
The Revelation of Continuously Organized, Co-Overexpressed Protein-Coding Genes with Roles in Cellular Communications in Breast Cancer
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression Analysis
2.2. CanCord34 Oncoprint
2.3. Statistical Evaluation of Expression
2.4. Correlation and Network Analyses
2.5. Prognostic Significance
2.6. Evidence of CanCord34 Genes as Secreted Molecules with a Role in Stemness
2.7. Functional Enrichment Analyses
2.8. Genomic Analysis and Regulatory Mechanism of CanCord34 Genes
2.9. Phenotypic Characterization Studies
2.10. Protein–Protein Interaction Analysis
2.11. Cell Culture and RNA Isolation
2.12. Semi-Quantitative and Realtime PCR
2.13. Analyses of Microvesicles from MSCs Exposed to Breast Cancer Cells
3. Results
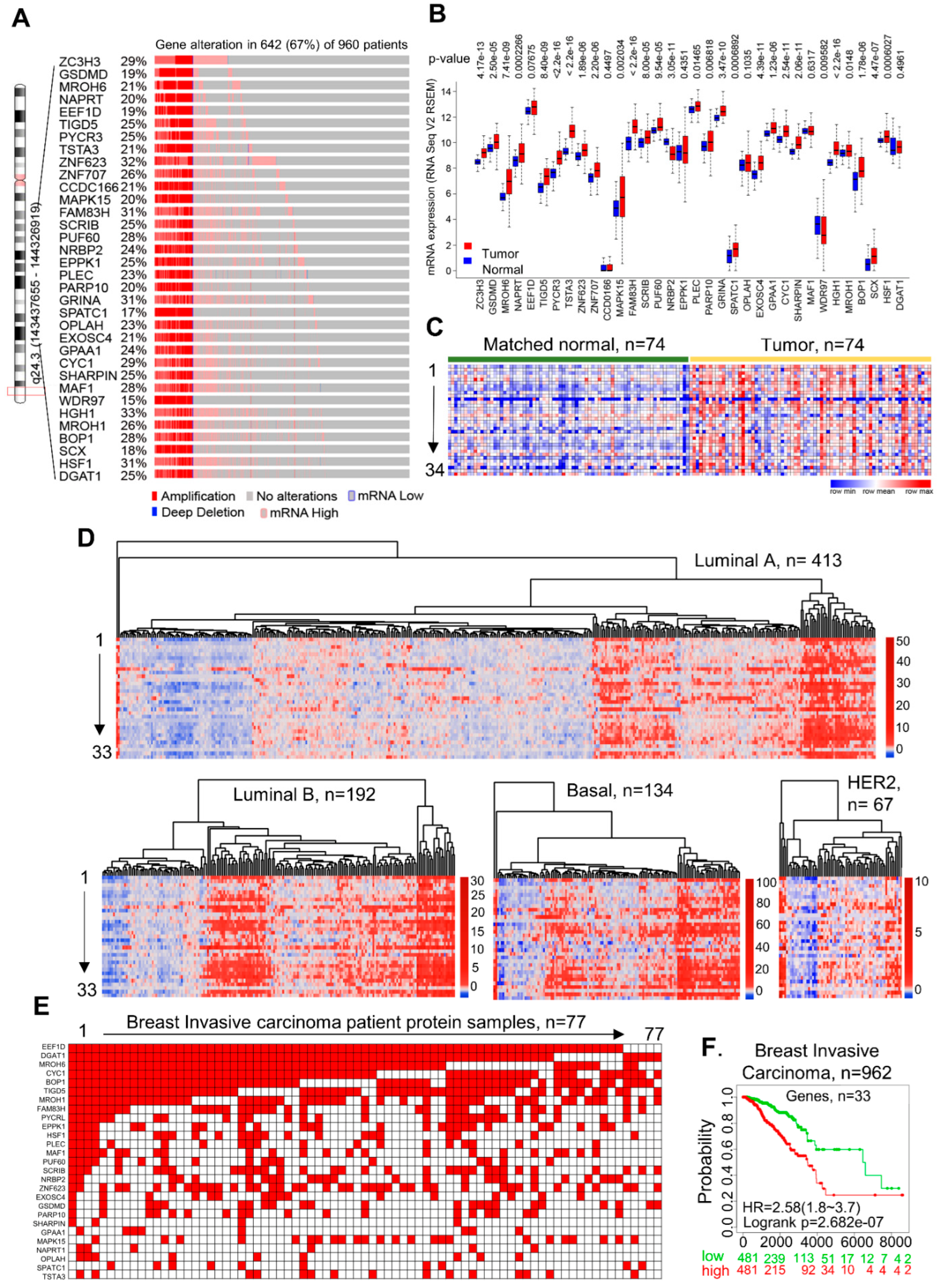
3.1. Discovery of CanCord34 Genes in Breast Cancer
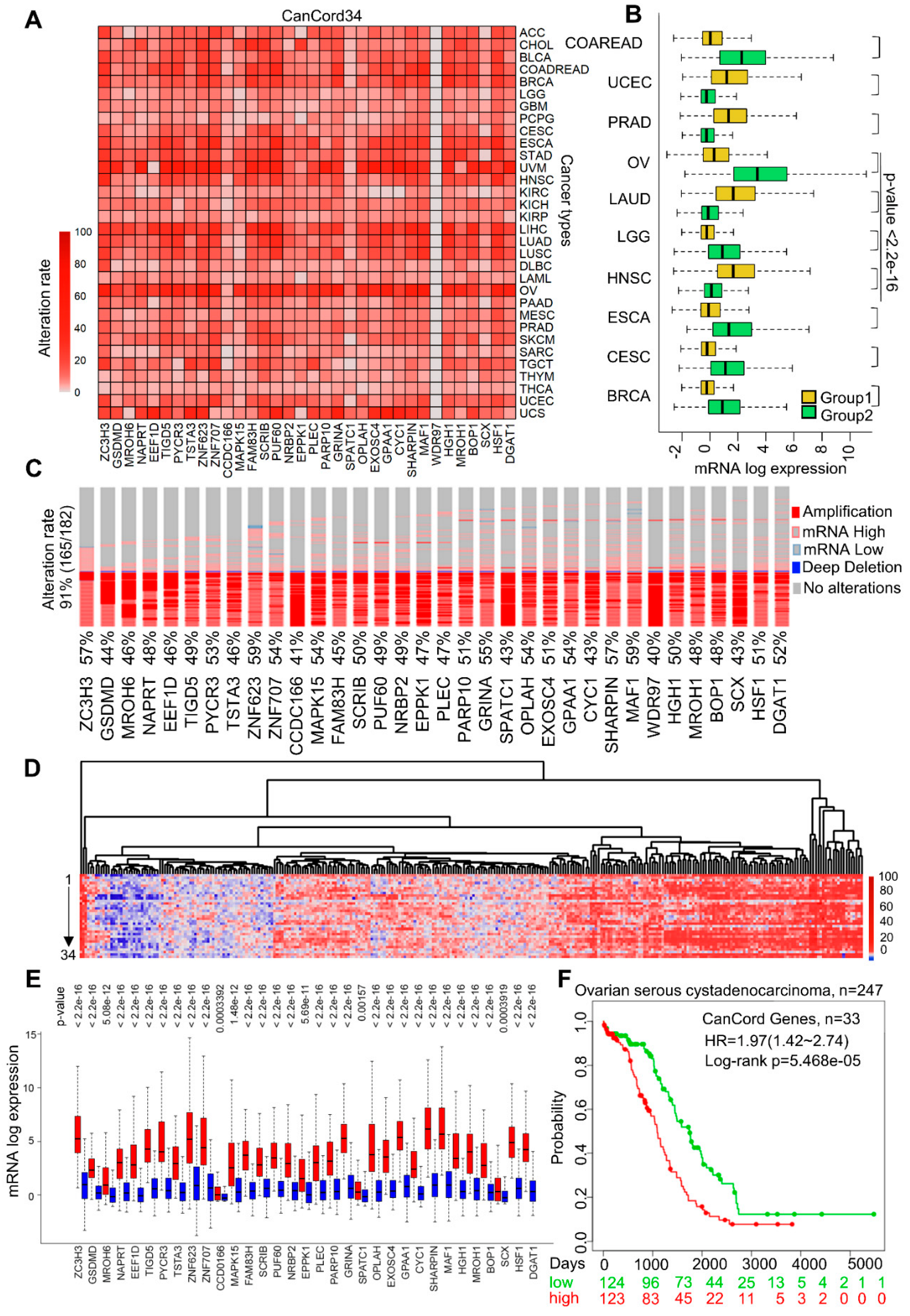
3.2. The Co-Overexpression of CanCord34 Family Members Is a Common Phenotype in Human Cancer
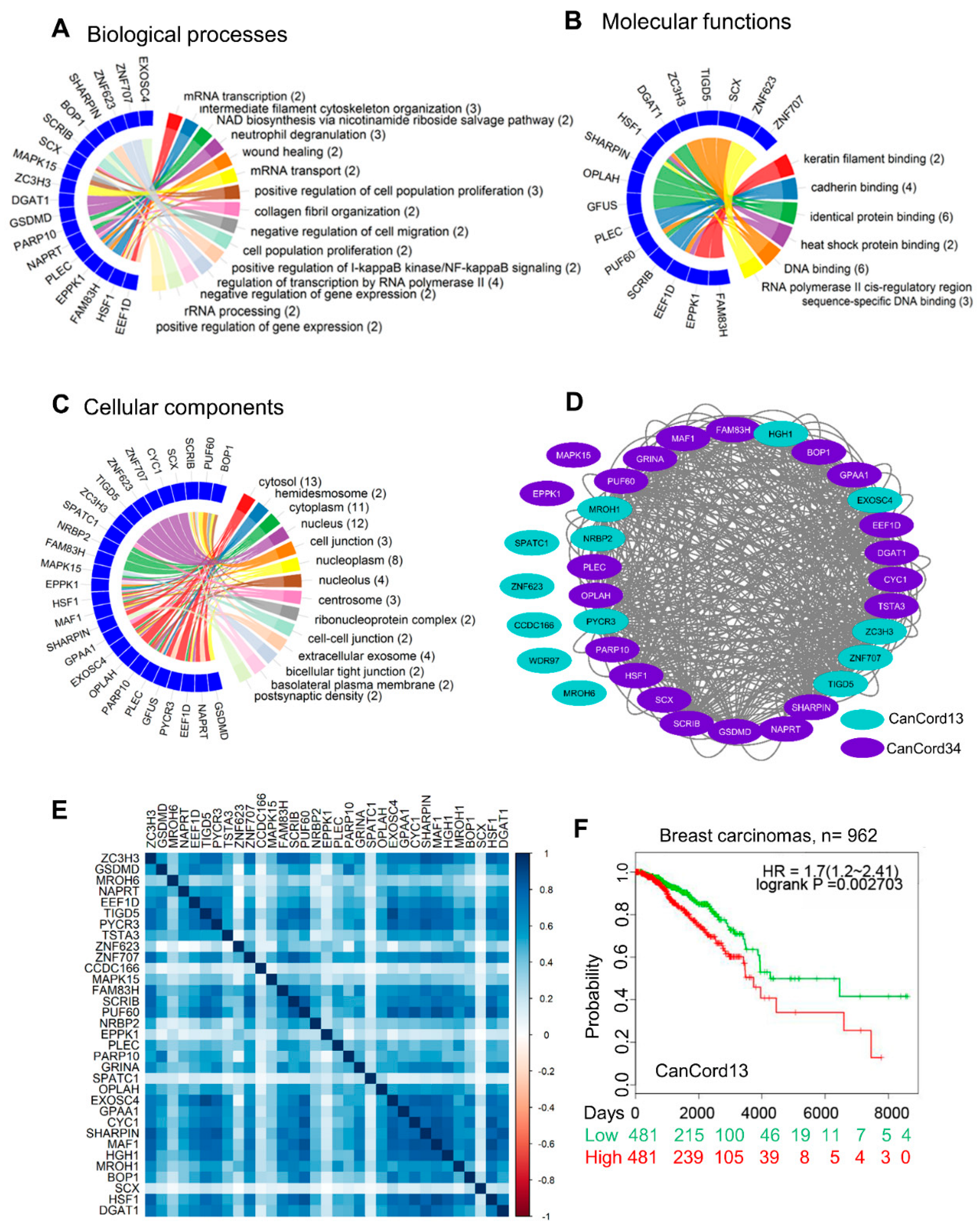
3.3. Towards an Understanding of the Functions of CanCord34 Genes
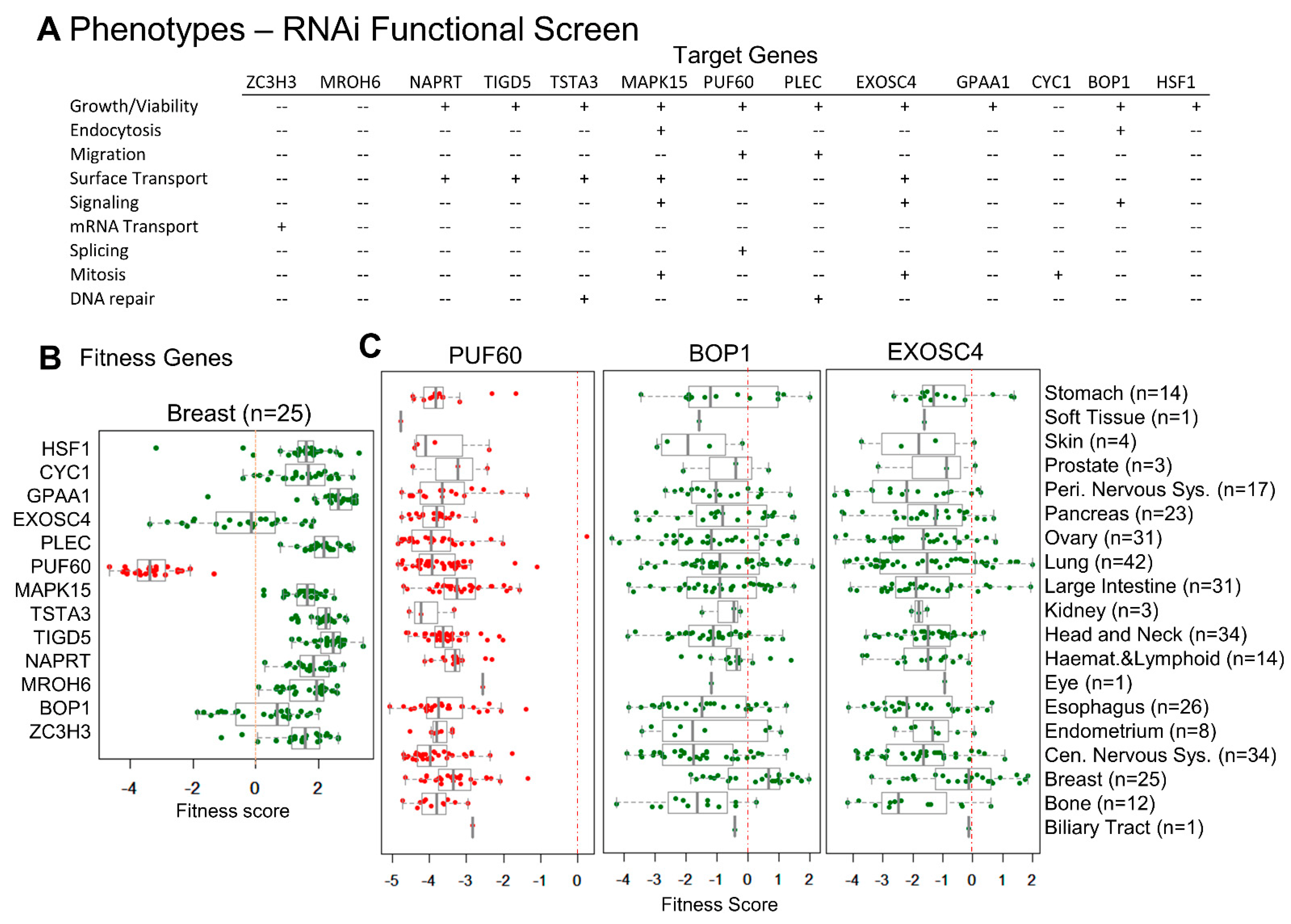
3.4. CanCord34 Genes’ Regulation of Cell Viability
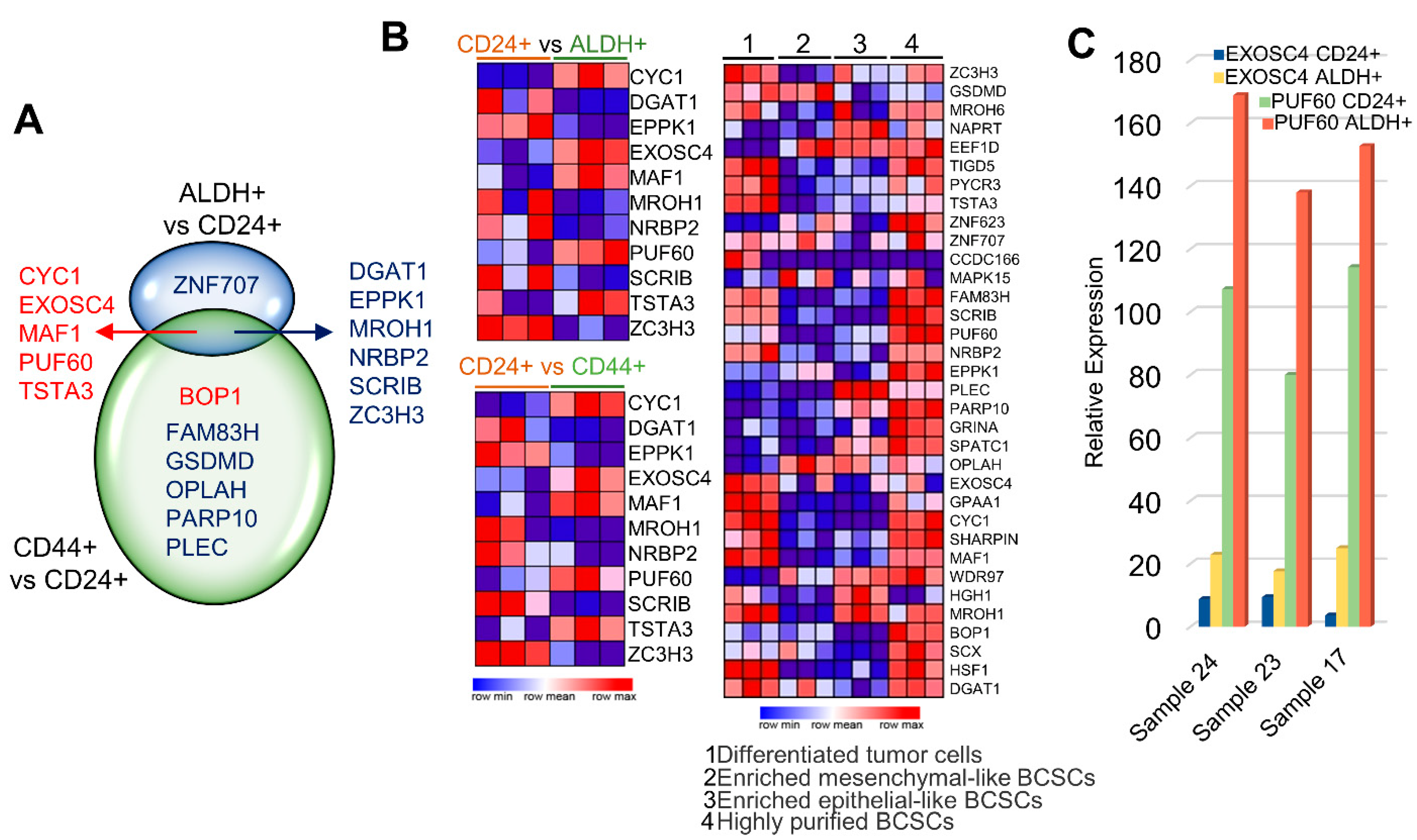
3.5. Upregulation of CanCord34 Family Members in Cancer Stem Cells
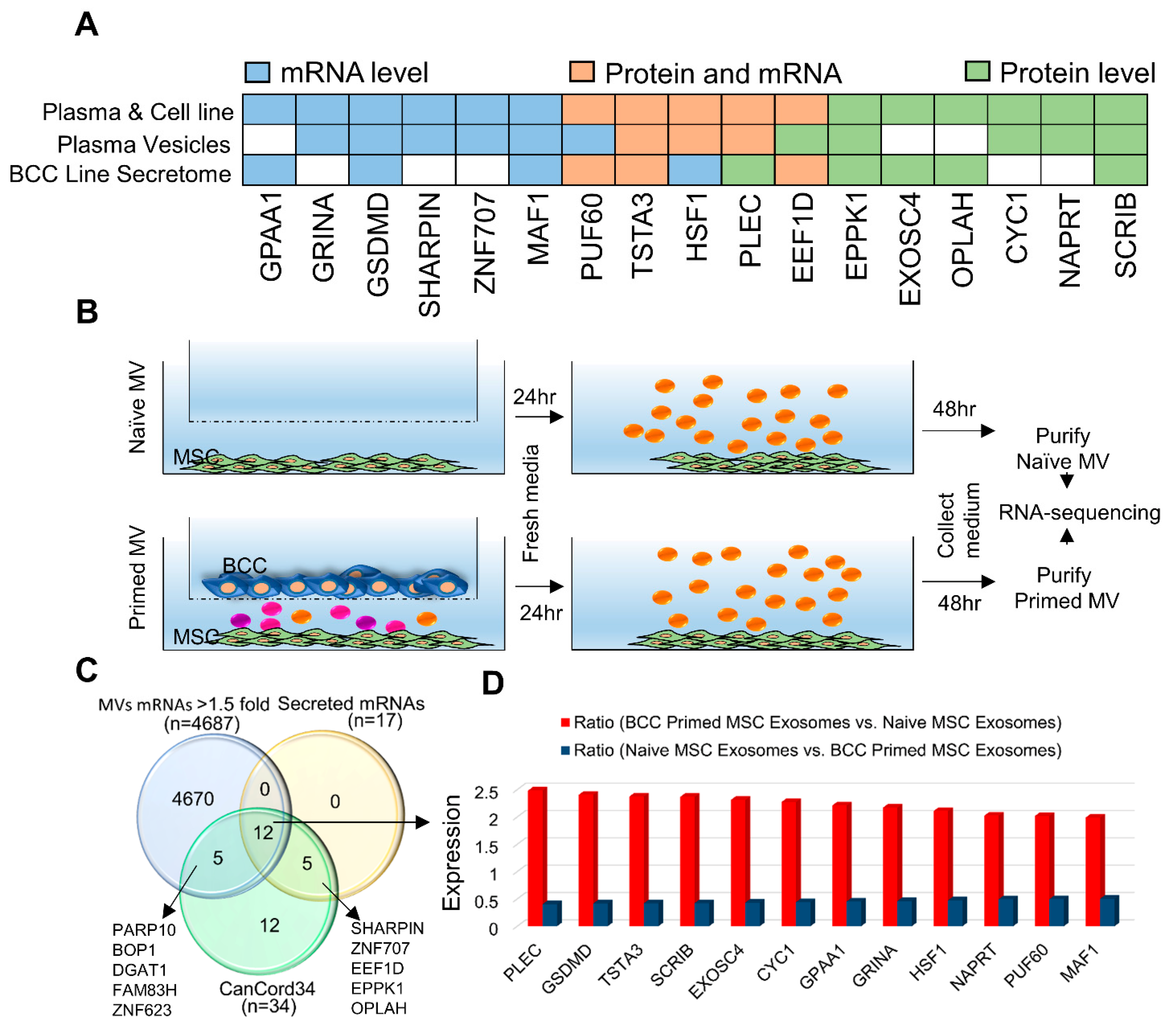
3.6. CanCord34 Family Members as Components of Extracellular Microvesicles and Vesicles/Exosomes
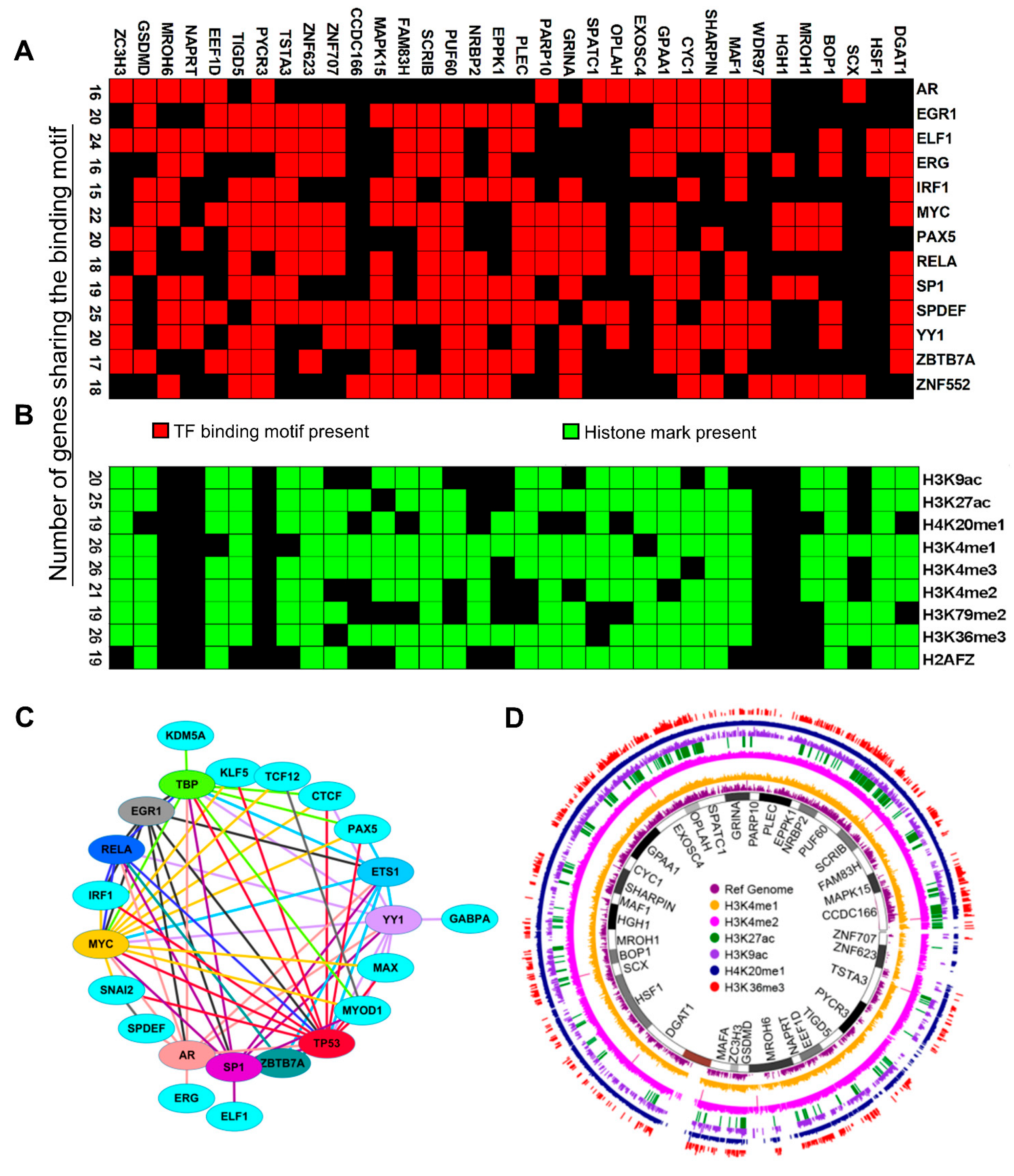
3.7. Search for Shared Mechanisms of the Co-Upregulation of CanCord34 Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Horne, S.D.; Pollick, S.A.; Heng, H.H.Q. Evolutionary Mechanism Unifies the Hallmarks of Cancer. Int. J. Cancer 2015, 136, 2012–2021. [Google Scholar] [CrossRef]
- Lonard, D.M.; Kumar, R.; O’Malley, B.W. Minireview: The SRC Family of Coactivators: An Entrée to Understanding a Subset of Polygenic Diseases? Mol. Endocrinol. 2010, 24, 279–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswaran, J.; Cyanam, D.; Mudvari, P.; Divijendra, S.; Reddy, N.; Pakala, S.B.; Nair, S.S.; Florea, L.; Fuqua, S.A.W.; Godbole, S.; et al. Transcriptomic Landscape of Breast Cancers through MRNA Sequencing. Sci. Rep. 2012, 2, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, S.; McLeod, H.L. Cancer Pharmacogenetics. Br. J. Cancer 2004, 90, 8–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasikci, Y.; Gronemeyer, H. Complexity against Current Cancer Research—Are We on the Wrong Track? Int. J. Cancer 2022, 150, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Anastassiou, D. Pan-Cancer Driver Copy Number Alterations Identified by Joint Expression/CNA Data Analysis. Sci. Rep. 2020, 10, 17199. [Google Scholar] [CrossRef] [PubMed]
