. [Google Scholar] [CrossRef]
- Cepeda, S.; Cantu, C.; Orozco, S.; Xiao, Y.; Brown, Z.; Semwal, M.K.; Venables, T.; Anderson, M.S.; Griffith, A.V. Age-Associated Decline in Thymic B Cell Expression of Aire and Aire-Dependent Self-Antigens. Cell Rep. 2018, 22, 1276–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.M.; Owen, K.; Witte, P.L. Aging and developmental transitions in the B cell lineage. Int. Immunol. 2002, 14, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Kline, G.H.; Hayden, T.A.; Klinman, N.R. B cell maintenance in aged mice reflects both increased B cell longevity and decreased B cell generation. J. Immunol. 1999, 162, 3342–3349. [Google Scholar] [PubMed]
- Nguyen, V.; Mendelsohn, A.; Larrick, J.W. Interleukin-7 and Immunosenescence. J. Immunol. Res. 2017, 2017, 4807853. [Google Scholar] [CrossRef] [Green Version]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [Green Version]
- Bonecchi, R.; Savino, B.; Borroni, E.M.; Mantovani, A.; Locati, M. Chemokine decoy receptors: Structure-function and biological properties. Curr. Top. Microbiol. Immunol. 2010, 341, 15–36. [Google Scholar] [CrossRef]
- Mantovani, A.; Bonecchi, R.; Locati, M. Tuning inflammation and immunity by chemokine sequestration: Decoys and more. Nat. Rev. Immunol. 2006, 6, 907–918. [Google Scholar] [CrossRef]
- Chen, K.; Bao, Z.; Tang, P.; Gong, W.; Yoshimura, T.; Wang, J.M. Chemokines in homeostasis and diseases. Cell Mol. Immunol. 2018, 15, 324–334. [Google Scholar] [CrossRef]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Mantovani, A.; Savino, B.; Locati, M.; Zammataro, L.; Allavena, P.; Bonecchi, R. The chemokine system in cancer biology and therapy. Cytokine Growth Factor Rev. 2010, 21, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. CXC chemokines in cancer angiogenesis and metastases. Adv. Cancer Res. 2010, 106, 91–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strieter, R.M.; Polverini, P.J.; Kunkel, S.L.; Arenberg, D.A.; Burdick, M.D.; Kasper, J.; Dzuiba, J.; Van Damme, J.; Walz, A.; Marriott, D.; et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J. Biol. Chem. 1995, 270, 27348–27357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez-Retamozo, V.; Etzrodt, M.; Newton, A.; Rauch, P.J.; Chudnovskiy, A.; Berger, C.; Ryan, R.J.; Iwamoto, Y.; Marinelli, B.; Gorbatov, R.; et al. Origins of tumor-associated macrophages and neutrophils. Proc. Natl. Acad. Sci. USA 2012, 109, 2491–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Sobierajska, K.; Ciszewski, W.M.; Sacewicz-Hofman, I.; Niewiarowska, J. Endothelial Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1234, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, J.; Zheng, P.; Li, H.; Zhao, S. The Origins and Generation of Cancer-Associated Mesenchymal Stromal Cells: An Innovative Therapeutic Target for Solid Tumors. Front. Oncol. 2021, 11, 723707. [Google Scholar] [CrossRef] [PubMed]
- Vetsika, E.K.; Koukos, A.; Kotsakis, A. Myeloid-Derived Suppressor Cells: Major Figures that Shape the Immunosuppressive and Angiogenic Network in Cancer. Cells 2019, 8, 1647. [Google Scholar] [CrossRef]
- Ozga, A.J.; Chow, M.T.; Luster, A.D. Chemokines and the immune response to cancer. Immunity 2021, 54, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Yoshie, O.; Nakayama, T. Multifaceted Roles of Chemokines and Chemokine Receptors in Tumor Immunity. Cancers 2021, 13, 6132. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E.; Mezzapelle, R. The Chemokine Receptor CXCR4 in Cell Proliferation and Tissue Regeneration. Front. Immunol. 2020, 11, 2109. [Google Scholar] [CrossRef]
