
Extracellular Vesicles from Human Umbilical Cord-Derived MSCs Affect Vessel Formation In Vitro and Promote VEGFR2-Mediated Cell Survival

Abstract
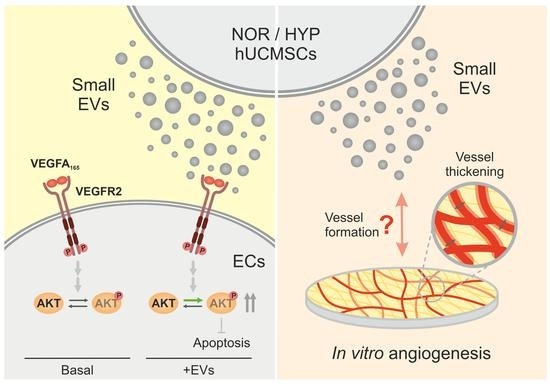
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cell Culture
2.3. Extracellular Vesicles Isolation and Analysis
2.3.1. Transmission Electron Microscopy (TEM)
2.3.2. Western Immunoblotting (WB) for EV Analysis
2.4. Acute Stimulation Studies
2.5. Cellular Lysates and Western Immunoblotting
2.6. 3D Collagen Gel Tube Formation Assay
2.7. Cell Proliferation Assay
2.8. Apoptosis Assay
2.9. Endothelial-Fibroblast Co-Culture Angiogenesis Assay
2.10. Statistical Analysis
3. Results
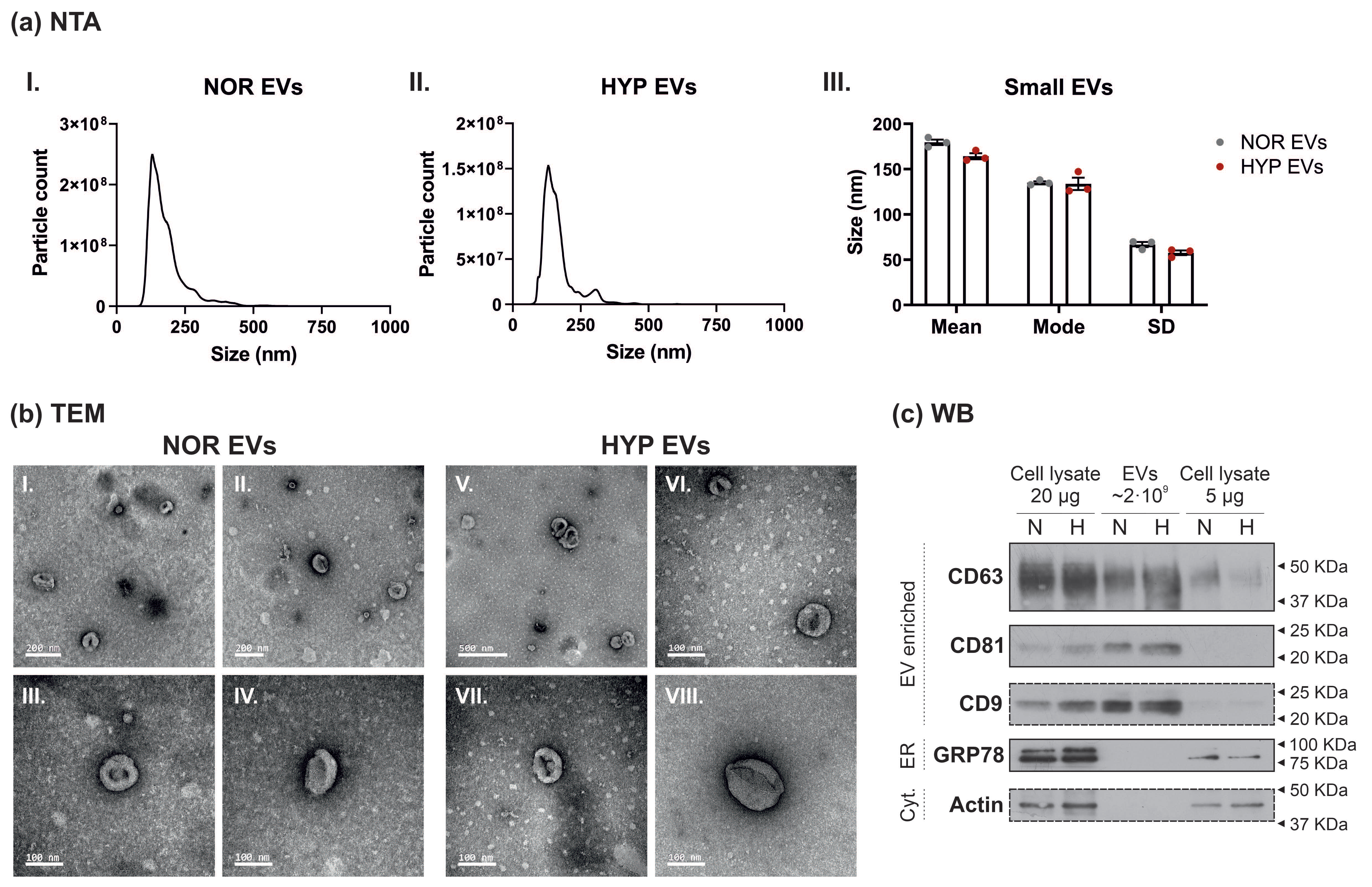
3.1. Characterisation of Small Extracellular Vesicles from hUCMSCs
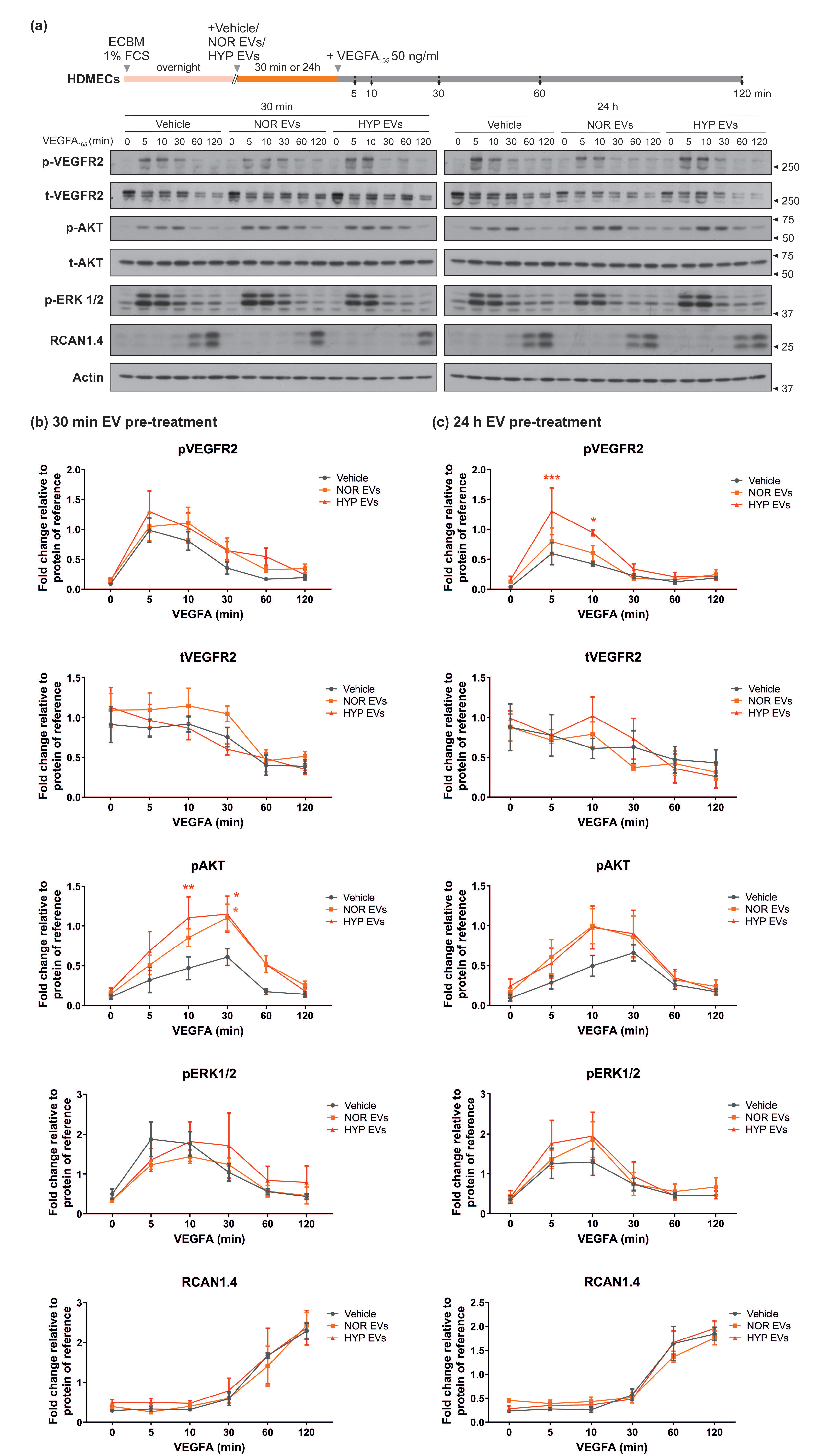
3.2. hUCMSC-Derived EVs Affect VEGFR2-Mediated Phosphorylation of AKT
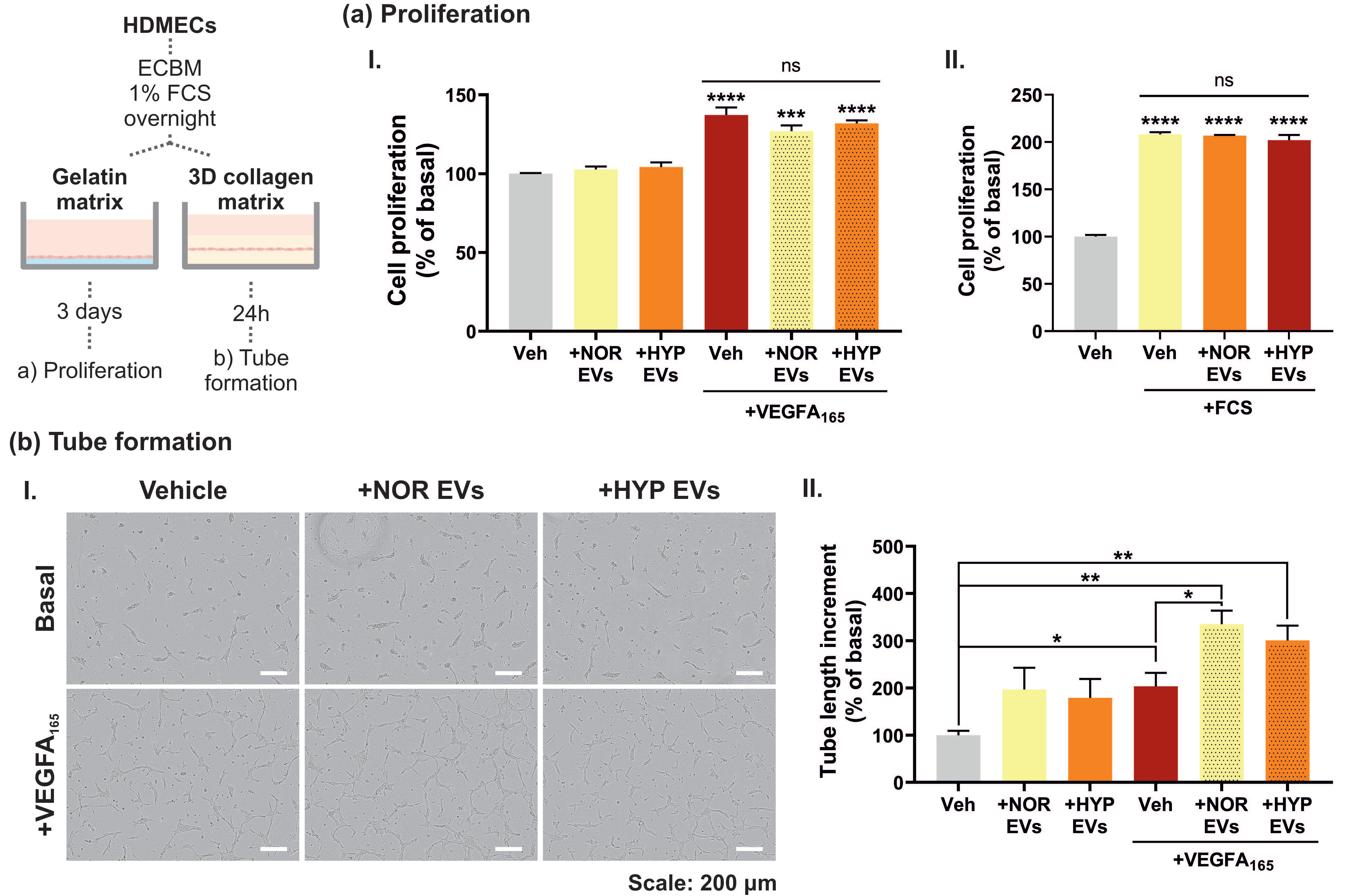
3.3. hUCMSC-Derived EVs Affect VEGFA-Mediated Tubular Morphogenesis but Not Cell Proliferation
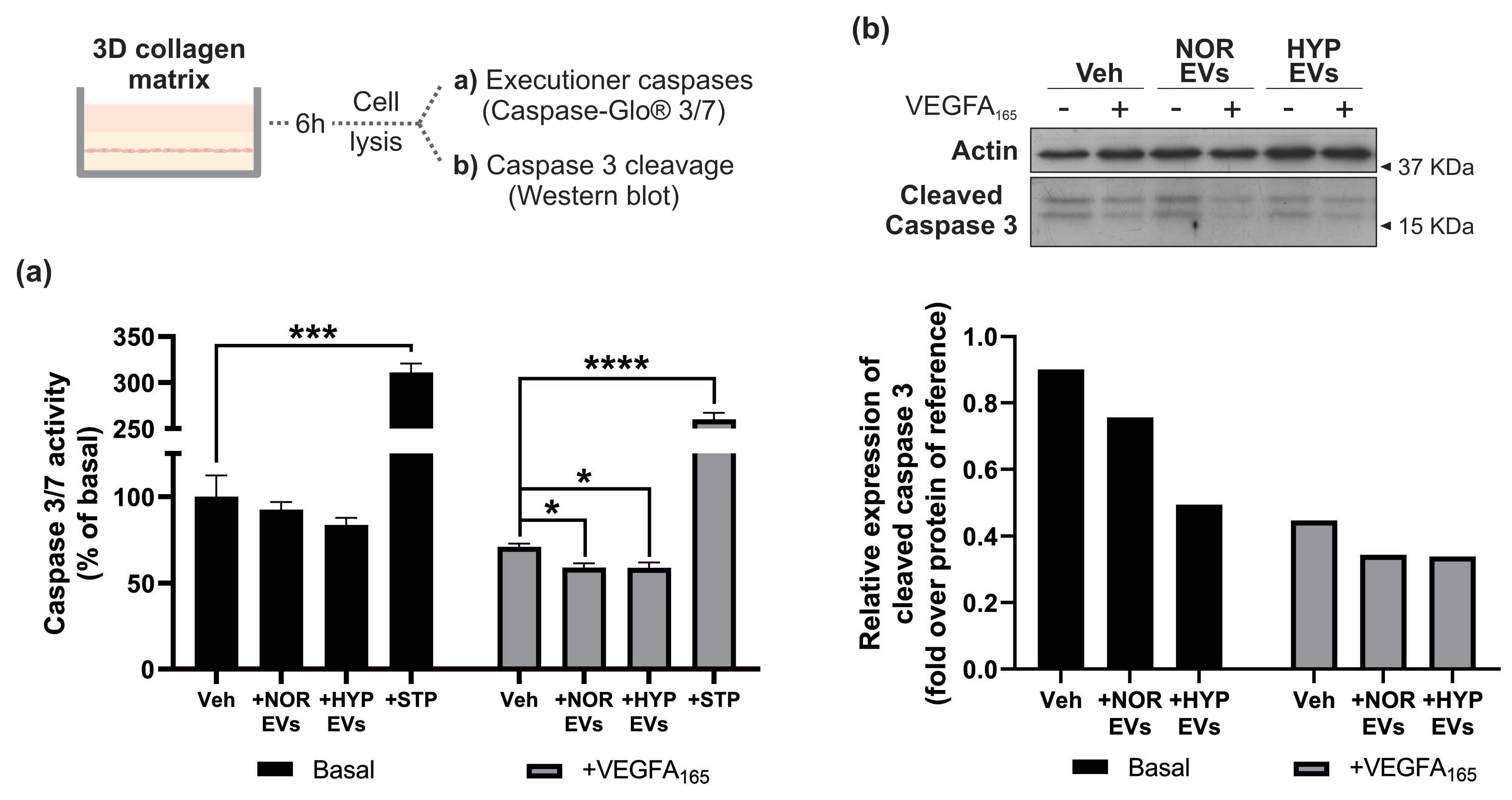
3.4. hUCMSC-Derived EVs Suppress Apoptosis during Tubular Morphogenesis
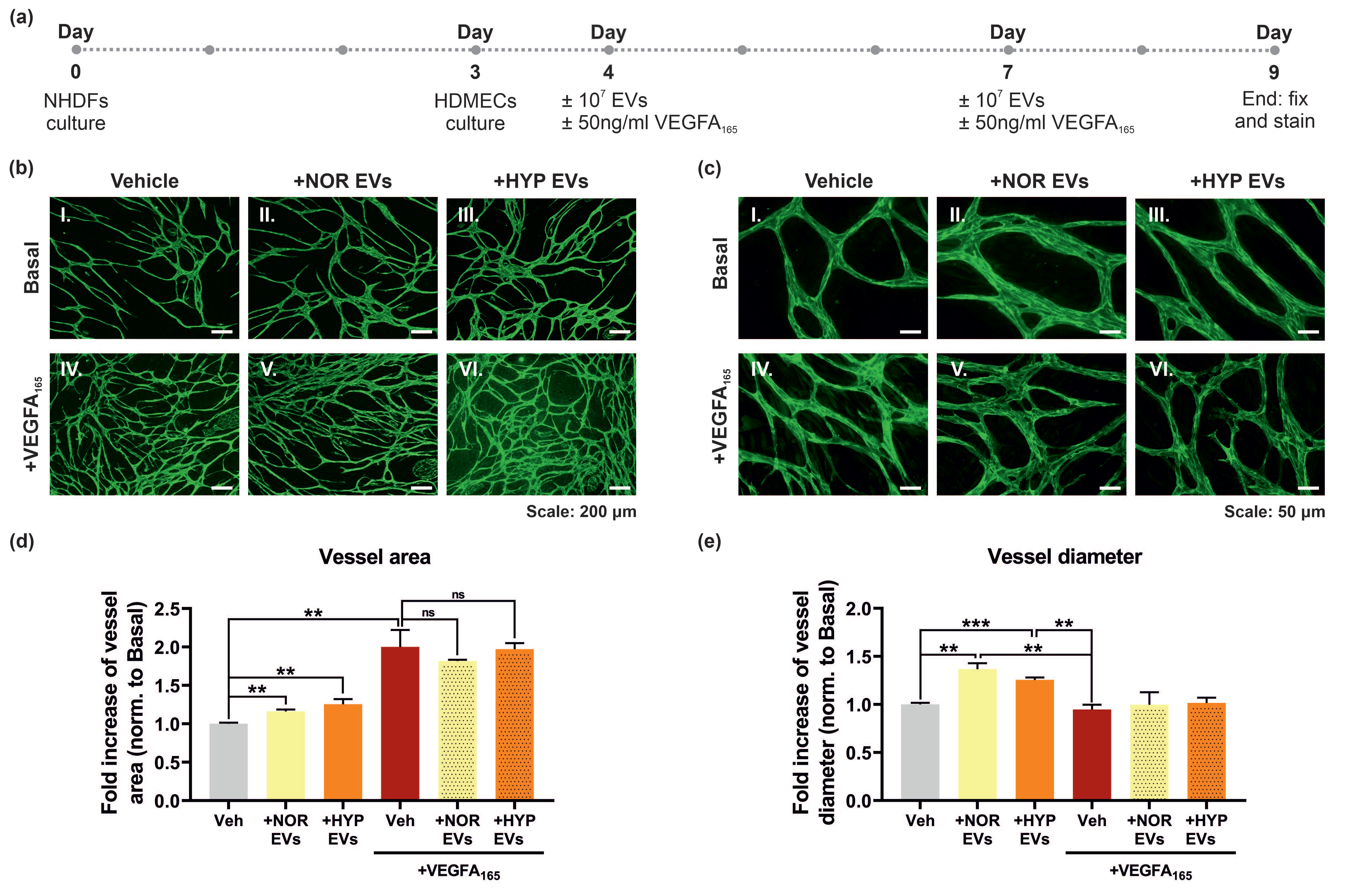
3.5. hUCMSC-Derived EVs Promote Vessel Formation and Impact Vessel Diameter in a Long-Term Angiogenesis Model In Vitro
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. CMLS 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Yáñez-Mó, M.; Siljander, P.R.M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Brennan, M.Á.; Lötvall, J.; Breakefield, X.O.; EL Andaloussi, S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11, eaav8521. [Google Scholar] [CrossRef]