- Ghoussaini, M.; Song, H.; Koessler, T.; al Olama, A.A.; Kote-Jarai, Z.; Driver, K.E.; Pooley, K.A.; Ramus, S.J.; Kjaer, S.K.; Hogdall, E.; et al. Multiple Loci with Different Cancer Specificities within the 8q24 Gene Desert. J. Natl. Cancer Inst. 2008, 100, 962–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easton, D.F.; Pooley, K.A.; Dunning, A.M.; Pharoah, P.D.P.; Thompson, D.; Ballinger, D.G.; Struewing, J.P.; Morrison, J.; Field, H.; Luben, R.; et al. Genome-Wide Association Study Identifies Novel Breast Cancer Susceptibility Loci. Nature 2007, 447, 1087–1093. [Google Scholar] [CrossRef] [Green Version]
- Grisanzio, C.; Freedman, M.L. Chromosome 8q24-Associated Cancers and MYC. Genes Cancer 2010, 1, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Landan, G.; Pomerantz, M.; Jaschek, R.; Herman, P.; Reich, D.; Yan, C.; Khalid, O.; Kantoff, P.; Oh, W.; et al. Functional Enhancers at the Gene-Poor 8q24 Cancer-Linked Locus. PLoS Genet. 2009, 5, e1000597. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.Q.; Williams, H.; Prince, T.L.; Ho, E.S. Overexpressed HSF1 Cancer Signature Genes Cluster in Human Chromosome 8q. Hum. Genom. 2017, 11, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, S.F.; Gruel, N.; Chapeaublanc, E.; Lescure, A.; Jones, T.; Reyal, F.; Vincent-Salomon, A.; Raynal, V.; Pierron, G.; Perez, F.; et al. A SiRNA Screen Identifies RAD21, EIF3H, CHRAC1 and TANC2 as Driver Genes within the 8q23, 8q24.3 and 17q23 Amplicons in Breast Cancer with Effects on Cell Growth, Survival and Transformation. Carcinogenesis 2014, 35, 670–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilal, E.; Vassallo, K.; Toppmeyer, D.; Barnard, N.; Rye, I.H.; Almendro, V.; Russnes, H.; Børresen-Dale, A.-L.; Levine, A.J.; Bhanot, G.; et al. Amplified Loci on Chromosomes 8 and 17 Predict Early Relapse in ER-Positive Breast Cancers. PLoS ONE 2012, 7, e38575. [Google Scholar] [CrossRef] [PubMed]
- Brusselaers, N.; Ekwall, K.; Durand-Dubief, M. Copy Number of 8q24.3 Drives HSF1 Expression and Patient Outcome in Cancer: An Individual Patient Data Meta-Analysis. Hum. Genom. 2019, 13, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Park, K.; Shin, E.; Kim, H.-J.; Kim, J.Y.; Kim, J.Y.; Gwak, G. Genomic Change of Chromosome 8 Predicts the Response to Taxane-Based Neoadjuvant Chemotherapy in Node-Positive Breast Cancer. Oncol. Rep. 2010, 24, 121–128. [Google Scholar]
- Letessier, A.; Sircoulomb, F.; Ginestier, C.; Cervera, N.; Monville, F.; Gelsi-Boyer, V.; Esterni, B.; Geneix, J.; Finetti, P.; Zemmour, C.; et al. Frequency, Prognostic Impact, and Subtype Association of 8p12, 8q24, 11q13, 12p13, 17q12, and 20q13 Amplifications in Breast Cancers. BMC Cancer 2006, 6, 245. [Google Scholar] [CrossRef] [Green Version]