- Liang, K.; Liu, Y.; Eer, D.; Liu, J.; Yang, F.; Hu, K. High CXC Chemokine Ligand 16 (CXCL16) Expression Promotes Proliferation and Metastasis of Lung Cancer via Regulating the NF-kappaB Pathway. Med. Sci. Monit. 2018, 24, 405–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, W.B.; Jokar, I.; Zou, A.; Lambert, D.; Dendukuri, P.; Cheng, N. CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J. Biol. Chem. 2012, 287, 36593–36608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.K.; Park, M.H.; Choi, D.Y.; Yoo, H.S.; Han, S.B.; Yoon, D.Y.; Hong, J.T. Deficiency of C-C chemokine receptor 5 suppresses tumor development via inactivation of NF-kappaB and upregulation of IL-1Ra in melanoma model. PLoS ONE 2012, 7, e33747. [Google Scholar] [CrossRef]
- Bonecchi, R.; Garlanda, C.; Mantovani, A.; Riva, F. Cytokine decoy and scavenger receptors as key regulators of immunity and inflammation. Cytokine 2016, 87, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ou, Z.L.; Hou, Y.F.; Luo, J.M.; Shen, Z.Z.; Ding, J.; Shao, Z.M. Enhanced expression of Duffy antigen receptor for chemokines by breast cancer cells attenuates growth and metastasis potential. Oncogene 2006, 25, 7201–7211. [Google Scholar] [CrossRef] [Green Version]
- Peiper, S.C.; Wang, Z.X.; Neote, K.; Martin, A.W.; Showell, H.J.; Conklyn, M.J.; Ogborne, K.; Hadley, T.J.; Lu, Z.H.; Hesselgesser, J.; et al. The Duffy antigen/receptor for chemokines (DARC) is expressed in endothelial cells of Duffy negative individuals who lack the erythrocyte receptor. J. Exp. Med. 1995, 181, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.; Zbrzezna, V.; Polyakova, J.; Pogo, A.O.; Hesselgesser, J.; Horuk, R. Expression of the Duffy antigen in K562 cells. Evidence that it is the human erythrocyte chemokine receptor. J. Biol. Chem. 1994, 269, 7835–7838. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.H.; Mason, S.J.; Dvorak, J.A.; McGinniss, M.H.; Rothman, I.K. Erythrocyte receptors for (Plasmodium knowlesi) malaria: Duffy blood group determinants. Science 1975, 189, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Comerford, I.; Nibbs, R.J. Post-translational control of chemokines: A role for decoy receptors? Immunol. Lett. 2005, 96, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Gutjahr, J.C.; Crawford, K.S.; Jensen, D.R.; Naik, P.; Peterson, F.C.; Samson, G.P.B.; Legler, D.F.; Duchene, J.; Veldkamp, C.T.; Rot, A.; et al. The dimeric form of CXCL12 binds to atypical chemokine receptor 1. Sci. Signal. 2021, 14, eabc9012. [Google Scholar] [CrossRef] [PubMed]
- Pruenster, M.; Mudde, L.; Bombosi, P.; Dimitrova, S.; Zsak, M.; Middleton, J.; Richmond, A.; Graham, G.J.; Segerer, S.; Nibbs, R.J.; et al. The Duffy antigen receptor for chemokines transports chemokines and supports their promigratory activity. Nat. Immunol. 2009, 10, 101–108. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA Cancer J. Clin. 2014, 64, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Ganz, P.A.; Goodwin, P.J. Breast Cancer Survivorship: Where Are We Today? Adv. Exp. Med. Biol. 2015, 862, 1–8. [Google Scholar] [CrossRef]
- Lee, J.S.; Wurfel, M.M.; Matute-Bello, G.; Frevert, C.W.; Rosengart, M.R.; Ranganathan, M.; Wong, V.W.; Holden, T.; Sutlief, S.; Richmond, A.; et al. The Duffy antigen modifies systemic and local tissue chemokine responses following lipopolysaccharide stimulation. J. Immunol. 2006, 177, 8086–8094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuma, N.; Akimitsu, N.; Hamamoto, H.; Kusuhara, H.; Sugiyama, Y.; Sekimizu, K. A role of the Duffy antigen for the maintenance of plasma chemokine concentrations. Biochem. Biophys. Res. Commun. 2003, 303, 137–139. [Google Scholar] [CrossRef]
- Pruenster, M.; Rot, A. Throwing light on DARC. Biochem. Soc. Trans. 2006, 34, 1005–1008. [Google Scholar] [CrossRef]