- Rashed, M.H.; Bayraktar, E.; Helal, G.K.; Abd-Ellah, M.F.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From Garbage Bins to Promising Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 538. [Google Scholar] [CrossRef] [Green Version]
- Yuana, Y.; Sturk, A.; Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Discher, D.E.; Péault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Levy, O.; Kuai, R.; Siren, E.M.J.; Bhere, D.; Milton, Y.; Nissar, N.; De Biasio, M.; Heinelt, M.; Reeve, B.; Abdi, R.; et al. Shattering barriers toward clinically meaningful MSC therapies. Sci. Adv. 2020, 6, eaba6884. [Google Scholar] [CrossRef]
- Barkholt, L.; Flory, E.; Jekerle, V.; Lucas-Samuel, S.; Ahnert, P.; Bisset, L.; Büscher, D.; Fibbe, W.; Foussat, A.; Kwa, M.; et al. Risk of tumorigenicity in mesenchymal stromal cell-based therapies--bridging scientific observations and regulatory viewpoints. Cytotherapy 2013, 15, 753–759. [Google Scholar] [CrossRef]
- Breitbach, M.; Bostani, T.; Roell, W.; Xia, Y.; Dewald, O.; Nygren, J.M.; Fries, J.W.; Tiemann, K.; Bohlen, H.; Hescheler, J.; et al. Potential risks of bone marrow cell transplantation into infarcted hearts. Blood 2007, 110, 1362–1369. [Google Scholar] [CrossRef] [Green Version]
- von Bahr, L.; Batsis, I.; Moll, G.; Hägg, M.; Szakos, A.; Sundberg, B.; Uzunel, M.; Ringden, O.; Le Blanc, K. Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long-term engraftment and no ectopic tissue formation. Stem Cells 2012, 30, 1575–1578. [Google Scholar] [CrossRef]