- Ahmadiyeh, N.; Pomerantz, M.M.; Grisanzio, C.; Herman, P.; Jia, L.; Almendro, V.; He, H.H.; Brown, M.; Liu, X.S.; Davis, M.; et al. 8q24 Prostate, Breast, and Colon Cancer Risk Loci Show Tissue-Specific Long-Range Interaction with MYC. Proc. Natl. Acad. Sci. USA 2010, 107, 9742–9746. [Google Scholar] [CrossRef] [Green Version]
- Brisbin, A.G.; Asmann, Y.W.; Song, H.; Tsai, Y.-Y.; Aakre, J.A.; Yang, P.; Jenkins, R.B.; Pharoah, P.; Schumacher, F.; Conti, D.V.; et al. Meta-Analysis of 8q24 for Seven Cancers Reveals a Locus between NOV and ENPP2 Associated with Cancer Development. BMC Med. Genet. 2011, 12, 156. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ni, S.; Xiang, Y.; Parvin, J.D.; Yang, Y.; Zhou, Y.; Huang, K. Gene Co-Expression Analysis Predicts Genetic Aberration Loci Associated with Colon Cancer Metastasis. Int. J. Comput. Biol. Drug Des. 2013, 6, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Bian, X.W.; Li, D.K.; Xu, C.; Wang, G.M.; Xia, Q.Y.; Xiong, Q. Large-Scale RNA-Seq Transcriptome Analysis of 4043 Cancers and 548 Normal Tissue Controls across 12 TCGA Cancer Types. Sci. Rep. 2015, 5, 13413. [Google Scholar] [CrossRef] [Green Version]
- Pärssinen, J.; Kuukasjärvi, T.; Karhu, R.; Kallioniemi, A. High-Level Amplification at 17q23 Leads to Coordinated Overexpression of Multiple Adjacent Genes in Breast Cancer. Br. J. Cancer 2007, 96, 1258–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, B.; Chin, S.-F.; Rueda, O.M.; Vollan, H.-K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.-J.; et al. The Somatic Mutation Profiles of 2,433 Breast Cancers Refines Their Genomic and Transcriptomic Landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The Genomic and Transcriptomic Architecture of 2000 Breast Tumours Reveals Novel Subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- rueda, O.M.; Sammut, S.-J.; Seoane, J.A.; Chin, S.-F.; Caswell-Jin, J.L.; Callari, M.; Batra, R.; Pereira, B.; Bruna, A.; raza Ali, H.; et al. Dynamics of Breast-Cancer Relapse Reveal Late-Recurring ER-Positive Genomic Subgroups. Nature 2019, 567, 399–404. [Google Scholar] [CrossRef]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [CrossRef] [PubMed]
- Rody, A.; Karn, T.; Liedtke, C.; Pusztai, L.; Ruckhaeberle, E.; Hanker, L.; Gaetje, R.; Solbach, C.; Ahr, A.; Metzler, D.; et al. A Clinically Relevant Gene Signature in Triple Negative and Basal-like Breast Cancer. Breast Cancer Res. 2011, 13, R97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jézéquel, P.; Loussouarn, D.; Guérin-Charbonnel, C.; Campion, L.; Vanier, A.; Gouraud, W.; Lasla, H.; Guette, C.; Valo, I.; Verrièle, V.; et al. Gene-Expression Molecular Subtyping of Triple-Negative Breast Cancer Tumours: Importance of Immune Response. Breast Cancer Res. 2015, 17, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jézéquel, P.; Kerdraon, O.; Hondermarck, H.; Guérin-Charbonnel, C.; Lasla, H.; Gouraud, W.; Canon, J.