
Reduced SKP2 Expression Adversely Impacts Genome Stability and Promotes Cellular Transformation in Colonic Epithelial Cells

{kind=link}
{kind=link}
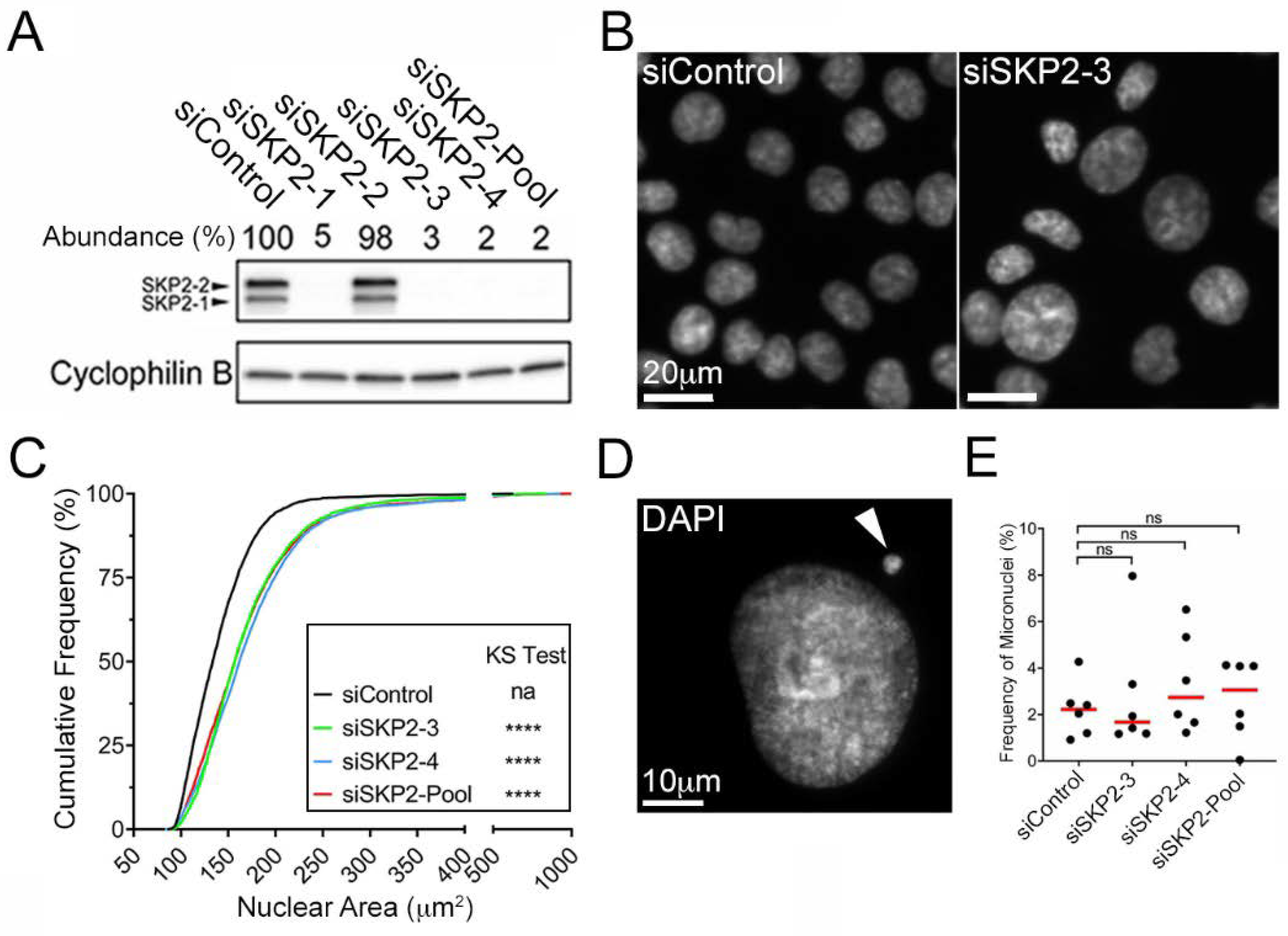{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. siRNA-Based F-Box Protein Screen
2.3. SKP2 Copy Number Alterations, Reduced Expression, and Survival Analyses
2.4. Silencing and Western Blot Analyses
2.5. QuantIM and CIN Analyses
2.6. Mitotic Chromosome Spread Enumeration
2.7. CRISPR/Cas9 Approaches
2.8. Soft Agar Colony Formation Assay
3. Results
3.1. An siRNA-Based F-Box Protein Screen Identifies SKP2 as a Strong Candidate CIN Gene
3.2. SKP2 Copy Number Losses Are Present in CRC and Correspond with Reduced Expression and Worse Patient Outcomes
3.3. Diminished SKP2 Expression Induces Increases in CIN-Associated Phenotypes in HCT116 Cells

3.4. SKP2 Is Required to Maintain Chromosome Stability in HCT116 Cells
3.5. Reduced SKP2 Expression Induces CIN in Non-Malignant, Non-Transformed Colonic Epithelial Cell Contexts
3.6. SKP2 Loss Corresponds with Dynamic CIN Phenotypes in Models of Disease Development
3.7. SKP2 Loss Promotes Anchorage-Independent Growth and Cellular Transformation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining ‘chromosomal instability’. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.L.; McManus, K.J. A novel multiplexed, image-based approach to detect phenotypes that underlie chromosome instability in human cells. PLoS ONE 2015, 10, e0123200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepage, C.C.; Morden, C.R.; Palmer, M.C.L.; Nachtigal, M.W.; McManus, K.J. Detecting chromosome instability in cancer: Approaches to resolve cell-to-cell heterogeneity. Cancers 2019, 11, 226. [Google Scholar] [CrossRef] [Green Version]
- Nowak, M.A.; Komarova, N.L.; Sengupta, A.; Jallepalli, P.V.; Shih Ie, M.; Vogelstein, B.; Lengauer, C. The role of chromosomal instability in tumor initiation. Proc. Natl. Acad. Sci. USA 2002, 99, 16226–16231. [Google Scholar] [CrossRef] [Green Version]
- Sieber, O.; Heinimann, K.; Tomlinson, I. Genomic instability—the engine of tumorigenesis? Nat. Rev. Cancer 2003, 3, 701–708. [Google Scholar] [CrossRef]
- McClelland, S.E. Role of chromosomal instability in cancer progression. Endocr. Relat. Cancer 2017, 24, T23–T31. [Google Scholar] [CrossRef] [Green Version]
- McClelland, S.E.; Burrell, R.A.; Swanton, C. Chromosomal instability: A composite phenotype that influences sensitivity to chemotherapy. Cell Cycle 2009, 8, 3262–3266. [Google Scholar] [CrossRef] [Green Version]
- Thompson, L.L.; Rutherford, K.A.; Lepage, C.C.; McManus, K.J. Aberrant SKP1 Expression: Diverse Mechanisms Impacting Genome and Chromosome Stability. Front. Cell Dev. Biol. 2022, 10, 859582. [Google Scholar] [CrossRef]
- Palmer, M.C.L.; Neudorf, N.M.; Farrell, A.C.; Razi, T.; Lichtensztejn, Z.; McManus, K.J. The F-box protein, FBXO7 is required to maintain chromosome stability in humans. Hum. Mol. Genet. 2021, 31, 1471–1486. [Google Scholar] [CrossRef]
