
PI3K Isoform-Specific Regulation of Leader and Follower Cell Function for Collective Migration and Proliferation in Response to Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Model and Treatments
2.2. Phase Microscopy and Wound Healing
2.3. Immunofluorescence, EdU-Labeling, and Confocal Microscopy
2.4. Western Blot and Wes Protein Analysis
2.5. Statistical Analysis
3. Results
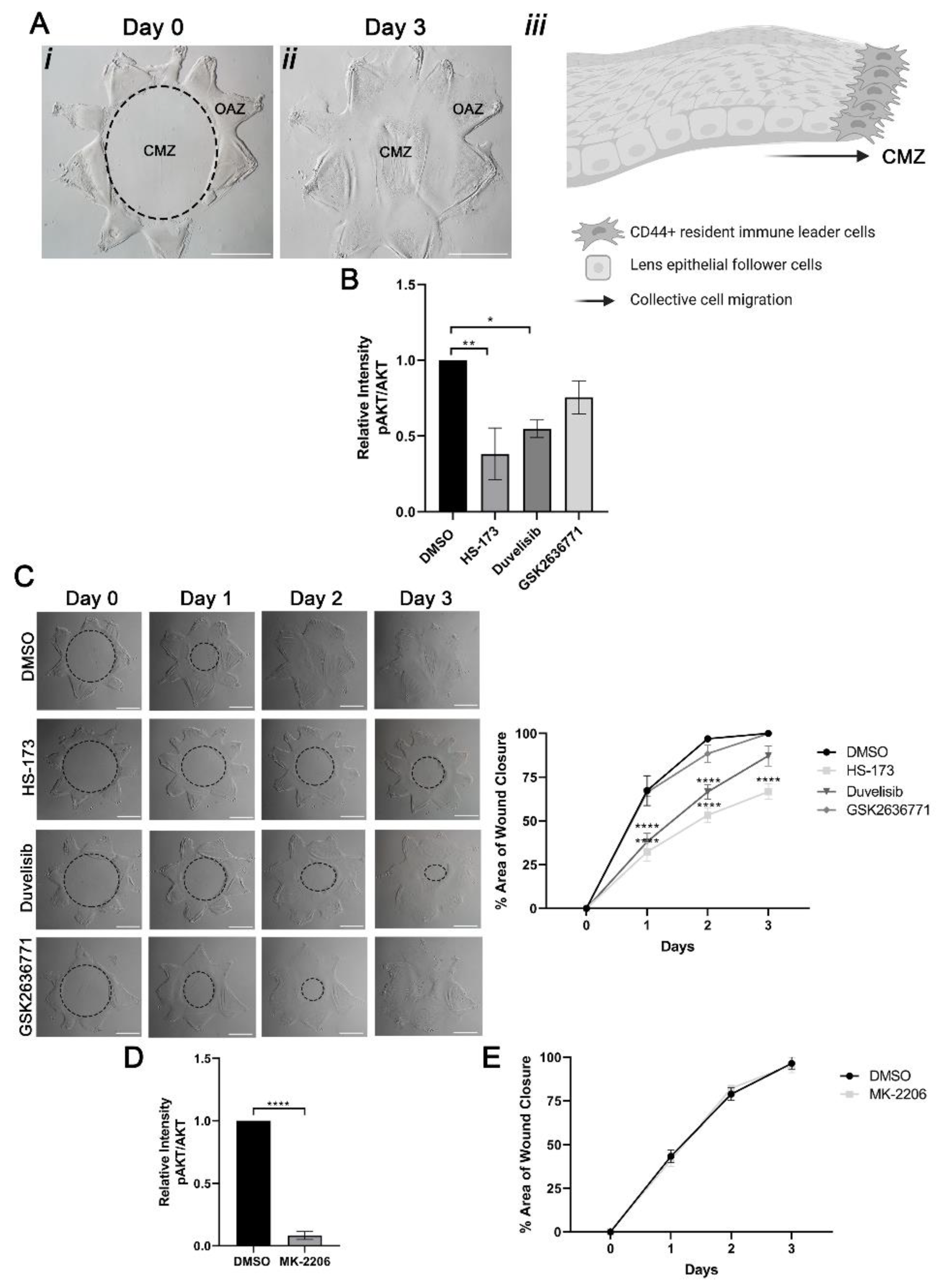
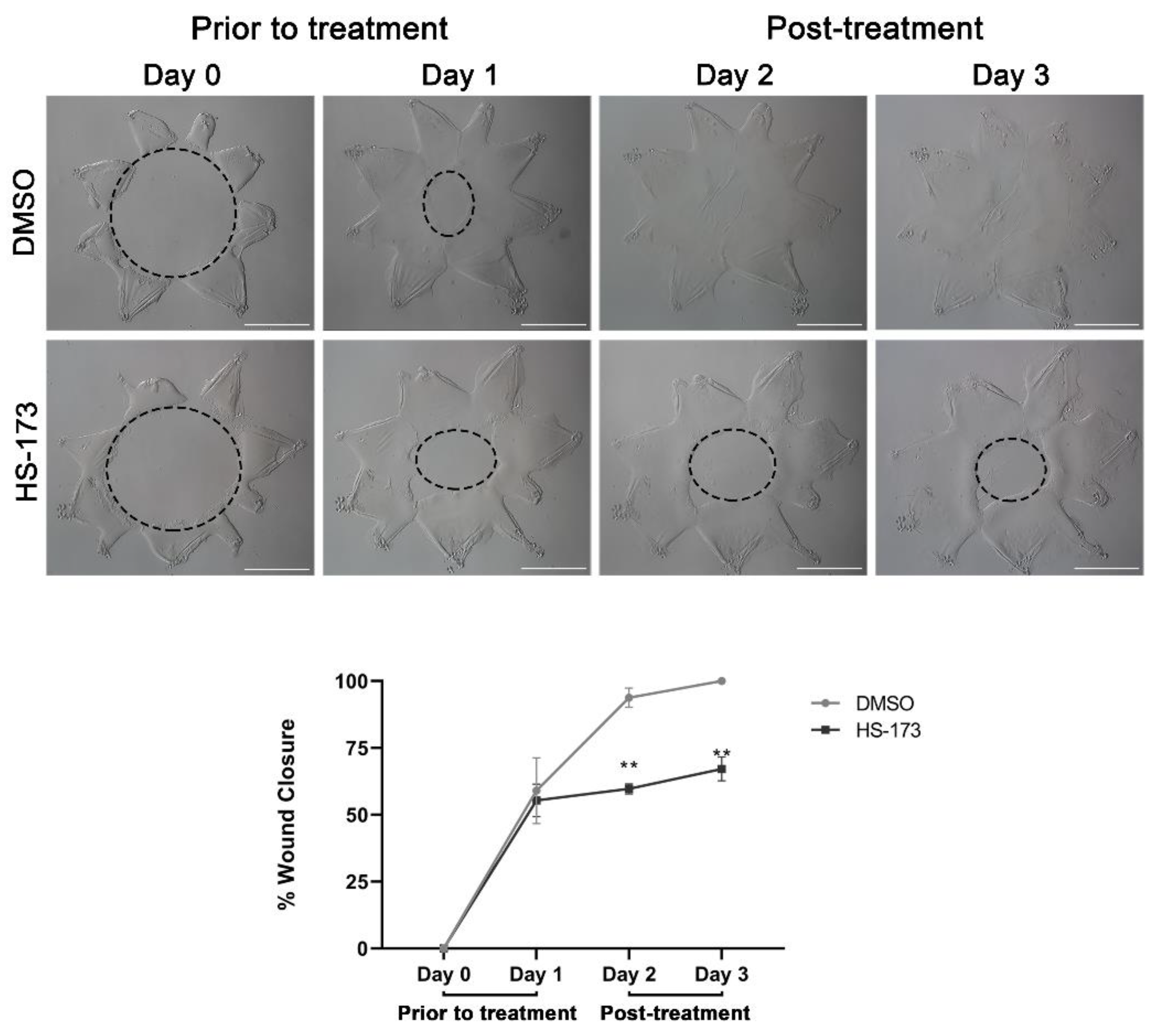
3.1. Collective Migration for Wound Healing in Response to Cataract Surgery Injury Occurs in a PI3K Isoform-Specific Manner Independent of Akt Signaling
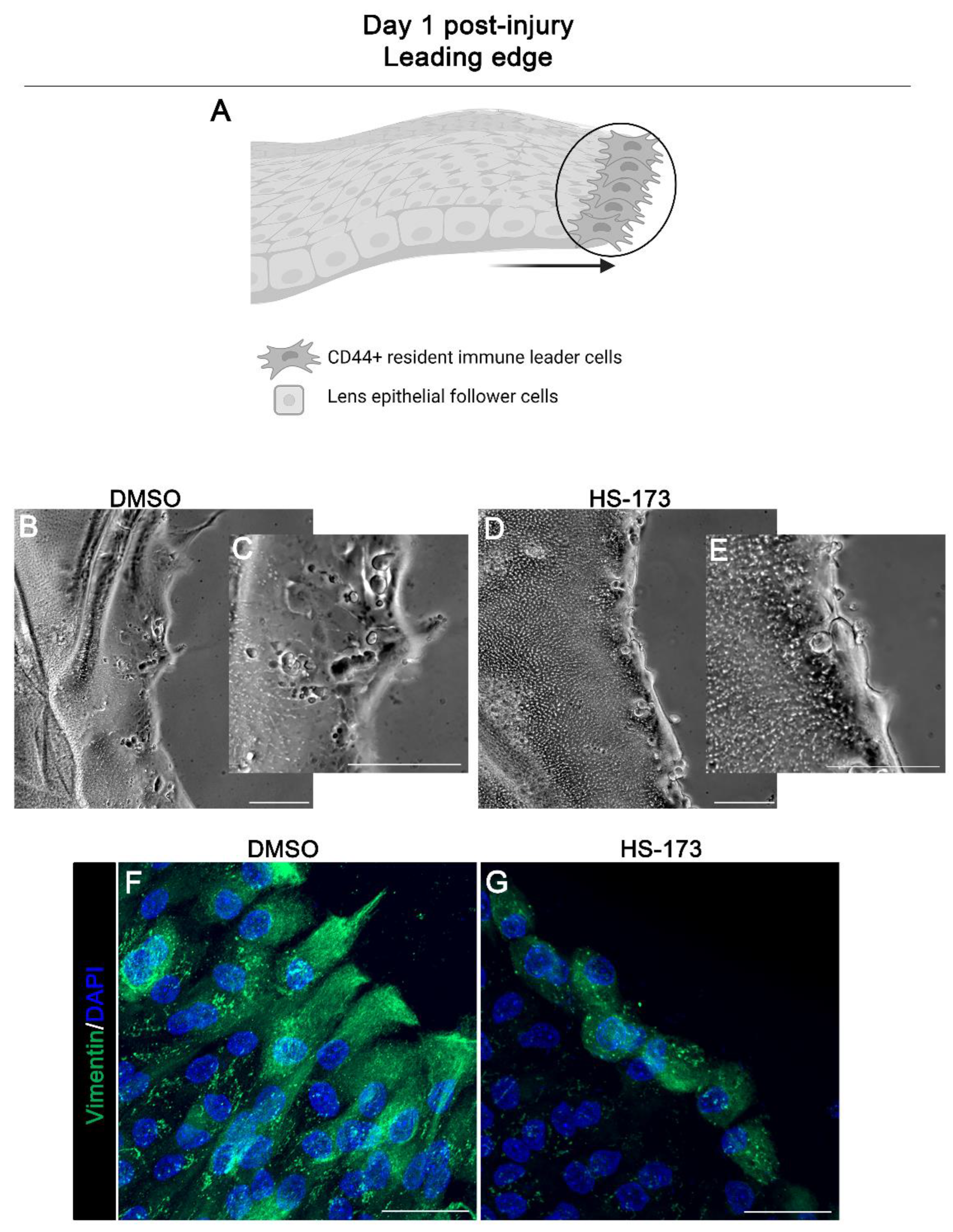
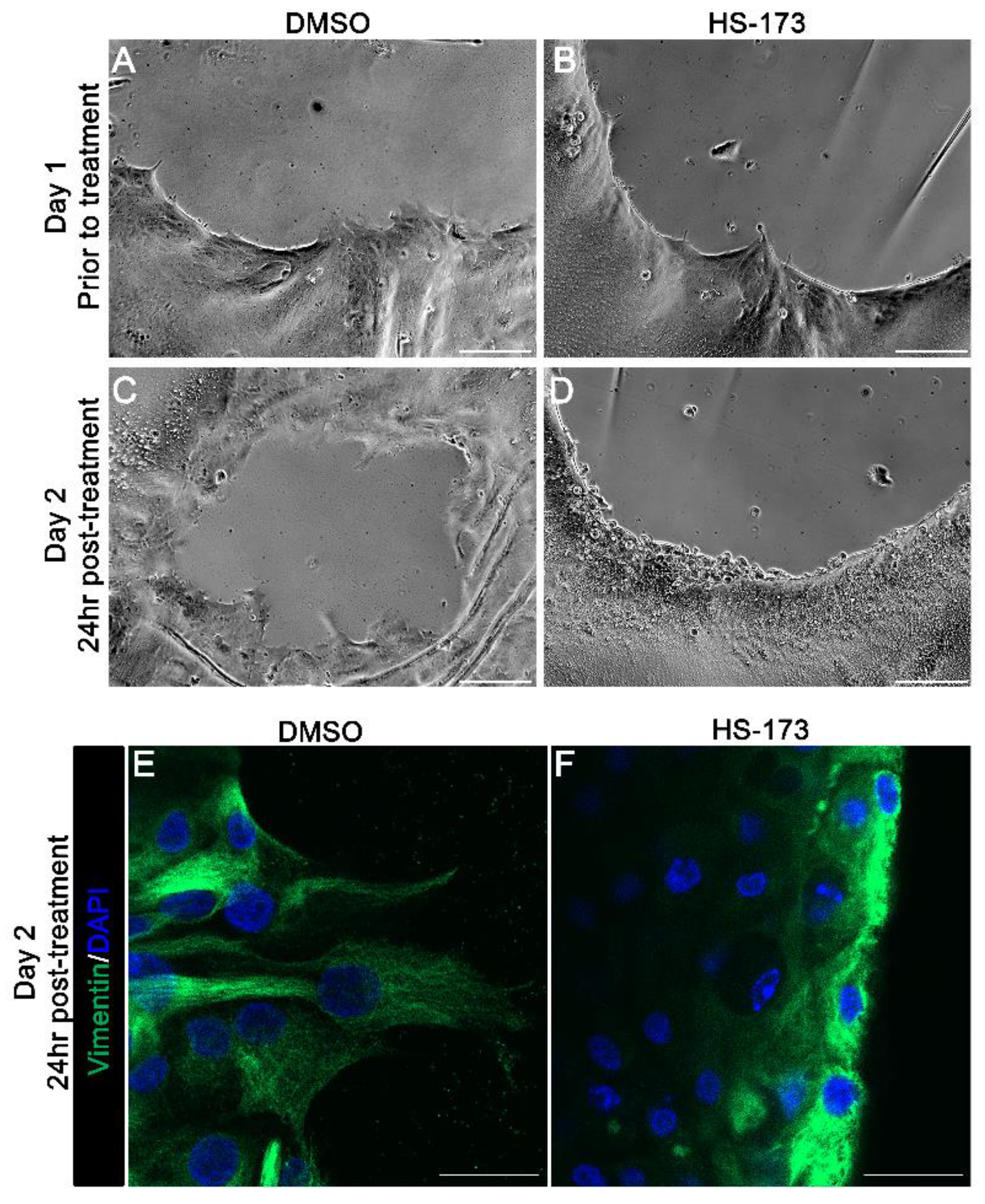
3.2. p110α Is Required to Promote and Maintain Extension of Vimentin-Rich Lamellipodial Protrusions by Leader Cells
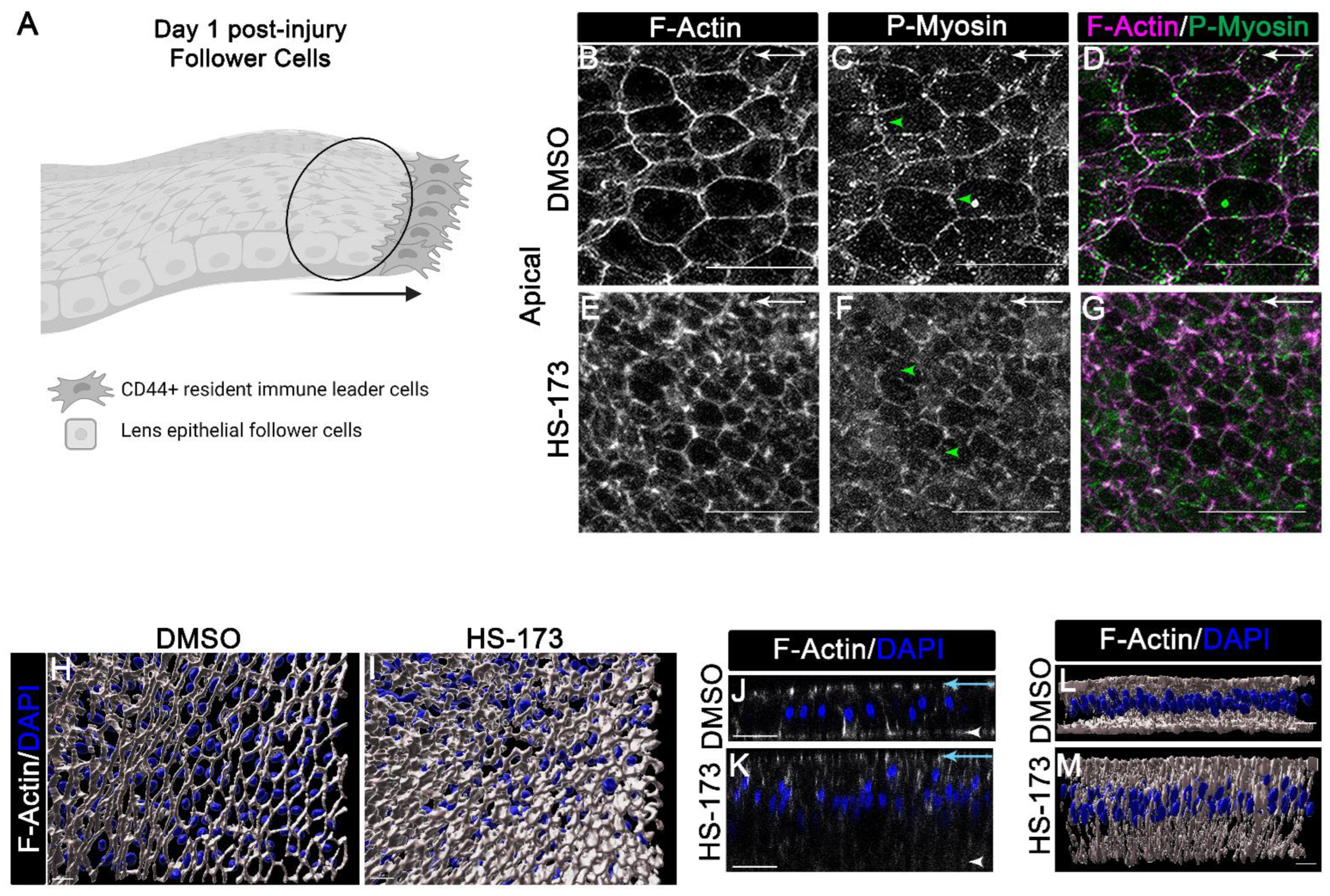
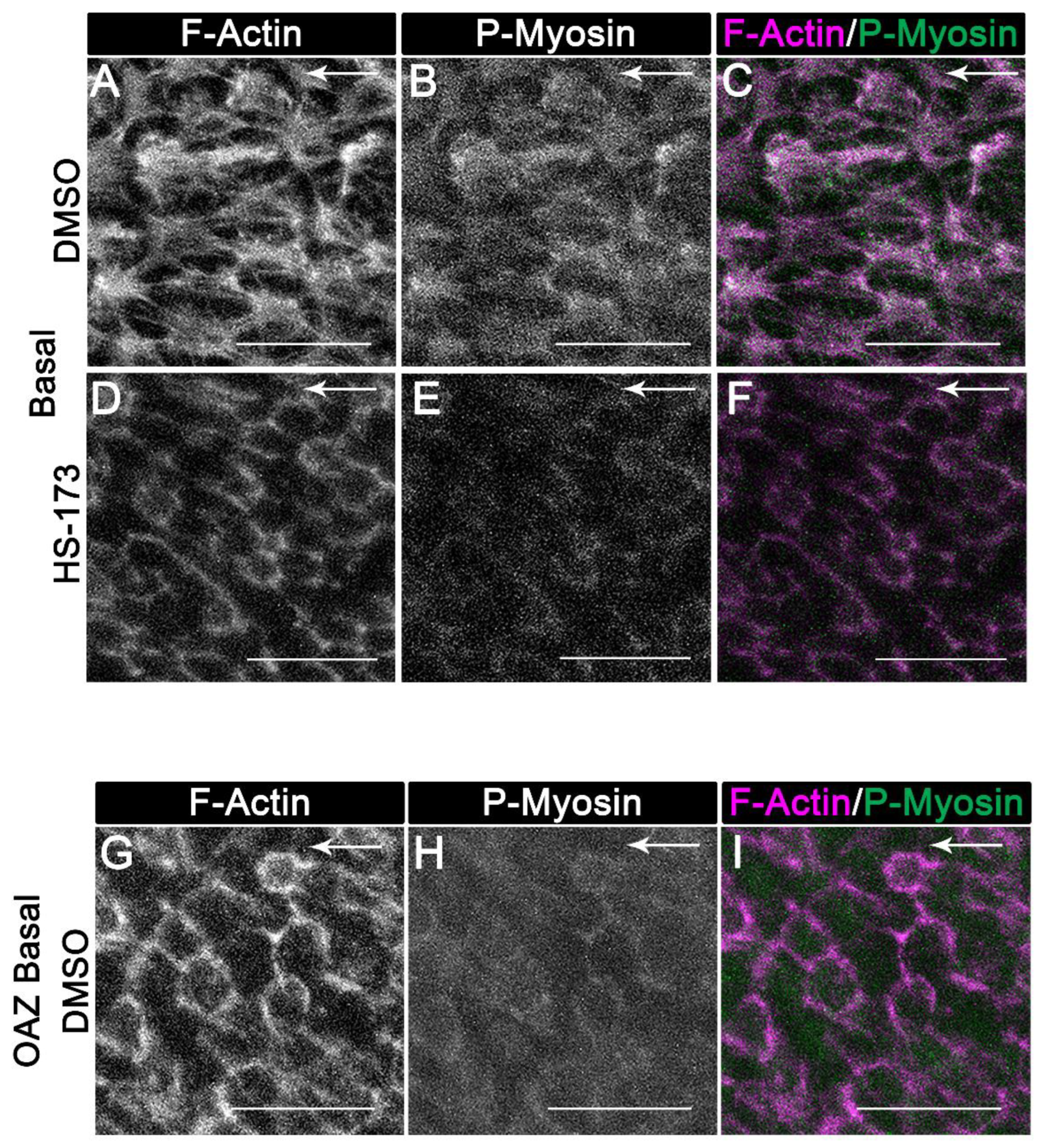
3.3. p110α Regulates the Reorganization of F-Actin along the Basal Surface of the Wounded Epithelial Cells
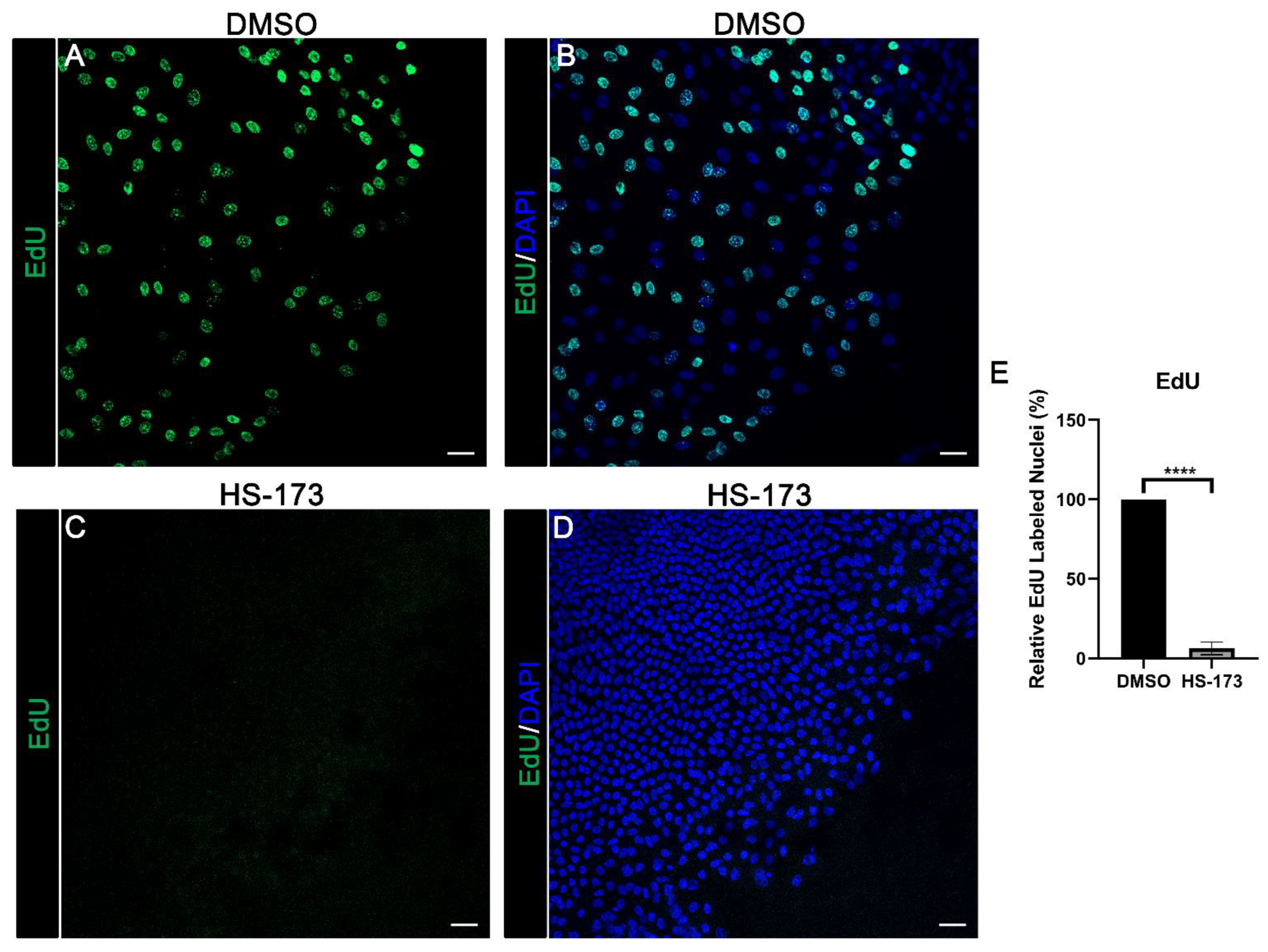
3.4. p110α Is Required for Epithelial Cell Proliferation in Response to Wounding
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jain, S.; Ladoux, B.; Mege, R.M. Mechanical plasticity in collective cell migration. Curr. Opin. Cell Biol. 2021, 72, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Yang, D.; Yi, W.; Cao, H.; Xiao, G. Roles of leader and follower cells in collective cell migration. Mol. Biol. Cell 2021, 32, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Rorth, P. Collective cell migration. Annu. Rev. Cell Dev. Biol. 2009, 25, 407–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capuana, L.; Bostrom, A.; Etienne-Manneville, S. Multicellular scale front-to-rear polarity in collective migration. Curr. Opin. Cell Biol. 2020, 62, 114–122. [Google Scholar] [CrossRef]
- Mayor, R.; Etienne-Manneville, S. The front and rear of collective cell migration. Nat. Rev. Mol. Cell Biol. 2016, 17, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Farooqui, R.; Fenteany, G. Multiple rows of cells behind an epithelial wound edge extend cryptic lamellipodia to collectively drive cell-sheet movement. J. Cell Sci. 2005, 118, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Menko, A.S.; Bleaken, B.M.; Walker, J.L. Regional-specific alterations in cell-cell junctions, cytoskeletal networks and myosin-mediated mechanical cues coordinate collectivity of movement of epithelial cells in response to injury. Exp. Cell Res. 2014, 322, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, M.; Hiver, S.; Yamamoto, T.; Shibata, T.; Upadhyayula, S.; Mimori-Kiyosue, Y.; Takeichi, M. Adherens junction regulates cryptic lamellipodia formation for epithelial cell migration. J. Cell Biol. 2020, 219, e202006196. [Google Scholar] [CrossRef]
- Khalil, A.A.; de Rooij, J. Cadherin mechanotransduction in leader-follower cell specification during collective migration. Exp. Cell Res. 2019, 376, 86–91. [Google Scholar] [CrossRef]