-L.; Gombos, A.; Dalenc, F.; Delaloge, S.; et al. Identification of Three Subtypes of Triple-Negative Breast Cancer with Potential Therapeutic Implications. Breast Cancer Res. 2019, 21, 65. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A Bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.S.; Hwang, S.; Zhang, S.-D. RMA with Quantile Normalization Mixes Biological Signals between Different Sample Groups in Microarray Data Analysis. In Proceedings of the 2014 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Belfast, UK, 2–5 November 2014; pp. 139–143. [Google Scholar]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics Connects Somatic Mutations to Signalling in Breast Cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martínez-Ledesma, E.; Martínez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Peña, J.G.; Treviño, V. SurvExpress: An Online Biomarker Validation Tool and Database for Cancer Gene Expression Data Using Survival Analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [Green Version]
- Kalra, H.; Simpson, R.J.; Ji, H.; Aikawa, E.; Altevogt, P.; Askenase, P.; Bond, V.C.; Borràs, F.E.; Breakefield, X.; Budnik, V.; et al. Vesiclepedia: A Compendium for Extracellular Vesicles with Continuous Community Annotation. PLOS Biol. 2012, 10, e1001450. [Google Scholar] [CrossRef] [Green Version]
- Colacino, J.A.; Azizi, E.; Brooks, M.D.; Harouaka, R.; Fouladdel, S.; McDermott, S.P.; Lee, M.; Hill, D.; Madden, J.; Boerner, J.; et al. Heterogeneity of Human Breast Stem and Progenitor Cells as Revealed by Transcriptional Profiling. Stem Cell Rep. 2018, 10, 1596–1609. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Liu, Y.; Deng, L.; Wang, D.; He, X.; Zhou, L.; Wicha, M.S.; Bai, F.; Liu, S. Transcriptional Profiles of Different States of Cancer Stem Cells in Triple-Negative Breast Cancer. Mol. Cancer 2018, 17, 1–6. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-Scale Gene Function Analysis with the PANTHER Classification System. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Zúñiga-León, E.; Carrasco-Navarro, U.; Fierro, F. NeVOmics: An Enrichment Tool for Gene Ontology and Functional Network Analysis and Visualization of Data from OMICs Technologies. Genes 2018, 9, 569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudis, M.; Cleary, M.L. Progenetix.Net: An Online Repository for Molecular Cytogenetic Aberration Data. Bioinformatics 2001, 17, 1228–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangwala, S.H.; Kuznetsov, A.; Ananiev, V.; Asztalos, A.; Borodin, E.; Evgeniev, V.; Joukov, V.; Lotov, V.; Pannu, R.; Rudnev, D.; et al. Accessing NCBI Data Using the NCBI Sequence Viewer and Genome Data Viewer (GDV). Genome Res. 2021, 31, 159–169. [Google Scholar] [CrossRef]