- Thompson, L.L.; Rutherford, K.A.; Lepage, C.C.; McManus, K.J. The SCF Complex Is Essential to Maintain Genome and Chromosome Stability. Int. J. Mol. Sci. 2021, 22, 8544. [Google Scholar] [CrossRef]
- Lepage, C.C.; Palmer, M.C.L.; Farrell, A.C.; Neudorf, N.M.; Lichtensztejn, Z.; Nachtigal, M.W.; McManus, K.J. Reduced SKP1 and CUL1 expression underlies increases in Cyclin E1 and chromosome instability in cellular precursors of high-grade serous ovarian cancer. Br. J. Cancer 2021, 124, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Bungsy, M.; Palmer, M.C.L.; Jeusset, L.M.; Neudorf, N.M.; Lichtensztejn, Z.; Nachtigal, M.W.; McManus, K.J. Reduced RBX1 expression induces chromosome instability and promotes cellular transformation in high-grade serous ovarian cancer precursor cells. Cancer Lett. 2021, 500, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.L.; Baergen, A.K.; Lichtensztejn, Z.; McManus, K.J. Reduced SKP1 expression induces chromosome instability through aberrant cyclin E1 protein turnover. Cancers 2020, 12, 531. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, K.I.; Hatakeyama, S.; Nakayama, K. Regulation of the cell cycle at the G1-S transition by proteolysis of cyclin E and p27Kip1. Biochem. Biophys Res. Commun. 2001, 282, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Nagahama, H.; Minamishima, Y.A.; Matsumoto, M.; Nakamichi, I.; Kitagawa, K.; Shirane, M.; Tsunematsu, R.; Tsukiyama, T.; Ishida, N.; et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000, 19, 2069–2081. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Herbst, A.; Tworkowski, K.A.; Salghetti, S.E.; Tansey, W.P. Skp2 regulates Myc protein stability and activity. Mol. Cell 2003, 11, 1177–1188. [Google Scholar] [CrossRef]
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; Nakayama, K.I. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004, 23, 2116–2125. [Google Scholar] [CrossRef] [Green Version]
- Duda, D.M.; Olszewski, J.L.; Tron, A.E.; Hammel, M.; Lambert, L.J.; Waddell, M.B.; Mittag, T.; DeCaprio, J.A.; Schulman, B.A. Structure of a Glomulin-RBX1-CUL1 Complex: Inhibition of a RING E3 Ligase through Masking of Its E2-Binding Surface. Mol. Cell 2012, 47, 371–382. [Google Scholar] [CrossRef]
- Deshaies, R.J. SCF and Cullin/RING H2-Based Ubiquitin Ligases. Annu. Rev. Cell Dev. Biol. 1999, 15, 435–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postow, L.; Funabiki, H. An SCF complex containing Fbxl12 mediates DNA damage-induced Ku80 ubiquitylation. Cell Cycle 2013, 12, 587–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system-Implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta Bioenerg. 2014, 1843, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorski, J.W.; Ueland, F.R.; Kolesar, J.M. CCNE1 Amplification as a Predictive Biomarker of Chemotherapy Resistance in Epithelial Ovarian Cancer. Diagnostics 2020, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Etemadmoghadam, D.; Weir, B.A.; Au-Yeung, G.; Alsop, K.; Mitchell, G.; George, J.; Australian Ovarian Cancer Study, G.; Davis, S.; D’Andrea, A.D.; Simpson, K.; et al. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc. Natl. Acad. Sci. USA 2013, 110, 19489–19494. [Google Scholar] [CrossRef] [Green Version]