- Lange, J.R.; Fabry, B. Cell and tissue mechanics in cell migration. Exp. Cell Res. 2013, 319, 2418–2423. [Google Scholar] [CrossRef]
- Aranjuez, G.; Burtscher, A.; Sawant, K.; Majumder, P.; McDonald, J.A. Dynamic myosin activation promotes collective morphology and migration by locally balancing oppositional forces from surrounding tissue. Mol. Biol. Cell 2016, 27, 1898–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di-Luoffo, M.; Ben-Meriem, Z.; Lefebvre, P.; Delarue, M.; Guillermet-Guibert, J. PI3K functions as a hub in mechanotransduction. Trends Biochem. Sci. 2021, 46, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Cain, R.J.; Ridley, A.J. Phosphoinositide 3-kinases in cell migration. Biol. Cell 2009, 101, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarris, M.; Sixt, M. Navigating in tissue mazes: Chemoattractant interpretation in complex environments. Curr. Opin. Cell Biol. 2015, 36, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourne, H.R.; Weiner, O. A chemical compass. Nature 2002, 419, 21. [Google Scholar] [CrossRef]
- Gambardella, L.; Vermeren, S. Molecular players in neutrophil chemotaxis--focus on PI3K and small GTPases. J. Leukoc Biol. 2013, 94, 603–612. [Google Scholar] [CrossRef]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K signaling in cancer: Beyond AKT. Curr. Opin Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Whitehead, M.A.; Pineiro, R. Molecules in medicine mini-review: Isoforms of PI3K in biology and disease. J. Mol. Med. 2016, 94, 5–11. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Campa, C.C.; Ciraolo, E.; Ghigo, A.; Germena, G.; Hirsch, E. Crossroads of PI3K and Rac pathways. Small GTPases 2015, 6, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukata, M.; Kaibuchi, K. Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nat. Rev. Mol. Cell Biol. 2001, 2, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Mizutani, T.; Kawabata, K.; Haga, H. Leader cells regulate collective cell migration via Rac activation in the downstream signaling of integrin beta1 and PI3K. Sci. Rep. 2015, 5, 7656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.L.; Bleaken, B.M.; Wolff, I.M.; Menko, A.S. Establishment of a Clinically Relevant Ex Vivo Mock Cataract Surgery Model for Investigating Epithelial Wound Repair in a Native Microenvironment. J. Vis. Exp. 2015, e52886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menko, A.S.; Bleaken, B.M.; Libowitz, A.A.; Zhang, L.; Stepp, M.A.; Walker, J.L. A central role for vimentin in regulating repair function during healing of the lens epithelium. Mol. Biol. Cell 2014, 25, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Menko, A.S.; DeDreu, J.; Logan, C.M.; Paulson, H.; Levin, A.V.; Walker, J.L. Resident immune cells of the avascular lens: Mediators of the injury and fibrotic response of the lens. FASEB J. 2021, 35, e21341. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.L.; Bleaken, B.M.; Romisher, A.R.; Alnwibit, A.A.; Menko, A.S. In wound repair vimentin mediates the transition of mesenchymal leader cells to a myofibroblast phenotype. Mol. Biol. Cell 2018, 29, 1555–1570. [Google Scholar] [CrossRef]