- Schrepfer, S.; Deuse, T.; Reichenspurner, H.; Fischbein, M.P.; Robbins, R.C.; Pelletier, M.P. Stem cell transplantation: The lung barrier. Transplant. Proc. 2007, 39, 573–576. [Google Scholar] [CrossRef]
- Ankrum, J.A.; Ong, J.F.; Karp, J.M. Mesenchymal stem cells: Immune evasive, not immune privileged. Nat. Biotechnol. 2014, 32, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Gwam, C.; Mohammed, N.; Ma, X. Stem cell secretome, regeneration, and clinical translation: A narrative review. Ann. Transl. Med. 2021, 9, 70. [Google Scholar] [CrossRef]
- Fuloria, S.; Subramaniyan, V.; Dahiya, R.; Dahiya, S.; Sudhakar, K.; Kumari, U.; Sathasivam, K.; Meenakshi, D.U.; Wu, Y.S.; Sekar, M.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles: Regenerative Potential and Challenges. Biology 2021, 10, 172. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, Y.; Li, H. Applications of extracellular vesicles in tissue regeneration. Biomicrofluidics 2020, 14, 011501. [Google Scholar] [CrossRef]
- Maacha, S.; Sidahmed, H.; Jacob, S.; Gentilcore, G.; Calzone, R.; Grivel, J.C.; Cugno, C. Paracrine Mechanisms of Mesenchymal Stromal Cells in Angiogenesis. Stem Cells Int. 2020, 2020, 4356359. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Ma, K.; Zhang, C.; Fu, X. Therapeutic angiogenesis using stem cell-derived extracellular vesicles: An emerging approach for treatment of ischemic diseases. Stem Cell Res. Ther. 2019, 10, 158. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 2007, 19, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Muñiz-García, A.; Romero, M.; Falcόn-Perez, J.M.; Murray, P.; Zorzano, A.; Mora, S. Hypoxia-induced HIF1α activation regulates small extracellular vesicle release in human embryonic kidney cells. Sci. Rep. 2022, 12, 1443. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. Curr. Protoc. Cell Biol. 2006, 30, 3.22.1–3.22.29. [Google Scholar] [CrossRef]
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis Analyzer for ImageJ—A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2020, 10, 11568. [Google Scholar] [CrossRef]
- Zudaire, E.; Gambardella, L.; Kurcz, C.; Vermeren, S. A computational tool for quantitative analysis of vascular networks. PLoS ONE 2011, 6, e27385. [Google Scholar] [CrossRef] [Green Version]
- Corliss, B.A.; Doty, R.W.; Mathews, C.; Yates, P.A.; Zhang, T.; Peirce, S.M. REAVER: A program for improved analysis of high-resolution vascular network images. Microcirculation 2020, 27, e12618. [Google Scholar] [CrossRef] [Green Version]
- Merino-González, C.; Zuñiga, F.A.; Escudero, C.; Ormazabal, V.; Reyes, C.; Nova-Lamperti, E.; Salomón, C.; Aguayo, C. Mesenchymal Stem Cell-Derived Extracellular Vesicles Promote Angiogenesis: Potencial Clinical Application. Front. Physiol. 2016, 7, 24. [Google Scholar] [CrossRef]
- Holmes, K.; Chapman, E.; See, V.; Cross, M.J. VEGF Stimulates RCAN1.4 Expression in Endothelial Cells via a Pathway Requiring Ca2+/Calcineurin and Protein Kinase C-δ. PLoS ONE 2010, 5, e11435. [Google Scholar] [CrossRef] [Green Version]