- Karst, A.M.; Jones, P.M.; Vena, N.; Ligon, A.H.; Liu, J.F.; Hirsch, M.S.; Etemadmoghadam, D.; Bowtell, D.D.; Drapkin, R. Cyclin E1 deregulation occurs early in secretory cell transformation to promote formation of fallopian tube-derived high-grade serous ovarian cancers. Cancer Res. 2014, 74, 1141–1152. [Google Scholar] [CrossRef] [Green Version]
- Aziz, K.; Limzerwala, J.F.; Sturmlechner, I.; Hurley, E.; Zhang, C.; Jeganathan, K.B.; Nelson, G.; Bronk, S.; Fierro Velasco, R.O.; van Deursen, E.J.; et al. Ccne1 Overexpression Causes Chromosome Instability in Liver Cells and Liver Tumor Development in Mice. Gastroenterology 2019, 157, 210–226.e212. [Google Scholar] [CrossRef]
- Nakayama, K.; Nagahama, H.; Minamishima, Y.A.; Miyake, S.; Ishida, N.; Hatakeyama, S.; Kitagawa, M.; Iemura, S.; Natsume, T.; Nakayama, K.I. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev. Cell 2004, 6, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [Green Version]
- Reed, S.E.; Spruck, C.H.; Sangfelt, O.; Van Drogen, F.; Mueller-Holzner, E.; Widschwendter, M.; Zetterberg, A.; Reed, S.I. Mutation of hCDC4 Leads to Cell Cycle Deregulation of Cyclin E in Cancer. Cancer Res. 2004, 64, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.; Aplin, A.E. Skp2 regulates G2/M progression in a p53-dependent manner. Mol. Biol. Cell 2008, 19, 4602–4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asmamaw, M.D.; Liu, Y.; Zheng, Y.C.; Shi, X.J.; Liu, H.M. Skp2 in the ubiquitin-proteasome system: A comprehensive review. Med. Res. Rev. 2020, 40, 1920–1949. [Google Scholar] [CrossRef]
- Cai, Z.; Moten, A.; Peng, D.; Hsu, C.C.; Pan, B.S.; Manne, R.; Li, H.Y.; Lin, H.K. The Skp2 Pathway: A Critical Target for Cancer Therapy. Semin. Cancer Biol. 2020, 67, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Hershko, D.D. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer 2008, 112, 1415–1424. [Google Scholar] [CrossRef]
- Jia, L.; Soengas, M.S.; Sun, Y. ROC1/RBX1 E3 ubiquitin ligase silencing suppresses tumor cell growth via sequential induction of G2-M arrest, apoptosis, and senescence. Cancer Res. 2009, 69, 4974–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbastabar, M.; Kheyrollah, M.; Azizian, K.; Bagherlou, N.; Tehrani, S.S.; Maniati, M.; Karimian, A. Multiple functions of p27 in cell cycle, apoptosis, epigenetic modification and transcriptional regulation for the control of cell growth: A double-edged sword protein. DNA Repair. 2018, 69, 63–72. [Google Scholar] [CrossRef]