- Walker, J.L.; Wolff, I.M.; Zhang, L.; Menko, A.S. Activation of SRC kinases signals induction of posterior capsule opacification. Invest. Ophthalmol. Vis. Sci. 2007, 48, 2214–2223. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.L.; Zhai, N.; Zhang, L.; Bleaken, B.M.; Wolff, I.; Gerhart, J.; George-Weinstein, M.; Menko, A.S. Unique precursors for the mesenchymal cells involved in injury response and fibrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 13730–13735. [Google Scholar] [CrossRef]
- Menko, A.S.; Walker, J.L. The Pro-Fibrotic Response to Lens Injury Is Signaled in a PI3K Isoform-Specific Manner. Biomolecules 2022, 12, 1181. [Google Scholar] [CrossRef] [PubMed]
- Gheyas, R.; Ortega-Alvarez, R.; Chauss, D.; Kantorow, M.; Menko, A.S. Suppression of PI3K signaling is linked to autophagy activation and the spatiotemporal induction of the lens organelle free zone. Exp. Cell Res. 2022, 412, 113043. [Google Scholar] [CrossRef] [PubMed]
- Sellitto, C.; Li, L.; Vaghefi, E.; Donaldson, P.J.; Lin, R.Z.; White, T.W. The Phosphoinosotide 3-Kinase Catalytic Subunit p110alpha is Required for Normal Lens Growth. Invest. Ophthalmol. Vis. Sci. 2016, 57, 3145–3151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.K.; Ryu, Y.L.; Jung, K.H.; Lee, H.; Lee, H.S.; Yan, H.H.; Park, H.J.; Ryu, J.K.; Suh, J.K.; Hong, S.; et al. HS-173, a novel PI3K inhibitor, attenuates the activation of hepatic stellate cells in liver fibrosis. Sci. Rep. 2013, 3, 3470. [Google Scholar] [CrossRef] [Green Version]
