

Arginine Reduces Glycation in γ2 Subunit of AMPK and Pathologies in Alzheimer’s Disease Model Mice

Abstract
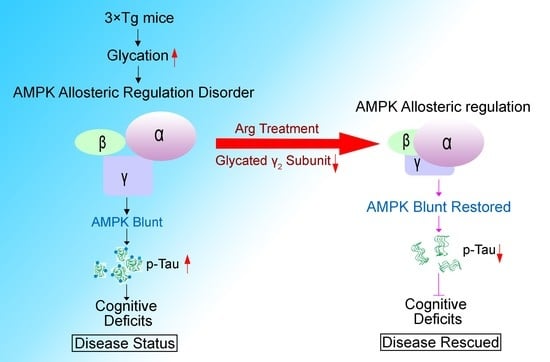
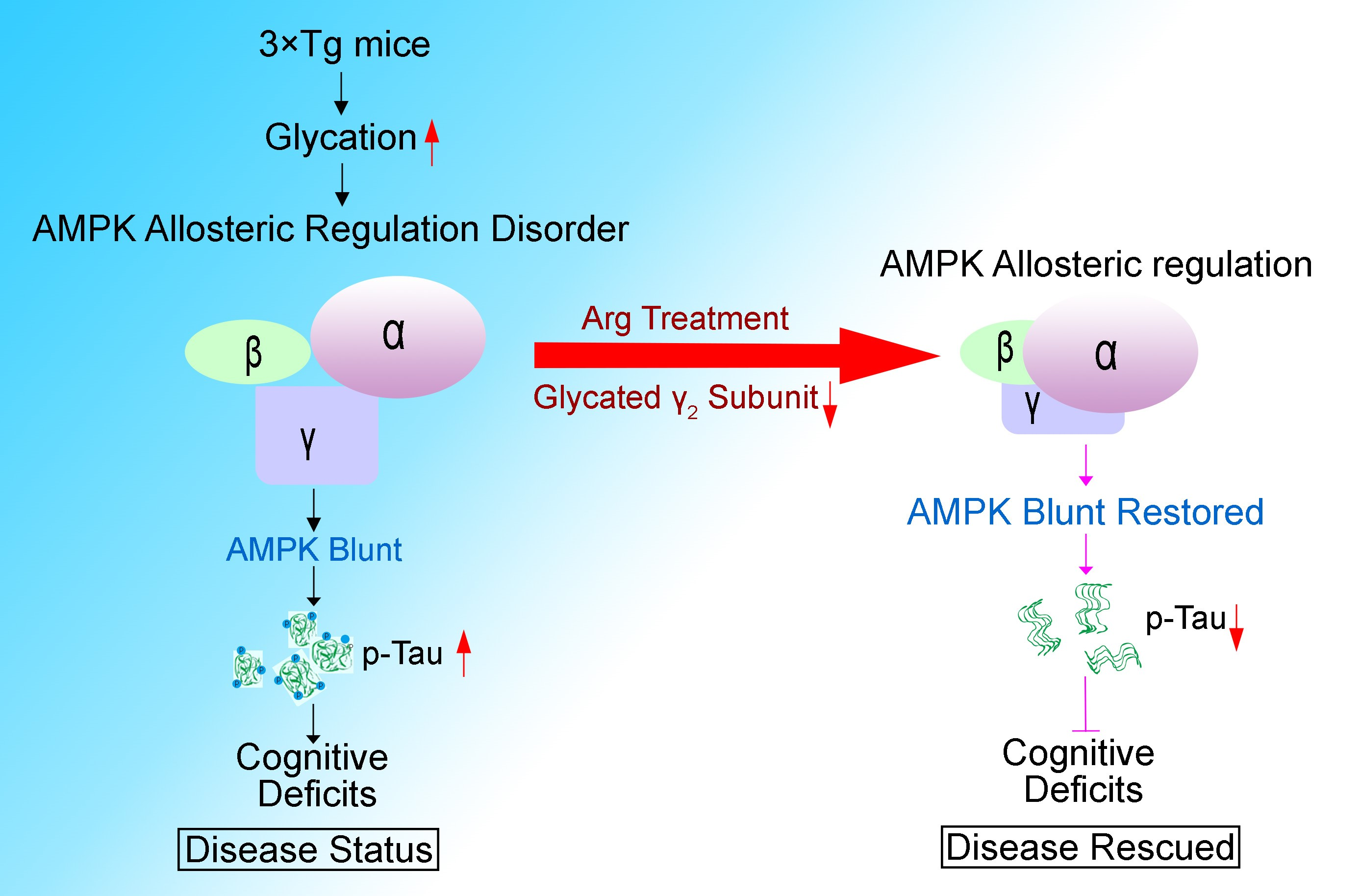
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibody and Drugs
2.2. Tube Experiment
2.3. Cell Culture and Treatment
2.4. Animals and Treatment
2.5. Morris Water Maze
2.6. Novel Object Location
2.7. Western Blot and Dot Blot
2.8. Immunoprecipitation
2.9. Statistical Analysis
3. Results
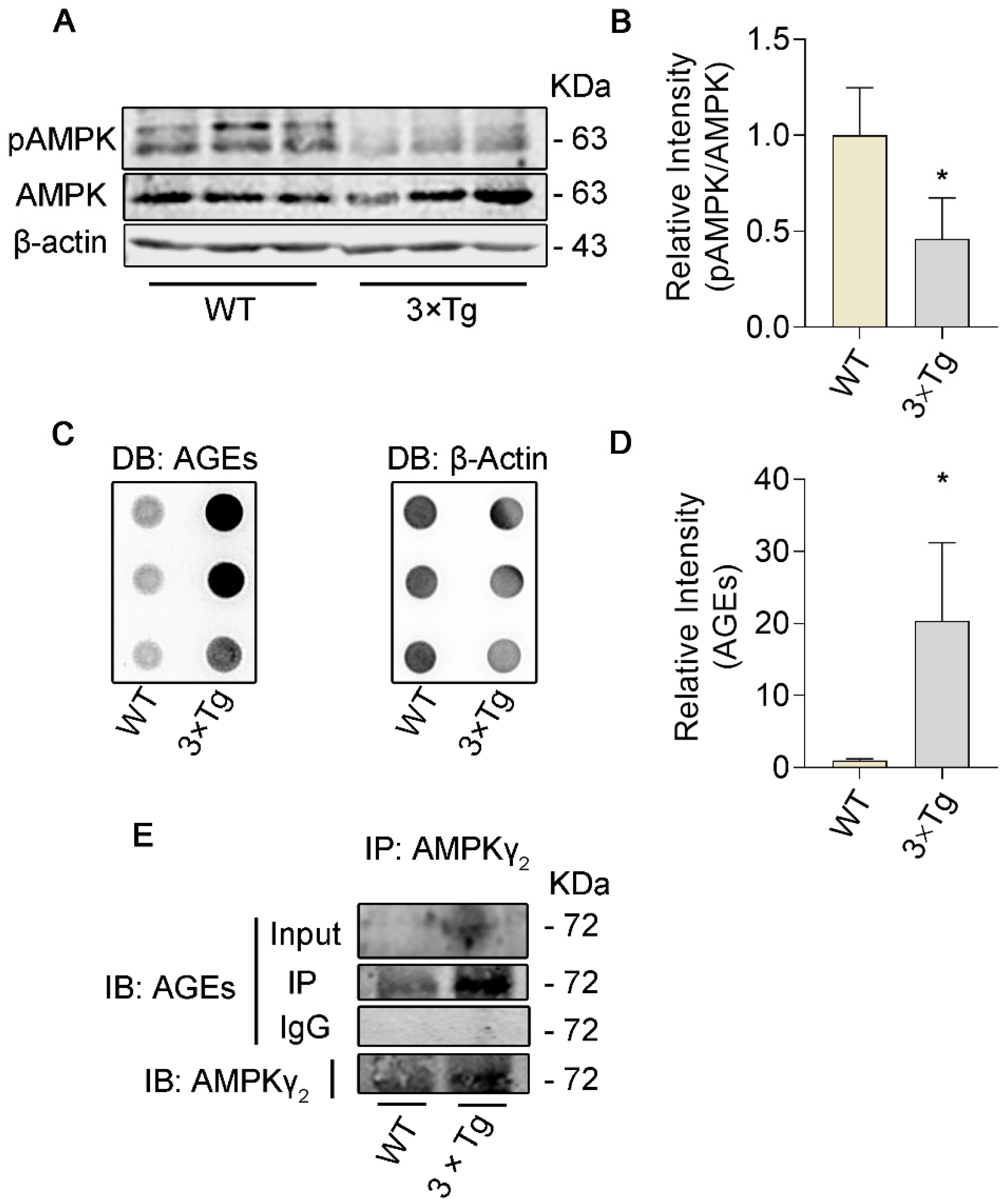
3.1. The Level of Glycation in γ2 Subunit of AMPK Increased and the Level of pT172−AMPK Decreased in 3 × Tg Mice
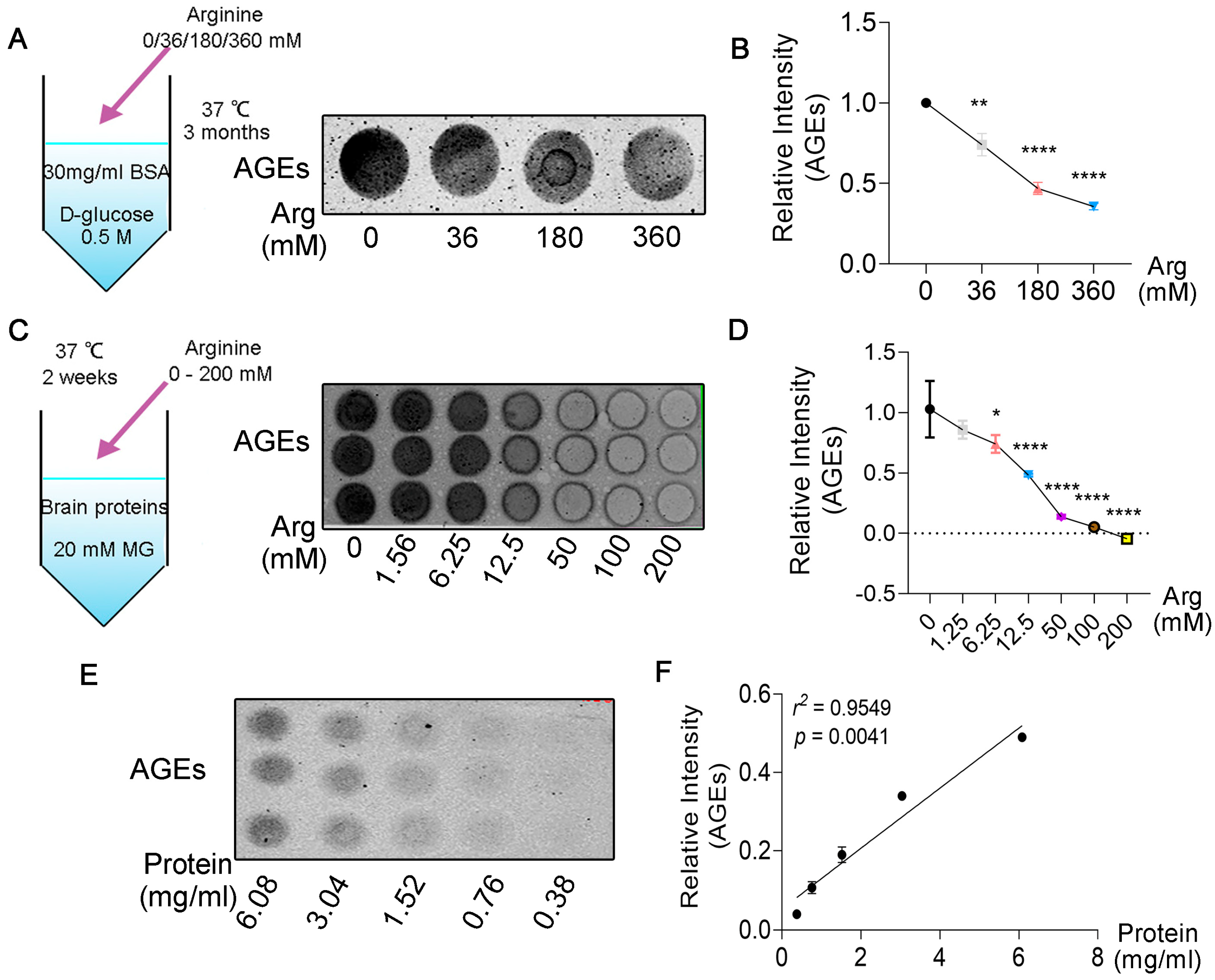
3.2. Arginine Decreases the Glycation of Proteins in a Dose−Dependent Manner
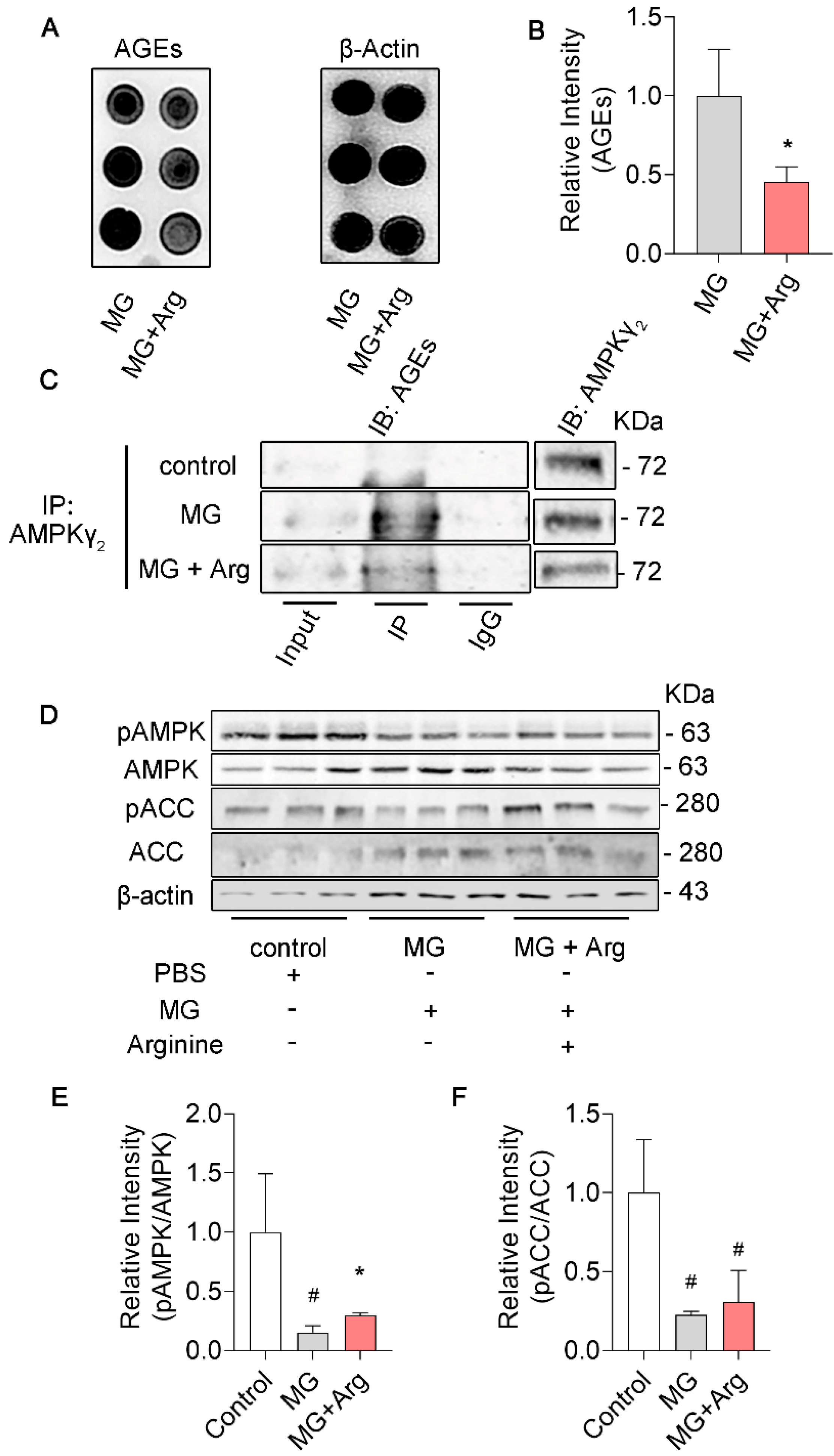
3.3. Arginine Mitigates Glycation in γ2 Subunit of AMPK Induced by MG and Maintains the AMPK Function in N2a Cells
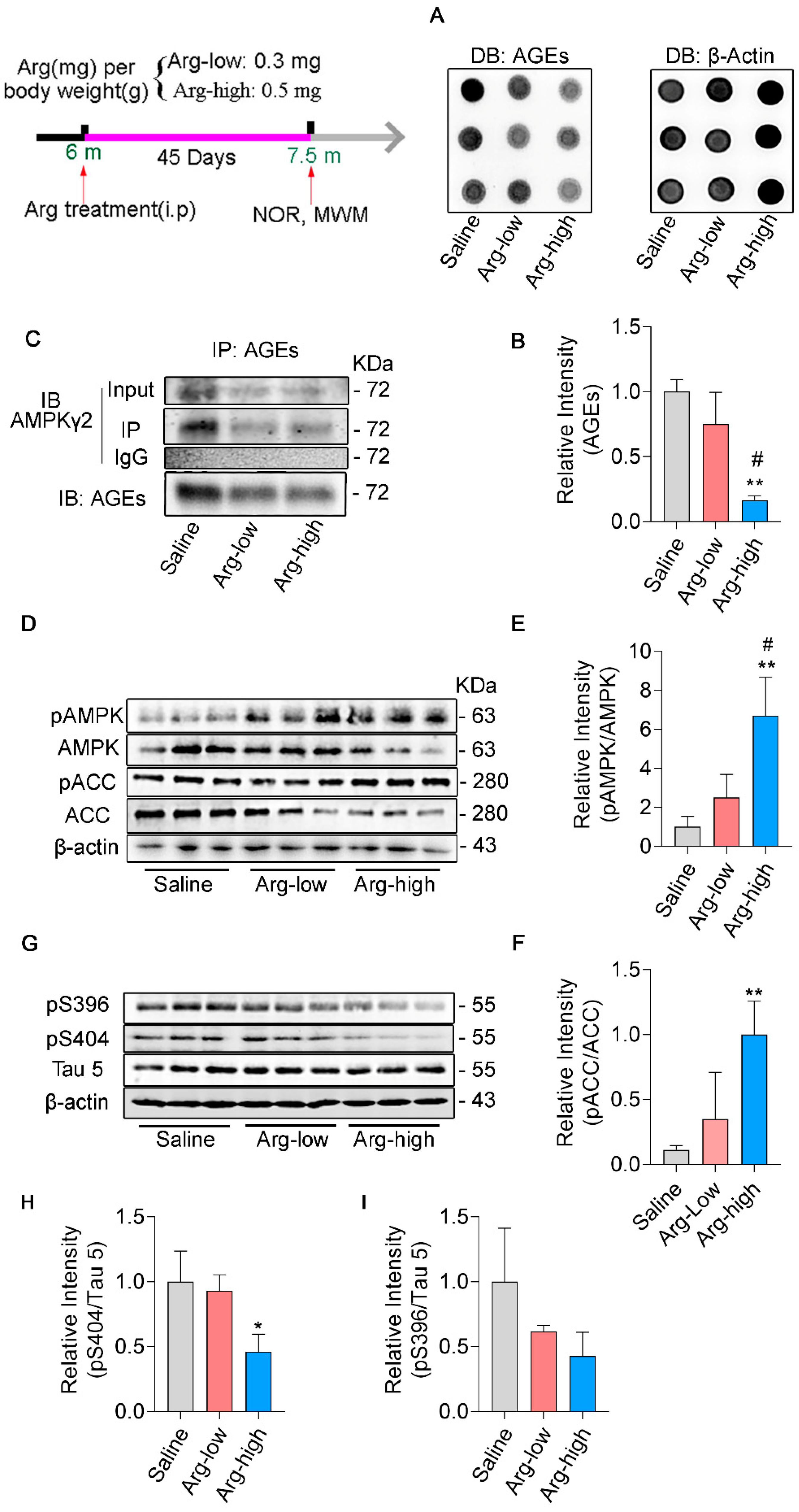
3.4. Arginine Defends γ2 Subunit of AMPK against Its Glycation and Improves AMPK Function in 3 × Tg Mice
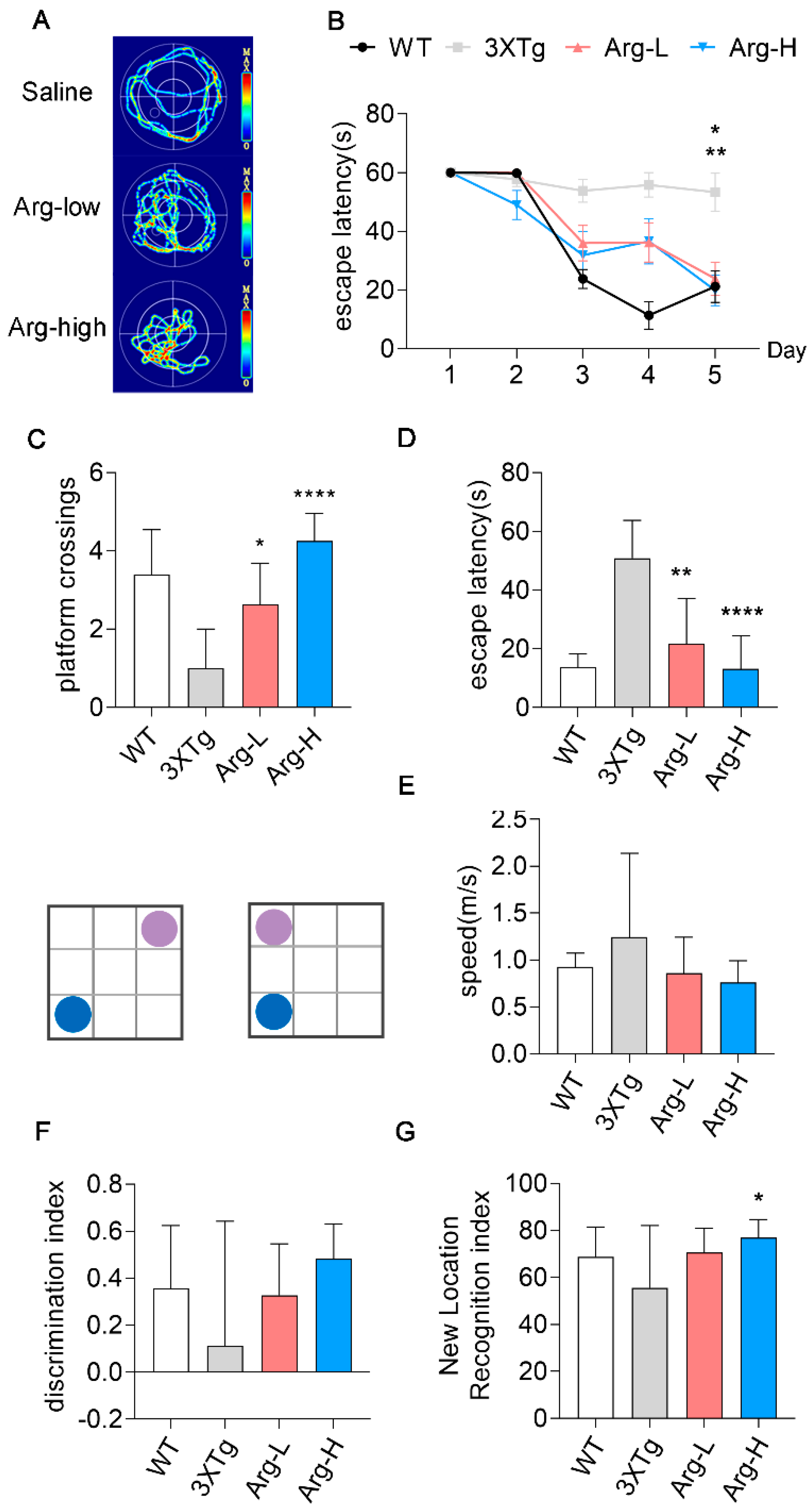
3.5. Arginine Attenuates the Impairments of Hippocampal−Dependent Spatial Learning in Memory in 3 × Tg Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guerrero, A.; De Strooper, B.; Arancibia-Cárcamo, I.L. Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci. 2021, 44, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Van Der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 2019, 21, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Kodamullil, A.T.; Hofmann-Apitius, M. Comorbidity Analysis between Alzheimer’s Disease and Type 2 Diabetes Mellitus (T2DM) Based on Shared Pathways and the Role of T2DM Drugs. J. Alzheimer Dis. 2017, 60, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [Green Version]
- Jash, K.; Gondaliya, P.; Kirave, P.; Kulkarni, B.; Sunkaria, A.; Kalia, K. Cognitive dysfunction: A growing link between diabetes and Alzheimer’s disease. Drug Dev. Res. 2019, 81, 144–164. [Google Scholar] [CrossRef]
- Szczepanik, J.C.; de Almeida, G.R.L.; Cunha, M.P.; Dafre, A.L. Repeated Methylglyoxal Treatment Depletes Dopamine in the Prefrontal Cortex, and Causes Memory Impairment and Depressive-Like Behavior in Mice. Neurochem. Res. 2019, 45, 354–370. [Google Scholar] [CrossRef]