- Bretones, G.; Acosta, J.C.; Caraballo, J.M.; Ferrandiz, N.; Gomez-Casares, M.T.; Albajar, M.; Blanco, R.; Ruiz, P.; Hung, W.C.; Albero, M.P.; et al. SKP2 oncogene is a direct MYC target gene and MYC down-regulates p27(KIP1) through SKP2 in human leukemia cells. J. Biol. Chem. 2011, 286, 9815–9825. [Google Scholar] [CrossRef] [Green Version]
- Shapira, M.; Ben-Izhak, O.; Linn, S.; Futerman, B.; Minkov, I.; Hershko, D.D. The prognostic impact of the ubiquitin ligase subunits Skp2 and Cks1 in colorectal carcinoma. Cancer 2005, 103, 1336–1346. [Google Scholar] [CrossRef]
- Latres, E.; Chiarle, R.; Schulman, B.A.; Pavletich, N.P.; Pellicer, A.; Inghirami, G.; Pagano, M. Role of the F-box protein Skp2 in lymphomagenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 2515–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Der Lehr, N.; Johansson, S.; Wu, S.; Bahram, F.; Castell, A.; Cetinkaya, C.; Hydbring, P.; Weidung, I.; Nakayama, K.; Nakayama, K.I.; et al. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol. Cell 2003, 11, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal. Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef]
- Roig, A.I.; Eskiocak, U.; Hight, S.K.; Kim, S.B.; Delgado, O.; Souza, R.F.; Spechler, S.J.; Wright, W.E.; Shay, J.W. Immortalized epithelial cells derived from human colon biopsies express stem cell markers and differentiate in vitro. Gastroenterology 2010, 138, 1012–1021.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kim, S.; Jia, G.; Buhmeida, A.; Dallol, A.; Wright, W.E.; Fornace, A.J.; Al-Qahtani, M.; Shay, J.W. Exome Sequencing of Normal and Isogenic Transformed Human Colonic Epithelial Cells (HCECs) Reveals Novel Genes Potentially Involved in the Early Stages of Colorectal Tumorigenesis. BMC Genom. 2015, 16 (Suppl. S1), S8. [Google Scholar] [CrossRef] [Green Version]
- Kolodner, R.D.; Hall, N.R.; Lipford, J.; Kane, M.F.; Morrison, P.T.; Finan, P.J.; Burn, J.; Chapman, P.; Earabino, C.; Merchant, E.; et al. Structure of the human MLH1 locus and analysis of a large hereditary nonpolyposis colorectal carcinoma kindred for mlh1 mutations. Cancer Res. 1995, 55, 242–248. [Google Scholar]
- Asbaghi, Y.; Thompson, L.L.; Lichtensztejn, Z.; McManus, K.J. KIF11 silencing and inhibition induces chromosome instability that may contribute to cancer. Genes Chromosomes Cancer 2017, 56, 668–680. [Google Scholar] [CrossRef]
- Guppy, B.J.; McManus, K.J. Mitotic accumulation of dimethylated lysine 79 of histone H3 is important for maintaining genome integrity during mitosis in human cells. Genetics 2015, 199, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Barber, T.D.; McManus, K.; Yuen, K.W.; Reis, M.; Parmigiani, G.; Shen, D.; Barrett, I.; Nouhi, Y.; Spencer, F.; Markowitz, S.; et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 3443–3448. [Google Scholar] [CrossRef] [Green Version]