- Zhou, K.-R.; Liu, S.; Sun, W.-J.; Zheng, L.-L.; Zhou, H.; Yang, J.-H.; Qu, L.-H. ChIPBase v2.0: Decoding Transcriptional Regulatory Networks of Non-Coding RNAs and Protein-Coding Genes from ChIP-Seq Data. Nucleic Acids Res. 2017, 45, D43–D50. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.I.; Schein, J.E.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.E.; Pelz, O.; Buhlmann, S.; Kerr, G.; Horn, T.; Boutros, M. GenomeRNAi: A Database for Cell-Based and in Vivo RNAi Phenotypes, 2013 Update. Nucleic Acids Res. 2013, 41, D1021–D1026. [Google Scholar] [CrossRef] [Green Version]
- Dwane, L.; Behan, F.M.; Gonçalves, E.; Lightfoot, H.; Yang, W.; van der Meer, D.; Shepherd, R.; Pignatelli, M.; Iorio, F.; Garnett, M.J. Project Score Database: A Resource for Investigating Cancer Cell Dependencies and Prioritizing Therapeutic Targets. Nucleic Acids Res. 2021, 49, D1365–D1372. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of Cancer Therapeutic Targets Using CRISPR–Cas9 Screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Pillai, P.M.; Paul, A.M.; Amjesh, R.; Leitzel, K.; Ali, S.M.; Sandiford, O.; Lipton, A.; Rameshwar, P.; Hortobagyi, G.N.; et al. Cellular Fitness Phenotypes of Cancer Target Genes from Oncobiology to Cancer Therapeutics. Cells 2021, 10, 433. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef] [Green Version]
- Sandiford, O.A.; Donnelly, R.J.; El-Far, M.H.; Burgmeyer, L.M.; Sinha, G.; Pamarthi, S.H.; Sherman, L.S.; Ferrer, A.I.; DeVore, D.E.; Patel, S.A.; et al. Mesenchymal Stem Cell-Secreted Extracellular Vesicles Instruct Stepwise Dedifferentiation of Breast Cancer Cells into Dormancy at the Bone Marrow Perivascular Region. Cancer Res. 2021, 81, 1567–1582. [Google Scholar] [CrossRef]
- Yan, H.; Luo, B.; Wu, X.; Guan, F.; Yu, X.; Zhao, L.; Ke, X.; Wu, J.; Yuan, J. Cisplatin Induces Pyroptosis via Activation of MEG3/NLRP3/Caspase-1/GSDMD Pathway in Triple-Negative Breast Cancer. Int. J. Biol. Sci. 2021, 17, 2606–2621. [Google Scholar] [CrossRef]
- Al Mamun, A.; Mimi, A.A.; Aziz, M.A.; Zaeem, M.; Ahmed, T.; Munir, F.; Xiao, J. Role of Pyroptosis in Cancer and Its Therapeutic Regulation. Eur. J. Pharmacol. 2021, 910, 174444. [Google Scholar] [CrossRef]
- Zhang, B.; Fan, Y.; Cao, P.; Tan, K. Multifaceted Roles of HSF1 in Cell Death: A State-of-the-Art Review. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188591. [Google Scholar] [CrossRef]