- Lotan, R.; Ganmore, I.; Livny, A.; Shelly, S.; Zacharia, M.; Uribarri, J.; Beisswenger, P.; Cai, W.; Beeri, M.S.; Troen, A.M. Design and Feasibility of a Randomized Controlled Pilot Trial to Reduce Exposure and Cognitive Risk Associated With Advanced Glycation End Products in Older Adults With Type 2 Diabetes. Front. Nutr. 2021, 8, 614149. [Google Scholar] [CrossRef]
- Kimura, T.; Takamatsu, J.; Araki, N.; Goto, M.; Kondo, A.; Miyakawa, T.; Horiuchi, S. Are advanced glycation end-products associated with amyloidosis in Alzheimer’s disease? Neuroreport 1995, 6, 866–868. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, J.; Wang, Z.; Huang, W.; Yang, Y.; Cai, Z.; Li, K. Role of the Glyoxalase System in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 66, 887–899. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Dite, T.A.; Ling, N.; Scott, J.; Hoque, A.; Galic, S.; Parker, B.L.; Ngoei, K.R.W.; Langendorf, C.; O’Brien, M.T.; Kundu, M.; et al. The autophagy initiator ULK1 sensitizes AMPK to allosteric drugs. Nat. Commun. 2017, 8, 571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Mukherjee, S.; Harikumar, K.G.; Strutzenberg, T.S.; Zhou, X.E.; Suino-Powell, K.; Xu, T.-H.; Sheldon, R.D.; Lamp, J.; Brunzelle, J.S.; et al. Structure of an AMPK complex in an inactive, ATP-bound state. Science 2021, 373, 413–419. [Google Scholar] [CrossRef]
- Yan, Y.; Zhou, X.E.; Xu, H.E.; Melcher, K. Structure and Physiological Regulation of AMPK. Int. J. Mol. Sci. 2018, 19, 3534. [Google Scholar] [CrossRef] [Green Version]
- Cheung, P.C.F.; Salt, I.; Davies, S.P.; Hardie, G.; Carling, D. Characterization of AMP-activated protein kinase γ-subunit isoforms and their role in AMP binding. Biochem. J. 2000, 346, 659–669. [Google Scholar] [CrossRef]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end-products in age-related diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Zambonin, L.; Hrelia, S. Role of Methylglyoxal in Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 238485. [Google Scholar] [CrossRef] [Green Version]