- Alghanem, A.F.; Wilkinson, E.L.; Emmett, M.S.; Aljasir, M.A.; Holmes, K.; Rothermel, B.A.; Simms, V.A.; Heath, V.L.; Cross, M.J. RCAN1.4 regulates VEGFR-2 internalisation, cell polarity and migration in human microvascular endothelial cells. Angiogenesis 2017, 20, 341–358. [Google Scholar] [CrossRef] [Green Version]
- Bruns, A.F.; Herbert, S.P.; Odell, A.F.; Jopling, H.M.; Hooper, N.M.; Zachary, I.C.; Walker, J.H.; Ponnambalam, S. Ligand-stimulated VEGFR2 signaling is regulated by co-ordinated trafficking and proteolysis. Traffic 2010, 11, 161–174. [Google Scholar] [CrossRef]
- Dimmeler, S.; Zeiher, A.M. Endothelial cell apoptosis in angiogenesis and vessel regression. Circ. Res. 2000, 87, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Fujio, Y.; Walsh, K. Akt Mediates Cytoprotection of Endothelial Cells by Vascular Endothelial Growth Factor in an Anchorage-dependent Manner*. J. Biol. Chem. 1999, 274, 16349–16354. [Google Scholar] [CrossRef] [Green Version]
- Gerber, H.-P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular Endothelial Growth Factor Regulates Endothelial Cell Survival through the Phosphatidylinositol 3′-Kinase/Akt Signal Transduction Pathway: REQUIREMENT FOR Flk-1/KDR ACTIVATION. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef] [Green Version]
- Pober, J.S.; Min, W.; Bradley, J.R. Mechanisms of endothelial dysfunction, injury, and death. Annu. Rev. Pathol. 2009, 4, 71–95. [Google Scholar] [CrossRef]