- Li, S.; Wu, H.; Huang, X.; Jian, Y.; Kong, L.; Xu, H.; Ouyang, Y.; Chen, X.; Wu, G.; Yu, L.; et al. BOP1 Confers Chemoresistance of Triple-Negative Breast Cancer by Promoting CBP-Mediated β-Catenin Acetylation. J. Pathol. 2021, 254, 265–278. [Google Scholar] [CrossRef]
- Vellky, J.E.; Ricke, E.A.; Huang, W.; Ricke, W.A. Expression, Localization, and Function of the Nucleolar Protein BOP1 in Prostate Cancer Progression. Am. J. Pathol. 2021, 191, 168–179. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, Y. Block of Proliferation 1 Promotes Cell Migration and Invasion in Human Colorectal Cancer Cells via the JNK Pathway. J. Clin. Lab. Anal. 2020, 34, e23283. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.-Y.; Cheng, I.K.-C.; Ching, A.K.-K.; Chu, J.-H.; Lai, P.B.-S.; Wong, N. Block of Proliferation 1 (BOP1) Plays an Oncogenic Role in Hepatocellular Carcinoma by Promoting Epithelial-to-Mesenchymal Transition. Hepatology 2011, 54, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, R.; Allmang, C.; Raijmakers, R.; van Aarssen, Y.; Egberts, W.V.; Petfalski, E.; van Venrooij, W.J.; Tollervey, D.; Pruijn, G.J.M. Three Novel Components of the Human Exosome. J. Biol. Chem. 2001, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanska, B.; Cheishvili, D.; Suderman, M.; Arakelian, A.; Huang, J.; Hallett, M.; Han, Z.-G.; Al-Mahtab, M.; Akbar, S.M.F.; Khan, W.A.; et al. Genome-Wide Study of Hypomethylated and Induced Genes in Patients with Liver Cancer Unravels Novel Anticancer Targets. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 3118–3132. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Tong, J.H.M.; Kang, W.; Lung, R.W.M.; Chak, W.P.; Chung, L.Y.; Wu, F.; Li, H.; Yu, J.; Chan, A.W.H.; et al. EXOSC4 Functions as a Potential Oncogene in Development and Progression of Colorectal Cancer. Mol. Carcinog. 2018, 57, 1780–1791. [Google Scholar] [CrossRef]
- An, J.; Luo, Z.; An, W.; Cao, D.; Ma, J.; Liu, Z. Identification of Spliceosome Components Pivotal to Breast Cancer Survival. RNA Biol. 2021, 18, 833–842. [Google Scholar] [CrossRef]
- Long, Q.; An, X.; Chen, M.; Wang, N.; Sui, S.; Li, Y.; Zhang, C.; Lee, K.; Wang, X.; Tian, T.; et al. PUF60/AURKA Axis Contributes to Tumor Progression and Malignant Phenotypes in Bladder Cancer. Front. Oncol. 2020, 10, 568015. [Google Scholar] [CrossRef]
- Long, Q.; Hua, Y.; He, L.; Zhang, C.; Sui, S.; Li, Y.; Qiu, H.; Tian, T.; An, X.; Luo, G.; et al. Poly(U) Binding Splicing Factor 60 Promotes Renal Cell Carcinoma Growth by Transcriptionally Upregulating Telomerase Reverse Transcriptase. Int. J. Biol. Sci. 2020, 16, 3002–3017. [Google Scholar] [CrossRef]