- Thornalley, P.J.; Battah, S.; Ahmed, N.; Karachalias, N.; Agalou, S.; Babaei-Jadidi, R.; Dawnay, A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 2003, 375, 581–592. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Santos, A.N.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef]
- Bulteau, A.-L.; Verbeke, P.; Petropoulos, I.; Chaffotte, A.-F.; Friguet, B. Proteasome Inhibition in Glyoxal-treated Fibroblasts and Resistance of Glycated Glucose-6-phosphate Dehydrogenase to 20 S Proteasome Degradation in Vitro. J. Biol. Chem. 2001, 276, 45662–45668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, R.A.; Oliveira, L.M.A.; Silva, M.; Ascenso, C.; Quintas, A.; Costa, G.; Coelho, A.V.; Silva, M.S.; Ferreira, A.E.N.; Freire, A.P.; et al. Protein glycation in vivo: Functional and structural effects on yeast enolase. Biochem. J. 2008, 416, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Li, T.; Ji, T.; Yi, W.; Yang, Z.; Wang, S.; Yang, Y.; Gu, C. AMPK: Potential Therapeutic Target for Ischemic Stroke. Theranostics 2018, 8, 4535–4551. [Google Scholar] [CrossRef] [PubMed]
- Demaré, S.; Kothari, A.; Calcutt, N.A.; Fernyhough, P. Metformin as a potential therapeutic for neurological disease: Mobilizing AMPK to repair the nervous system. Expert Rev. Neurother. 2020, 21, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, C.; Zhou, H.; Feng, Y.; Tang, F.; Hoi, M.P.M.; He, C.; Ma, D.; Zhao, C.; Lee, S.M.Y. Inhibitory Effects of Betulinic Acid on LPS-Induced Neuroinflammation Involve M2 Microglial Polarization via CaMKKβ-Dependent AMPK Activation. Front. Mol. Neurosci. 2018, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Dineley, K.T.; Jahrling, J.B.; Denner, L. Insulin resistance in Alzheimer’s disease. Neurobiol. Dis. 2014, 72, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Huang, N.; Liu, J.; Huang, J.; Shi, J.; Jin, F. AMPK: A bridge between diabetes mellitus and Alzheimer’s disease. Behav. Brain Res. 2021, 400, 113043. [Google Scholar] [CrossRef]
- Camacho-Castillo, L.; Phillips-Farfán, B.V.; Rosas-Mendoza, G.; Baires-López, A.; Toral-Ríos, D.; Campos-Peña, V.; Carvajal, K. Increased oxidative stress contributes to enhance brain amyloidogenesis and blunts energy metabolism in sucrose-fed rat: Effect of AMPK activation. Sci. Rep. 2021, 11, 19547. [Google Scholar] [CrossRef]