- Middleton, J.; Neil, S.; Wintle, J.; Clark-Lewis, I.; Moore, H.; Lam, C.; Auer, M.; Hub, E.; Rot, A. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell 1997, 91, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Middleton, J.; Patterson, A.M.; Gardner, L.; Schmutz, C.; Ashton, B.A. Leukocyte extravasation: Chemokine transport and presentation by the endothelium. Blood 2002, 100, 3853–3860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rot, A.; Hub, E.; Middleton, J.; Pons, F.; Rabeck, C.; Thierer, K.; Wintle, J.; Wolff, B.; Zsak, M.; Dukor, P. Some aspects of IL-8 pathophysiology. III: Chemokine interaction with endothelial cells. J. Leukoc. Biol. 1996, 59, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Atherton, A.; Born, G.V. Quantitative investigations of the adhesiveness of circulating polymorphonuclear leucocytes to blood vessel walls. J. Physiol. 1972, 222, 447–474. [Google Scholar] [CrossRef] [Green Version]
- von Andrian, U.H.; Chambers, J.D.; McEvoy, L.M.; Bargatze, R.F.; Arfors, K.E.; Butcher, E.C. Two-step model of leukocyte-endothelial cell interaction in inflammation: Distinct roles for LECAM-1 and the leukocyte beta 2 integrins in vivo. Proc. Natl. Acad. Sci. USA 1991, 88, 7538–7542. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Ashkenazi, A.; Chaudhuri, A. Duffy antigen/receptor for chemokines (DARC) attenuates angiogenesis by causing senescence in endothelial cells. Angiogenesis 2007, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.T.; Lamb, P.W.; Rinker-Schaeffer, C.W.; Vukanovic, J.; Ichikawa, T.; Isaacs, J.T.; Barrett, J.C. KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science 1995, 268, 884–886. [Google Scholar] [CrossRef] [Green Version]
- Furuta, E.; Bandyopadhyay, S.; Iiizumi, M.; Mohinta, S.; Zhan, R.; Watabe, K. The role of tumor metastasis suppressors in cancers of breast and prostate. Front. Biosci. 2006, 11, 2845–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranti, C.K. Controlling cell surface dynamics and signaling: How CD82/KAI1 suppresses metastasis. Cell Signal. 2009, 21, 196–211. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Zhan, R.; Chaudhuri, A.; Watabe, M.; Pai, S.K.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; Takano, Y.; et al. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nat. Med. 2006, 12, 933–938. [Google Scholar] [CrossRef]
- Wang, J.; Ou, Z.L.; Hou, Y.F.; Luo, J.M.; Chen, Y.; Zhou, J.; Shen, Z.Z.; Ding, J.; Shao, Z.M. [Duffy antigen receptor for chemokines attenuates breast cancer growth and metastasis: An experiment with nude mice]. Zhonghua Yi Xue Za Zhi 2005, 85, 2033–2037. [Google Scholar]
- Bule, P.; Aguiar, S.I.; Aires-Da-Silva, F.; Dias, J.N.R. Chemokine-Directed Tumor Microenvironment Modulation in Cancer Immunotherapy. Int. J. Mol. Sci. 2021, 22, 9804. [Google Scholar] [CrossRef]
- Chow, M.T.; Luster, A.D. Chemokines in cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.R. The chemokine system and cancer. J. Pathol. 2012, 226, 148–157. [Google Scholar] [CrossRef]
- Caronni, N.; Savino, B.; Bonecchi, R. Myeloid cells in cancer-related inflammation. Immunobiology 2015, 220, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Massara, M.; Bonavita, O.; Mantovani, A.; Locati, M.; Bonecchi, R. Atypical chemokine receptors in cancer: Friends or foes? J. Leukoc. Biol. 2016, 99, 927–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit. Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Kryczek, I.; Zou, L.; Rodriguez, P.; Zhu, G.; Wei, S.; Mottram, P.; Brumlik, M.; Cheng, P.; Curiel, T.; Myers, L.; et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J. Exp. Med. 2006, 203, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Kryczek, I.; Chen, L.; Zou, W.; Welling, T.H. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 2009, 69, 8067–8075. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Luboshits, G.; Shina, S.; Kaplan, O.; Engelberg, S.; Nass, D.; Lifshitz-Mercer, B.; Chaitchik, S.; Keydar, I.; Ben-Baruch, A. Elevated expression of the CC chemokine regulated on activation, normal T cell expressed and secreted (RANTES) in advanced breast carcinoma. Cancer Res. 1999, 59, 4681–4687. [Google Scholar] [PubMed]