- Sun, D.; Lei, W.; Hou, X.; Li, H.; Ni, W. PUF60 Accelerates the Progression of Breast Cancer through Downregulation of PTEN Expression. Cancer Manag. Res. 2019, 11, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Yu, Y.; Akilli Öztürk, Ö.; Holland, J.D.; Besser, D.; Fritzmann, J.; Wulf-Goldenberg, A.; Eckert, K.; Fichtner, I.; Birchmeier, W. New Wnt/β-Catenin Target Genes Promote Experimental Metastasis and Migration of Colorectal Cancer Cells through Different Signals. Gut 2016, 65, 1690–1701. [Google Scholar] [CrossRef]
- Mistry, D.S.; Chen, Y.; Sen, G.L. Progenitor Function in Self-Renewing Human Epidermis Is Maintained by the Exosome. Cell Stem Cell 2012, 11, 127–135. [Google Scholar] [CrossRef] [PubMed]
- McIver, S.C.; Katsumura, K.R.; Davids, E.; Liu, P.; Kang, Y.-A.; Yang, D.; Bresnick, E.H. Exosome Complex Orchestrates Developmental Signaling to Balance Proliferation and Differentiation during Erythropoiesis. eLife 2016, 5, e17877. [Google Scholar] [CrossRef] [PubMed]
- Skamagki, M.; Zhang, C.; Ross, C.A.; Ananthanarayanan, A.; Liu, Z.; Mu, Q.; Basu, U.; Wang, J.; Zhao, R.; Li, H.; et al. RNA Exosome Complex-Mediated Control of Redox Status in Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 1053–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbar Samadani, A.; Keymoradzdeh, A.; Shams, S.; Soleymanpour, A.; Elham Norollahi, S.; Vahidi, S.; Rashidy-Pour, A.; Ashraf, A.; Mirzajani, E.; Khanaki, K.; et al. Mechanisms of Cancer Stem Cell Therapy. Clin. Chim. Acta 2020, 510, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Brooks, M.; Wicha, M.S. Epithelial-Mesenchymal Plasticity of Breast Cancer Stem Cells: Implications for Metastasis and Therapeutic Resistance. Curr. Pharm. Des. 2015, 21, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Qadir, F.; Aziz, M.A.; Sari, C.P.; Ma, H.; Dai, H.; Wang, X.; Raithatha, D.; da Silva, L.G.L.; Hussain, M.; Poorkasreiy, S.P.; et al. Transcriptome Reprogramming by Cancer Exosomes: Identification of Novel Molecular Targets in Matrix and Immune Modulation. Mol. Cancer 2018, 17, 97. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Wu, Y.; Shen, H.; Lv, M.; Chen, W.; Zhang, X.; Zhong, S.; Tang, J.; Zhao, J. Exosomes in Development, Metastasis and Drug Resistance of Breast Cancer. Cancer Sci. 2015, 106, 959–964. [Google Scholar] [CrossRef] [Green Version]
- Piao, Y.J.; Kim, H.S.; Hwang, E.H.; Woo, J.; Zhang, M.; Moon, W.K. Breast Cancer Cell-Derived Exosomes and Macrophage Polarization Are Associated with Lymph Node Metastasis. Oncotarget 2018, 9, 7398–7410. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Deivendran, S.; Santhoshkumar, T.R.; Pillai, M.R. Signaling Coupled Epigenomic Regulation of Gene Expression. Oncogene 2017, 36, 5917–5926. [Google Scholar] [CrossRef]