- Wani, A.; Al Rihani, S.B.; Sharma, A.; Weadick, B.; Govindarajan, R.; Khan, S.U.; Sharma, P.R.; Dogra, A.; Nandi, U.; Reddy, C.N.; et al. Crocetin promotes clearance of amyloid-β by inducing autophagy via the STK11/LKB1-mediated AMPK pathway. Autophagy 2021, 17, 3813–3832. [Google Scholar] [CrossRef]
- Yang, T.-T.; Shih, Y.-S.; Chen, Y.-W.; Kuo, Y.-M.; Lee, C.-W. Glucose regulates amyloid β production via AMPK. J. Neural Transm. 2015, 122, 1381–1390. [Google Scholar] [CrossRef]
- Kim, B.; Figueroa-Romero, C.; Pacut, C.; Backus, C.; Feldman, E.L. Insulin Resistance Prevents AMPK-induced Tau Dephosphorylation through Akt-mediated Increase in AMPKSer-485 Phosphorylation. J. Biol. Chem. 2015, 290, 19146–19157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peixoto, C.A.; de Oliveira, W.H.; Araújo, S.M.D.R.; Nunes, A.K.S. AMPK activation: Role in the signaling pathways of neuroinflammation and neurodegeneration. Exp. Neurol. 2017, 298, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Yoon, G.H.; Chung, S.S.; Abid, M.N.; Kim, T.H.; Lee, H.Y.; Kim, M.O. Novel osmotin inhibits SREBP2 via the AdipoR1/AMPK/SIRT1 pathway to improve Alzheimer’s disease neuropathological deficits. Mol. Psychiatry 2017, 22, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.; Sadeghnia, H.R.; Salehabadi, S.; Alavi, H.; Gorji, A. The effect of l-arginine and l-NAME on pentylenetetrazole induced seizures in ovariectomized rats, an in vivo study. Seizure 2009, 18, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Tisljar, M.; Grabarević, Z.; Artuković, B.; Dzaja, P.; Cenan, S.; Zelenika, T.A.; Cooper, R.G.; Dinarina-Sablić, M. The impact of L-NAME and L-arginine chronic toxicity induced lesions on ascites-pulmonary hypertension syndrome development in broiler chickens. Coll. Antropol. 2011, 35, 547–556. [Google Scholar] [PubMed]
- Mitani, Y.; Maruyama, K.; Sakurai, M. Prolonged administration of l-arginine ameliorates chronic pulmonary hyper-tension and pulmonary vascular remodeling in rats. Circulation 1997, 96, 689–697. [Google Scholar] [CrossRef]