- Cahill, D.P.; Lengauer, C.; Yu, J.; Riggins, G.J.; Willson, J.K.; Markowitz, S.D.; Kinzler, K.W.; Vogelstein, B. Mutations of mitotic checkpoint genes in human cancers. Nature 1998, 392, 300–303. [Google Scholar] [CrossRef]
- Rajagopalan, H.; Jallepalli, P.V.; Rago, C.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Inactivation of hCDC4 can cause chromosomal instability. Nature 2004, 428, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Sajesh, B.V.; Bailey, M.; Lichtensztejn, Z.; Hieter, P.; McManus, K.J. Synthetic lethal targeting of superoxide dismutase 1 selectively kills RAD54B-deficient colorectal cancer cells. Genetics 2013, 195, 757–767. [Google Scholar]
- Sajesh, B.V.; Lichtensztejn, Z.; McManus, K.J. Sister chromatid cohesion defects are associated with chromosome instability in Hodgkin lymphoma cells. BMC Cancer 2013, 13, 391. [Google Scholar] [CrossRef] [Green Version]
- Lepage, C.C.; Thompson, L.L.; Larson, B.; McManus, K.J. An Automated, Single Cell Quantitative Imaging Microscopy Approach to Assess Micronucleus Formation, Genotoxicity and Chromosome Instability. Cells 2020, 9, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. 2014, 92, e51998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, I.; Kotb, W.F.; Friedrich, K.H.; Schluns, K.; Bocking, A.; Dietel, M. Core classification of lung cancer: Correlating nuclear size and mitoses with ploidy and clinicopathological parameters. Lung Cancer 2009, 65, 312–318. [Google Scholar] [CrossRef]
- Zeimet, A.G.; Fiegl, H.; Goebel, G.; Kopp, F.; Allasia, C.; Reimer, D.; Steppan, I.; Mueller-Holzner, E.; Ehrlich, M.; Marth, C. DNA ploidy, nuclear size, proliferation index and DNA-hypomethylation in ovarian cancer. Gynecol. Oncol. 2011, 121, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e516. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.R.; Poirier, A.; Woods, R.R.; Ellison, L.F.; Billette, J.M.; Demers, A.A.; Zhang, S.X.; Yao, C.; Finley, C.; Fitzgerald, N.; et al. Projected estimates of cancer in Canada in 2022. Cmaj 2022, 194, E601–E607. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L. Microscopy-Based High-Content Analyses Identify Novel Chromosome Instability Genes including SKP1. 2018. Available online: https://mspace.lib.umanitoba.ca/handle/1993/33282 (accessed on 1 April 2022).
- Bhatia, A.; Kumar, Y. Cancer cell micronucleus: An update on clinical and diagnostic applications. APMIS 2013, 121, 569–581. [Google Scholar] [CrossRef]
- Ye, C.J.; Sharpe, Z.; Alemara, S.; Mackenzie, S.; Liu, G.; Abdallah, B.; Horne, S.; Regan, S.; Heng, H.H. Micronuclei and Genome Chaos: Changing the System Inheritance. Genes 2019, 10, 366. [Google Scholar] [CrossRef]
- The Human Protein Atlas. SKP2 Protein Information. Available online: https://www.proteinatlas.org/ENSG00000145604-SKP2 (accessed on 1 April 2022).
- UniprotKB. SKP2: Family & Domains. Available online: https://www.uniprot.org/uniprot/Q13309 (accessed on 1 April 2022).