- Kabir, J.; Lobo, M.; Zachary, I. Staurosporine induces endothelial cell apoptosis via focal adhesion kinase dephosphorylation and focal adhesion disassembly independent of focal adhesion kinase proteolysis. Biochem. J. 2002, 367, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Sorrell, J.M.; Baber, M.A.; Caplan, A.I. A Self-Assembled Fibroblast-Endothelial Cell Co-Culture System That Supports in vitro Vasculogenesis by both Human Umbilical Vein Endothelial Cells and Human Dermal Microvascular Endothelial Cells. Cells Tissues Organs 2007, 186, 157–168. [Google Scholar] [CrossRef]
- Roberts, O.L.; Holmes, K.; Müller, J.; Cross, D.A.E.; Cross, M.J. ERK5 is required for VEGF-mediated survival and tubular morphogenesis of primary human microvascular endothelial cells. J. Cell Sci. 2010, 123, 3189–3200. [Google Scholar] [CrossRef]
- Racchetti, G.; Meldolesi, J. Extracellular Vesicles of Mesenchymal Stem Cells: Therapeutic Properties Discovered with Extraordinary Success. Biomedicines 2021, 9, 667. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling—In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bove, A.M.; Simone, G.; Ma, B. Molecular Bases of VEGFR-2-Mediated Physiological Function and Pathological Role. Front. Cell Dev. Biol. 2020, 8, 599281. [Google Scholar] [CrossRef] [PubMed]