- Lueptow, L.M. Novel Object Recognition Test for the Investigation of Learning and Memory in Mice. J. Vis. Exp. 2017, 2017, e55718. [Google Scholar] [CrossRef]
- Zhou, X.-W.; Li, X.; Bjorkdahl, C.; Sjogren, M.J.; Alafuzoff, I.; Soininen, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Pei, J.-J. Assessments of the accumulation severities of amyloid β-protein and hyperphosphorylated tau in the medial temporal cortex of control and Alzheimer’s brains. Neurobiol. Dis. 2006, 22, 657–668. [Google Scholar] [CrossRef]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef]
- Krautwald, M.; Münch, G. Advanced glycation end products as biomarkers and gerontotoxins—A basis to explore methylglyoxal-lowering agents for Alzheimer’s disease? Exp. Gerontol. 2010, 45, 744–751. [Google Scholar] [CrossRef]
- Mirmiranpour, H.; Khaghani, S.; Bathaie, S.Z.; Nakhjavani, M.; Kebriaeezadeh, A.; Ebadi, M.; Gerayesh-Nejad, S.; Zangooei, M. The Preventive Effect of L-Lysine on Lysozyme Glycation in Type 2 Diabetes. Acta Med. Iran. 2016, 54, 24–31. [Google Scholar] [PubMed]
- Berggreen, C.; Gormand, A.; Omar, B.; Degerman, E.; Göransson, O. Protein kinase B activity is required for the effects of insulin on lipid metabolism in adipocytes. Am. J. Physiol. Metab. 2009, 296, E635–E646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhun, B.S.; Jin, Q.; Oh, Y.T.; Kim, S.S.; Kong, Y.; Cho, Y.H.; Ha, J.; Baik, H.H.; Kang, I. 5-Aminoimidazole-4-carboxamide riboside suppresses lipopolysaccharide-induced TNF-α production through inhibition of phosphatidylinositol 3-kinase/Akt activation in RAW 264.7 murine macrophages. Biochem. Biophys. Res. Commun. 2004, 318, 372–380. [Google Scholar] [CrossRef]
- Hardie, D.G.; Carling, D.; Carlson, M. The Amp-Activated/Snf1 Protein Kinase Subfamily: Metabolic Sensors of the Eukaryotic Cell? Annu. Rev. Biochem. 1998, 67, 821–855. [Google Scholar] [CrossRef] [PubMed]
- Zemla, R.; Basu, J. Hippocampal function in rodents. Curr. Opin. Neurobiol. 2017, 43, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Rocha, M.; Wang, D.; Avila-Quintero, V.; Bloch, M.H.; Kaffman, A. Deficits in hippocampal-dependent memory across different rodent models of early life stress: Systematic review and meta-analysis. Transl. Psychiatry 2021, 11, 231. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Liu, B.; Xu, W.; Wang, L.; Shi, F.; Li, N.; Lei, Y.; Wang, J.; Tian, Q.; Zhou, X. Inhibition of mTORC1 improves STZ-induced AD-like impairments in mice. Brain Res. Bull. 2020, 162, 166–179. [Google Scholar] [CrossRef]