- Petrie, R.J.; Doyle, A.D.; Yamada, K.M. Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Pankov, R.; Endo, Y.; Even-Ram, S.; Araki, M.; Clark, K.; Cukierman, E.; Matsumoto, K.; Yamada, K.M. A Rac switch regulates random versus directionally persistent cell migration. J. Cell Biol. 2005, 170, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Laufer, J.M.; Hauser, M.A.; Kindinger, I.; Purvanov, V.; Pauli, A.; Legler, D.F. Chemokine Receptor CCR7 Triggers an Endomembrane Signaling Complex for Spatial Rac Activation. Cell Rep. 2019, 29, 995–1009.e6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.J.; Tao, B.B.; Wang, M.J.; Jin, H.M.; Zhu, Y.C. PI3K p110alpha isoform-dependent Rho GTPase Rac1 activation mediates H2S-promoted endothelial cell migration via actin cytoskeleton reorganization. PLoS ONE 2012, 7, e44590. [Google Scholar] [CrossRef]
- Zeller, K.S.; Idevall-Hagren, O.; Stefansson, A.; Velling, T.; Jackson, S.P.; Downward, J.; Tengholm, A.; Johansson, S. PI3-kinase p110alpha mediates beta1 integrin-induced Akt activation and membrane protrusion during cell attachment and initial spreading. Cell Signal. 2010, 22, 1838–1848. [Google Scholar] [CrossRef]