- Azenshtein, E.; Luboshits, G.; Shina, S.; Neumark, E.; Shahbazian, D.; Weil, M.; Wigler, N.; Keydar, I.; Ben-Baruch, A. The CC chemokine RANTES in breast carcinoma progression: Regulation of expression and potential mechanisms of promalignant activity. Cancer Res. 2002, 62, 1093–1102. [Google Scholar] [PubMed]
- Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, S.; Stoppacciaro, A.; Ballerini, F.; Marchesi, M.; Prat, M.; Stella, M.C.; Sozzani, S.; Allavena, P.; Mantovani, A.; Ruco, L.P. Papillary carcinoma of the thyroid: Hepatocyte growth factor (HGF) stimulates tumor cells to release chemokines active in recruiting dendritic cells. Am. J. Pathol. 2000, 156, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Gobert, M.; Treilleux, I.; Bendriss-Vermare, N.; Bachelot, T.; Goddard-Leon, S.; Arfi, V.; Biota, C.; Doffin, A.C.; Durand, I.; Olive, D.; et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009, 69, 2000–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Chen, Y.S.; Yao, Y.D.; Chen, J.Q.; Chen, J.N.; Huang, S.Y.; Zeng, Y.J.; Yao, H.R.; Zeng, S.H.; Fu, Y.S.; et al. CCL18 from tumor-associated macrophages promotes angiogenesis in breast cancer. Oncotarget 2015, 6, 34758–34773. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Sadanandam, A.; Singh, R.K. Chemokines in tumor angiogenesis and metastasis. Cancer Metastasis Rev. 2007, 26, 453–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sozzani, S.; Del Prete, A.; Bonecchi, R.; Locati, M. Chemokines as effector and target molecules in vascular biology. Cardiovasc. Res. 2015, 107, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Szabo, M.C.; Soo, K.S.; Zlotnik, A.; Schall, T.J. Chemokine class differences in binding to the Duffy antigen-erythrocyte chemokine receptor. J. Biol. Chem. 1995, 270, 25348–25351. [Google Scholar] [CrossRef] [PubMed]
- Darash-Yahana, M.; Pikarsky, E.; Abramovitch, R.; Zeira, E.; Pal, B.; Karplus, R.; Beider, K.; Avniel, S.; Kasem, S.; Galun, E.; et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004, 18, 1240–1242. [Google Scholar] [CrossRef]
- Rundle, C.H.; Mohan, S.; Edderkaoui, B. Duffy antigen receptor for chemokines regulates post-fracture inflammation. PLoS ONE 2013, 8, e77362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, Z.L.; Wang, J.; Hou, Y.F.; Luo, J.M.; Shen, Z.Z.; Shao, Z.M. Downregulation of Duffy antigen receptor for chemokine (DARC) is associated with lymph node metastasis in human breast cancer. Zhonghua Zhong Liu Za Zhi 2006, 28, 586–589. [Google Scholar] [PubMed]
- Shen, H.; Schuster, R.; Stringer, K.F.; Waltz, S.E.; Lentsch, A.B. The Duffy antigen/receptor for chemokines (DARC) regulates prostate tumor growth. FASEB J. 2006, 20, 59–64. [Google Scholar] [CrossRef]
- Horton, L.W.; Yu, Y.; Zaja-Milatovic, S.; Strieter, R.M.; Richmond, A. Opposing roles of murine duffy antigen receptor for chemokine and murine CXC chemokine receptor-2 receptors in murine melanoma tumor growth. Cancer Res. 2007, 67, 9791–9799. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Liu, M.; Hu, Y.; An, W.; Liang, X.; Yu, W.; Piao, F. Expression of Duffy antigen receptor for chemokines (DARC) is down-regulated in colorectal cancer. J. Recept. Signal. Transduct. Res. 2015, 35, 462–467. [Google Scholar] [CrossRef]
- Kaminska, M.; Ciszewski, T.; Lopacka-Szatan, K.; Miotla, P.; Staroslawska, E. Breast cancer risk factors. Prz. Menopauzalny 2015, 14, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.; Tripathi, S.; Hughley, R.; He, Q.; Bae, S.; Karanam, B.; Martini, R.; Newman, L.; Colomb, W.; Grizzle, W.; et al. AR negative triple negative or “quadruple negative” breast cancers in African American women have an enriched basal and immune signature. PLoS ONE 2018, 13, e0196909. [Google Scholar] [CrossRef] [Green Version]