- Wang, L.; Li, N.; Shi, F.-X.; Xu, W.-Q.; Cao, Y.; Lei, Y.; Wang, J.-Z.; Tian, Q.; Zhou, X.-W. Upregulation of AMPK Ameliorates Alzheimer’s Disease-Like Tau Pathology and Memory Impairment. Mol. Neurobiol. 2020, 57, 3349–3361. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Perluigi, M.; Butterfield, D.A. The interplay among oxidative stress, brain insulin resistance and AMPK dysfunction contribute to neurodegeneration in type 2 diabetes and Alzheimer disease. Free Radic. Biol. Med. 2021, 176, 16–33. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef] [Green Version]
- Vingtdeux, V.; Davies, P.; Dickson, D.W.; Marambaud, P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 2011, 121, 337–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Soininen, H.; Hiltunen, M. AMP-activated protein kinase: A potential player in Alzheimer’s disease. J. Neurochem. 2011, 118, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.-Y.; Cooper, M.E. The Role of Advanced Glycation End Products in Progression and Complications of Diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Rahmadi, A.; Steiner, N.; Münch, G. Advanced glycation endproducts as gerontotoxins and biomarkers for carbonyl-based degenerative processes in Alzheimer’s disease. Clin. Chem. Lab. Med. (CCLM) 2011, 49, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.K.; Nandi, S.K.; Chakraborty, A.; Nagaraj, R.H.; Biswas, A. Differential role of arginine mutations on the structure and functions of α-crystallin. Biochim. Biophys. Acta (BBA) Gen. Subj. 2016, 1860, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, K.C.B.; Shiu, S.W.M.; Wong, Y.; Tam, X. Serum advanced glycation end products (AGEs) are associated with insulin resistance. Diabetes/Metab. Res. Rev. 2011, 27, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Ottum, M.S.; Mistry, A.M. Advanced glycation end-products: Modifiable environmental factors profoundly mediate insulin resistance. J. Clin. Biochem. Nutr. 2015, 57, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Unoki-Kubota, H.; Yamagishi, S.-I. Advanced Glycation End Products and Insulin Resistance. Curr. Pharm. Des. 2008, 14, 987–989. [Google Scholar] [CrossRef]