- Wan, G.; Pehlke, C.; Pepermans, R.; Cannon, J.L.; Lidke, D.; Rajput, A. The H1047R point mutation in p110 alpha changes the morphology of human colon HCT116 cancer cells. Cell Death Discov. 2015, 1, 15044. [Google Scholar] [CrossRef]
- Yip, S.C.; El-Sibai, M.; Coniglio, S.J.; Mouneimne, G.; Eddy, R.J.; Drees, B.E.; Neilsen, P.O.; Goswami, S.; Symons, M.; Condeelis, J.S.; et al. The distinct roles of Ras and Rac in PI 3-kinase-dependent protrusion during EGF-stimulated cell migration. J. Cell Sci. 2007, 120, 3138–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valgeirsdottir, S.; Claesson-Welsh, L.; Bongcam-Rudloff, E.; Hellman, U.; Westermark, B.; Heldin, C.H. PDGF induces reorganization of vimentin filaments. J. Cell Sci. 1998, 111 Pt 14, 1973–1980. [Google Scholar] [CrossRef]
- Barberis, L.; Pasquali, C.; Bertschy-Meier, D.; Cuccurullo, A.; Costa, C.; Ambrogio, C.; Vilbois, F.; Chiarle, R.; Wymann, M.; Altruda, F.; et al. Leukocyte transmigration is modulated by chemokine-mediated PI3Kgamma-dependent phosphorylation of vimentin. Eur. J. Immunol. 2009, 39, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Sotsios, Y.; Whittaker, G.C.; Westwick, J.; Ward, S.G. The CXC chemokine stromal cell-derived factor activates a Gi-coupled phosphoinositide 3-kinase in T lymphocytes. J. Immunol. 1999, 163, 5954–5963. [Google Scholar] [PubMed]
- Curnock, A.P.; Logan, M.K.; Ward, S.G. Chemokine signalling: Pivoting around multiple phosphoinositide 3-kinases. Immunology 2002, 105, 125–136. [Google Scholar] [CrossRef]