- Martini, R.; Delpe, P.; Chu, T.R.; Arora, K.; Lord, B.; Verma, A.; Bedi, D.; Karanam, B.; Elhussin, I.; Chen, Y.; et al. African Ancestry Associated Gene Expression Profiles in Triple Negative Breast Cancer Underlie Altered Tumor Biology and Clinical Outcome in Women of African Descent. Cancer Discov. 2022, 12, 2530–2551. [Google Scholar] [CrossRef]
- Myers, D.J.; Walls, A.L. Atypical Breast Hyperplasia; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- van Seijen, M.; Lips, E.H.; Thompson, A.M.; Nik-Zainal, S.; Futreal, A.; Hwang, E.S.; Verschuur, E.; Lane, J.; Jonkers, J.; Rea, D.W.; et al. Ductal carcinoma in situ: To treat or not to treat, that is the question. Br. J. Cancer 2019, 121, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Partridge, L.; Fuentealba, M.; Kennedy, B.K. The quest to slow ageing through drug discovery. Nat. Rev. Drug Discov. 2020, 19, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisimov, V.N. Metformin: Do we finally have an anti-aging drug? Cell Cycle 2013, 12, 3483–3489. [Google Scholar] [CrossRef] [Green Version]
- Popovich, I.G.; Anisimov, V.N.; Zabezhinski, M.A.; Semenchenko, A.V.; Tyndyk, M.L.; Yurova, M.N.; Blagosklonny, M.V. Lifespan extension and cancer prevention in HER-2/neu transgenic mice treated with low intermittent doses of rapamycin. Cancer Biol. Ther. 2014, 15, 586–592. [Google Scholar] [CrossRef] [Green Version]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, J.Y.; Lee, K.Y.; Kim, J.R.; Choi, H.C. Interaction between mTOR pathway inhibition and autophagy induction attenuates adriamycin-induced vascular smooth muscle cell senescence through decreased expressions of p53/p21/p16. Exp. Gerontol. 2018, 109, 51–58. [Google Scholar] [CrossRef]
- Wang, R.; Sunchu, B.; Perez, V.I. Rapamycin and the inhibition of the secretory phenotype. Exp. Gerontol. 2017, 94, 89–92. [Google Scholar] [CrossRef]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef]
- Perica, K.; Varela, J.C.; Oelke, M.; Schneck, J. Adoptive T cell immunotherapy for cancer. Rambam. Maimonides Med. J. 2015, 6, e0004. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Franzin, R.; Stasi, A.; Castellano, G.; Gesualdo, L. Methods for Characterization of Senescent Circulating and Tumor-Infiltrating T-Cells: An Overview from Multicolor Flow Cytometry to Single-Cell RNA Sequencing. Methods Mol. Biol. 2021, 2325, 79–95. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jinna, N.; Rida, P.; Su, T.; Gong, Z.; Yao, S.; LaBarge, M.; Natarajan, R.; Jovanovic-Talisman, T.; Ambrosone, C.; Seewaldt, V. The DARC Side of Inflamm-Aging: Duffy Antigen Receptor for Chemokines (DARC/ACKR1) as a Potential Biomarker of Aging, Immunosenescence, and Breast Oncogenesis among High-Risk Subpopulations. Cells 2022, 11, 3818. https://doi.org/10.3390/cells11233818
Jinna N, Rida P, Su T, Gong Z, Yao S, LaBarge M, Natarajan R, Jovanovic-Talisman T, Ambrosone C, Seewaldt V. The DARC Side of Inflamm-Aging: Duffy Antigen Receptor for Chemokines (DARC/ACKR1) as a Potential Biomarker of Aging, Immunosenescence, and Breast Oncogenesis among High-Risk Subpopulations. Cells. 2022; 11(23):3818. https://doi.org/10.3390/cells11233818
Chicago/Turabian StyleJinna, Nikita, Padmashree Rida, Tianyi Su, Zhihong Gong, Song Yao, Mark LaBarge, Rama Natarajan, Tijana Jovanovic-Talisman, Christine Ambrosone, and Victoria Seewaldt. 2022. "The DARC Side of Inflamm-Aging: Duffy Antigen Receptor for Chemokines (DARC/ACKR1) as a Potential Biomarker of Aging, Immunosenescence, and Breast Oncogenesis among High-Risk Subpopulations" Cells 11, no. 23: 3818. https://doi.org/10.3390/cells11233818