- Mellberg, S.; Dimberg, A.; Bahram, F.; Hayashi, M.; Rennel, E.; Ameur, A.; Westholm, J.O.; Larsson, E.; Lindahl, P.; Cross, M.J.; et al. Transcriptional profiling reveals a critical role for tyrosine phosphatase VE-PTP in regulation of VEGFR2 activity and endothelial cell morphogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 1490–1502. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Ruan, Y.; Zhao, S.; Ning, J.; Rao, T.; Yu, W.; Zhou, X.; Liu, C.; Qi, Y.; Cheng, F. MicroRNA-205 inhibits the apoptosis of renal tubular epithelial cells via the PTEN/Akt pathway in renal ischemia-reperfusion injury. Am. J. Transl. Res. 2019, 11, 7364–7375. [Google Scholar]
- Ferreira, A.D.F.; Cunha, P.D.S.; Carregal, V.M.; da Silva, P.C.; de Miranda, M.C.; Kunrath-Lima, M.; de Melo, M.I.A.; Faraco, C.C.F.; Barbosa, J.L.; Frezard, F.; et al. Extracellular Vesicles from Adipose-Derived Mesenchymal Stem/Stromal Cells Accelerate Migration and Activate AKT Pathway in Human Keratinocytes and Fibroblasts Independently of miR-205 Activity. Stem Cells Int. 2017, 2017, 9841035. [Google Scholar] [CrossRef] [Green Version]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.E.; Timmers, L.; van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G.; et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013, 10, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Feng, Y.; Wang, X.; Ding, J.; Li, H.; Guan, H.; Chen, Z. Human umbilical vein endothelial cells-derived exosomes enhance cardiac function after acute myocardial infarction by activating the PI3K/AKT signaling pathway. Bioengineered 2022, 13, 8850–8865. [Google Scholar] [CrossRef]