- Edgar, B.A.; Orr-Weaver, T.L. Endoreplication cell cycles: More for less. Cell 2001, 105, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Gascoyne, D.M.; Hixon, M.L.; Gualberto, A.; Vivanco, M.D. Loss of mitotic spindle checkpoint activity predisposes to chromosomal instability at early stages of fibrosarcoma development. Cell Cycle 2003, 2, 238–245. [Google Scholar] [CrossRef] [Green Version]
- Olaharski, A.J.; Sotelo, R.; Solorza-Luna, G.; Gonsebatt, M.E.; Guzman, P.; Mohar, A.; Eastmond, D.A. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis 2006, 27, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Mullany, L.K.; Wong, K.K.; Marciano, D.C.; Katsonis, P.; King-Crane, E.R.; Ren, Y.A.; Lichtarge, O.; Richards, J.S. Specific TP53 Mutants Overrepresented in Ovarian Cancer Impact CNV, TP53 Activity, Responses to Nutlin-3a, and Cell Survival. Neoplasia 2015, 17, 789–803. [Google Scholar] [CrossRef] [Green Version]
- Shu, Z.; Row, S.; Deng, W.M. Endoreplication: The Good, the Bad, and the Ugly. Trends Cell Biol. 2018, 28, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Dulić, V.; Lees, E.; Reed, S.I. Association of human cyclin E with a periodic G1-S phase protein kinase. Science 1992, 257, 1958–1961. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, M.; Theodoras, A.M.; Schumacher, J.; Roberts, J.M.; Pagano, M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol. Cell Biol. 1995, 15, 2612–2624. [Google Scholar] [CrossRef] [Green Version]
- Hall, L.L.; Th’ng, J.P.; Guo, X.W.; Teplitz, R.L.; Bradbury, E.M. A brief staurosporine treatment of mitotic cells triggers premature exit from mitosis and polyploid cell formation. Cancer Res. 1996, 56, 3551–3559. [Google Scholar] [PubMed]
- Storchova, Z.; Pellman, D. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 2004, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; de Lange, T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef]
- Muleris, M.; Salmon, R.J.; Dutrillaux, B. Cytogenetics of colorectal adenocarcinomas. Cancer Genet. Cytogenet. 1990, 46, 143–156. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, D.; Yang, Z.; Zhang, X. Tumor Budding, Micropapillary Pattern, and Polyploidy Giant Cancer Cells in Colorectal Cancer: Current Status and Future Prospects. Stem Cells Int. 2016, 2016, 4810734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, Z.A.; Mays, D.; Pietenpol, J.A. Defective G1-S cell cycle checkpoint function sensitizes cells to microtubule inhibitor-induced apoptosis. Cancer Res. 1999, 59, 3831–3837. [Google Scholar] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Fodde, R.; Smits, R.; Clevers, H. APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 2001, 1, 55–67. [Google Scholar] [CrossRef]
- Leslie, A.; Carey, F.A.; Pratt, N.R.; Steele, R.J. The colorectal adenoma-carcinoma sequence. Br. J. Surg. 2002, 89, 845–860. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic Alterations during Colorectal tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, K.B.; Burds, A.A.; Swedlow, J.R.; Bekir, S.S.; Sorger, P.K.; Näthke, I.S. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat. Cell Biol. 2001, 3, 429–432. [Google Scholar] [CrossRef]
- Zumbrunn, J.; Kinoshita, K.; Hyman, A.A.; Näthke, I.S. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Curr. Biol. 2001, 11, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Chan, C.H.; Li, C.F.; Yang, W.L.; Gao, Y.; Lee, S.W.; Feng, Z.; Huang, H.Y.; Tsai, K.K.; Flores, L.G.; Shao, Y.; et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 2012, 149, 1098–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penin, R.M.; Fernandez-Figueras, M.T.; Puig, L.; Rex, J.; Ferrandiz, C.; Ariza, A. Over-expression of p45(SKP2) in Kaposi’s sarcoma correlates with higher tumor stage and extracutaneous involvement but is not directly related to p27(KIP1) down-regulation. Mod. Pathol. 2002, 15, 1227–1235. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Murphy, M.; Ross, J.; Sheehan, C.; Carlson, J.A. Skp2 and p27kip1 expression in melanocytic nevi and melanoma: An inverse relationship. J. Cutan. Pathol. 2004, 31, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Philipp-Staheli, J.; Payne, S.R.; Kemp, C.J. p27(Kip1): Regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp. Cell Res. 2001, 264, 148–168. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neudorf, N.M.; Thompson, L.L.; Lichtensztejn, Z.; Razi, T.; McManus, K.J. Reduced SKP2 Expression Adversely Impacts Genome Stability and Promotes Cellular Transformation in Colonic Epithelial Cells. Cells 2022, 11, 3731. https://doi.org/10.3390/cells11233731
Neudorf NM, Thompson LL, Lichtensztejn Z, Razi T, McManus KJ. Reduced SKP2 Expression Adversely Impacts Genome Stability and Promotes Cellular Transformation in Colonic Epithelial Cells. Cells. 2022; 11(23):3731. https://doi.org/10.3390/cells11233731
Chicago/Turabian StyleNeudorf, Nicole M., Laura L. Thompson, Zelda Lichtensztejn, Tooba Razi, and Kirk J. McManus. 2022. "Reduced SKP2 Expression Adversely Impacts Genome Stability and Promotes Cellular Transformation in Colonic Epithelial Cells" Cells 11, no. 23: 3731. https://doi.org/10.3390/cells11233731