- Nair, S.S.; Mishra, S.K.; Yang, Z.; Balasenthil, S.; Kumar, R.; Vadlamudi, R.K. Potential Role of a Novel Transcriptional Coactivator PELP1 in Histone H1 Displacement in Cancer Cells. Cancer Res. 2004, 64, 6416–6423. [Google Scholar] [CrossRef] [Green Version]
- O’malley, B.W.; Kumar, R. Nuclear Receptor Coregulators in Cancer Biology. Cancer Res. 2009, 69, 8217–8222. [Google Scholar] [CrossRef] [PubMed]
- Manavathi, B.; Acconcia, F.; Rayala, S.K.; Kumar, R. An Inherent Role of Microtubule Network in the Action of Nuclear Receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 15981–15986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miki, Y.; Iwabuchi, E.; Ono, K.; Sasano, H.; Ito, K. Exploring Protein-Protein Interaction in the Study of Hormone-Dependent Cancers. Int. J. Mol. Sci. 2018, 19, 3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cockerill, P.N. Receptor Signaling Directs Global Recruitment of Pre-Existing Transcription Factors to Inducible Elements. Yale J. Biol. Med. 2016, 89, 591–596. [Google Scholar] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and Recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [Green Version]
- Henrich, L.M.; Smith, J.A.; Kitt, D.; Errington, T.M.; Nguyen, B.; Traish, A.M.; Lannigan, D.A. Extracellular Signal-Regulated Kinase 7, a Regulator of Hormone-Dependent Estrogen Receptor Destruction. Mol. Cell. Biol. 2003, 23, 5979–5988. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.T.Y.; Xu, Y.-M. Regulation of Human Mitogen-Activated Protein Kinase 15 (Extracellular Signal-Regulated Kinase 7/8) and Its Functions: A Recent Update. J. Cell. Physiol. 2018, 234, 75–88. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, A.M.; Amjesh, R.; George, B.; Sankaran, D.; Sandiford, O.A.; Rameshwar, P.; Pillai, M.R.; Kumar, R. The Revelation of Continuously Organized, Co-Overexpressed Protein-Coding Genes with Roles in Cellular Communications in Breast Cancer. Cells 2022, 11, 3806. https://doi.org/10.3390/cells11233806
Paul AM, Amjesh R, George B, Sankaran D, Sandiford OA, Rameshwar P, Pillai MR, Kumar R. The Revelation of Continuously Organized, Co-Overexpressed Protein-Coding Genes with Roles in Cellular Communications in Breast Cancer. Cells. 2022; 11(23):3806. https://doi.org/10.3390/cells11233806
Chicago/Turabian StylePaul, Aswathy Mary, Revikumar Amjesh, Bijesh George, Deivendran Sankaran, Oleta A. Sandiford, Pranela Rameshwar, Madhavan Radhakrishna Pillai, and Rakesh Kumar. 2022. "The Revelation of Continuously Organized, Co-Overexpressed Protein-Coding Genes with Roles in Cellular Communications in Breast Cancer" Cells 11, no. 23: 3806. https://doi.org/10.3390/cells11233806