- Shrikanth, C.; Nandini, C. AMPK in microvascular complications of diabetes and the beneficial effects of AMPK activators from plants. Phytomedicine 2020, 73, 152808. [Google Scholar] [CrossRef]
- Joshi, T.; Singh, A.K.; Haratipour, P.; Sah, A.N.; Pandey, A.K.; Naseri, R.; Juyal, V.; Farzaei, M.H. Targeting AMPK signaling pathway by natural products for treatment of diabetes mellitus and its complications. J. Cell. Physiol. 2019, 234, 17212–17231. [Google Scholar] [CrossRef]
- Jiang, B.; Le, L.; Liu, H.; Xu, L.; He, C.; Hu, K.; Peng, Y.; Xiao, P. Marein protects against methylglyoxal-induced apoptosis by activating the AMPK pathway in PC12 cells. Free Radic. Res. 2016, 50, 1173–1187. [Google Scholar] [CrossRef] [PubMed]
- Matos, A.L.D.S.A.; Oakhill, J.S.; Moreira, J.; Loh, K.; Galic, S.; Scott, J.W. Allosteric regulation of AMP-activated protein kinase by adenylate nucleotides and small-molecule drugs. Biochem. Soc. Trans. 2019, 47, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Chen, Z.; Van Denderen, B.J.; Morton, C.J.; Parker, M.W.; Witters, L.A.; Stapleton, D.; Kemp, B.E. Intrasteric control of AMPK via the 1 subunit AMP allosteric regulatory site. Protein Sci. 2004, 13, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Chen, Y.Y. Ampk and autophagy. In Autophagy: Biology and Diseases: Basic Science; Qin, Z.H., Ed.; Advances in Experimental Medicine and Biology 1206; Springer-Singapore Pte Ltd.: Singapore, 2019; pp. 85–108. [Google Scholar]
- Yu, E.; Ruiz-Canela, M.; Razquin, C.; Guasch-Ferré, M.; Toledo, E.; Wang, D.D.; Papandreou, C.; Dennis, C.; Clish, C.; Liang, L.; et al. Changes in arginine are inversely associated with type 2 diabetes: A case-cohort study in the PREDIMED trial. Diabetes Obes. Metab. 2019, 21, 397–401. [Google Scholar] [CrossRef]
- Yi, J.; Horky, L.L.; Friedlich, A.L.; Shi, Y.; Rogers, J.T.; Huang, X. L-arginine and alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2009, 2, 211–238. [Google Scholar]