- Walker, J.L.; Menko, A.S. Immune cells in lens injury repair and fibrosis. Exp. Eye Res. 2021, 209, 108664. [Google Scholar] [CrossRef]
- Sit, S.T.; Manser, E. Rho GTPases and their role in organizing the actin cytoskeleton. J. Cell Sci. 2011, 124, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Cain, R.J.; Vanhaesebroeck, B.; Ridley, A.J. The PI3K p110alpha isoform regulates endothelial adherens junctions via Pyk2 and Rac1. J. Cell Biol. 2010, 188, 863–876. [Google Scholar] [CrossRef] [Green Version]
- Vasilyev, A.; Liu, Y.; Hellman, N.; Pathak, N.; Drummond, I.A. Mechanical stretch and PI3K signaling link cell migration and proliferation to coordinate epithelial tubule morphogenesis in the zebrafish pronephros. PLoS ONE 2012, 7, e39992. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basta, M.D.; Menko, A.S.; Walker, J.L. PI3K Isoform-Specific Regulation of Leader and Follower Cell Function for Collective Migration and Proliferation in Response to Injury. Cells 2022, 11, 3515. https://doi.org/10.3390/cells11213515
Basta MD, Menko AS, Walker JL. PI3K Isoform-Specific Regulation of Leader and Follower Cell Function for Collective Migration and Proliferation in Response to Injury. Cells. 2022; 11(21):3515. https://doi.org/10.3390/cells11213515
Chicago/Turabian StyleBasta, Morgan D., A. Sue Menko, and Janice L. Walker. 2022. "PI3K Isoform-Specific Regulation of Leader and Follower Cell Function for Collective Migration and Proliferation in Response to Injury" Cells 11, no. 21: 3515. https://doi.org/10.3390/cells11213515