- Han, Y.; Ren, J.; Bai, Y.; Pei, X.; Han, Y. Exosomes from hypoxia-treated human adipose-derived mesenchymal stem cells enhance angiogenesis through VEGF/VEGF-R. Int. J. Biochem. Cell Biol. 2019, 109, 59–68. [Google Scholar] [CrossRef]
- Thurston, G.; Wang, Q.; Baffert, F.; Rudge, J.; Papadopoulos, N.; Jean-Guillaume, D.; Wiegand, S.; Yancopoulos, G.D.; McDonald, D.M. Angiopoietin 1 causes vessel enlargement, without angiogenic sprouting, during a critical developmental period. Development 2005, 132, 3317–3326. [Google Scholar] [CrossRef] [Green Version]
- Bister, N.; Pistono, C.; Huremagic, B.; Jolkkonen, J.; Giugno, R.; Malm, T. Hypoxia and extracellular vesicles: A review on methods, vesicular cargo and functions. J. Extracell. Vesicles 2020, 10, e12002. [Google Scholar] [CrossRef]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñiz-García, A.; Wilm, B.; Murray, P.; Cross, M.J. Extracellular Vesicles from Human Umbilical Cord-Derived MSCs Affect Vessel Formation In Vitro and Promote VEGFR2-Mediated Cell Survival. Cells 2022, 11, 3750. https://doi.org/10.3390/cells11233750
Muñiz-García A, Wilm B, Murray P, Cross MJ. Extracellular Vesicles from Human Umbilical Cord-Derived MSCs Affect Vessel Formation In Vitro and Promote VEGFR2-Mediated Cell Survival. Cells. 2022; 11(23):3750. https://doi.org/10.3390/cells11233750
Chicago/Turabian StyleMuñiz-García, Ana, Bettina Wilm, Patricia Murray, and Michael J. Cross. 2022. "Extracellular Vesicles from Human Umbilical Cord-Derived MSCs Affect Vessel Formation In Vitro and Promote VEGFR2-Mediated Cell Survival" Cells 11, no. 23: 3750. https://doi.org/10.3390/cells11233750