- Kan, M.J.; Lee, J.E.; Wilson, J.G.; Everhart, A.L.; Brown, C.M.; Hoofnagle, A.N.; Jansen, M.; Vitek, M.P.; Gunn, M.D.; Colton, C.A. Arginine Deprivation and Immune Suppression in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 5969–5982. [Google Scholar] [CrossRef] [Green Version]
- Gensert, J.M.; Ratan, R.R. The Metabolic Coupling of Arginine Metabolism to Nitric Oxide Generation by Astrocytes. Antioxid. Redox Signal. 2006, 8, 919–928. [Google Scholar] [CrossRef]
- Volz, T.J.; Schenk, J.O. L-arginine increases dopamine transporter activity in rat striatum via a nitric oxide synthase-dependent mechanism. Synapse 2004, 54, 173–182. [Google Scholar] [CrossRef]
- Hirata, K.; Akita, Y.; Povalko, N.; Nishioka, J.; Yatsuga, S.; Matsuishi, T.; Koga, Y. Effect of l-arginine on synaptosomal mitochondrial function. Brain Dev. 2008, 30, 238–245. [Google Scholar] [CrossRef]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.-J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef]
- Mahali, S.K.; Verma, N.; Manna, S.K. Advanced Glycation End Products Induce Lipogenesis: Regulation by Natural Xanthone through Inhibition of ERK and NF-κB. J. Cell. Physiol. 2014, 229, 1972–1980. [Google Scholar] [CrossRef] [PubMed]
- Mengstie, M.A.; Abebe, E.C.; Teklemariam, A.B.; Mulu, A.T.; Agidew, M.M.; Azezew, M.T.; Zewde, E.A.; Teshome, A.A. Endogenous advanced glycation end products in the pathogenesis of chronic diabetic complications. Front. Mol. Biosci. 2022, 9, 1002710. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, R.; Lei, Y.; Shi, F.; Tian, Q.; Zhou, X. Arginine Reduces Glycation in γ2 Subunit of AMPK and Pathologies in Alzheimer’s Disease Model Mice. Cells 2022, 11, 3520. https://doi.org/10.3390/cells11213520
Zhu R, Lei Y, Shi F, Tian Q, Zhou X. Arginine Reduces Glycation in γ2 Subunit of AMPK and Pathologies in Alzheimer’s Disease Model Mice. Cells. 2022; 11(21):3520. https://doi.org/10.3390/cells11213520
Chicago/Turabian StyleZhu, Rui, Ying Lei, Fangxiao Shi, Qing Tian, and Xinwen Zhou. 2022. "Arginine Reduces Glycation in γ2 Subunit of AMPK and Pathologies in Alzheimer’s Disease Model Mice" Cells 11, no. 21: 3520. https://doi.org/10.3390/cells11213520