
Integration of O-GlcNAc into Stress Response Pathways

Abstract
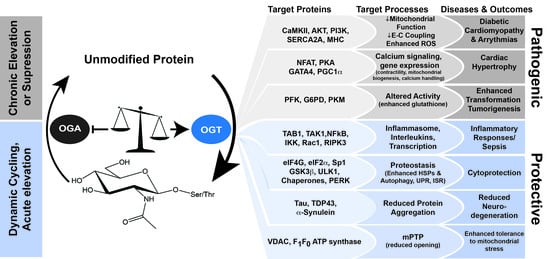
:
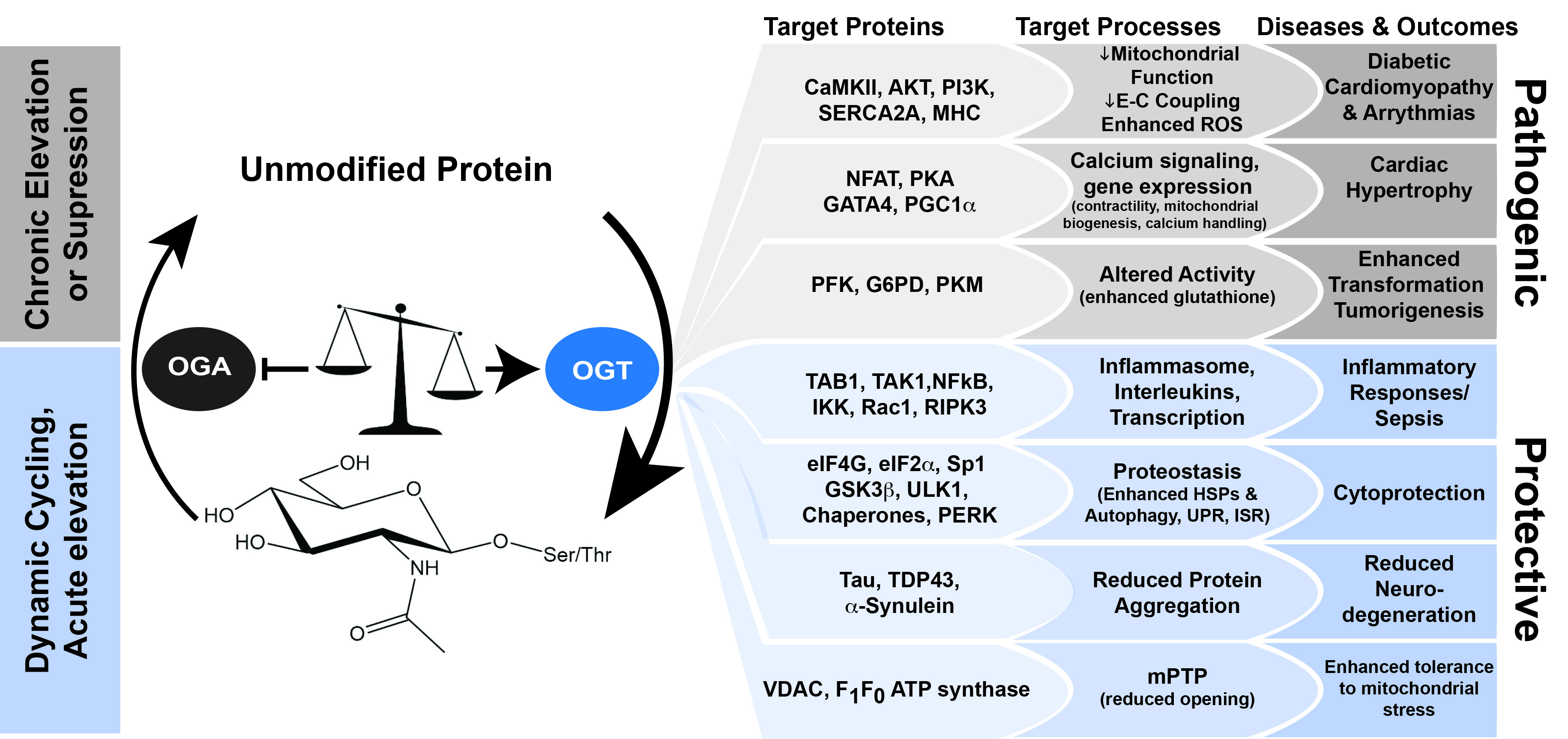{kind=link}
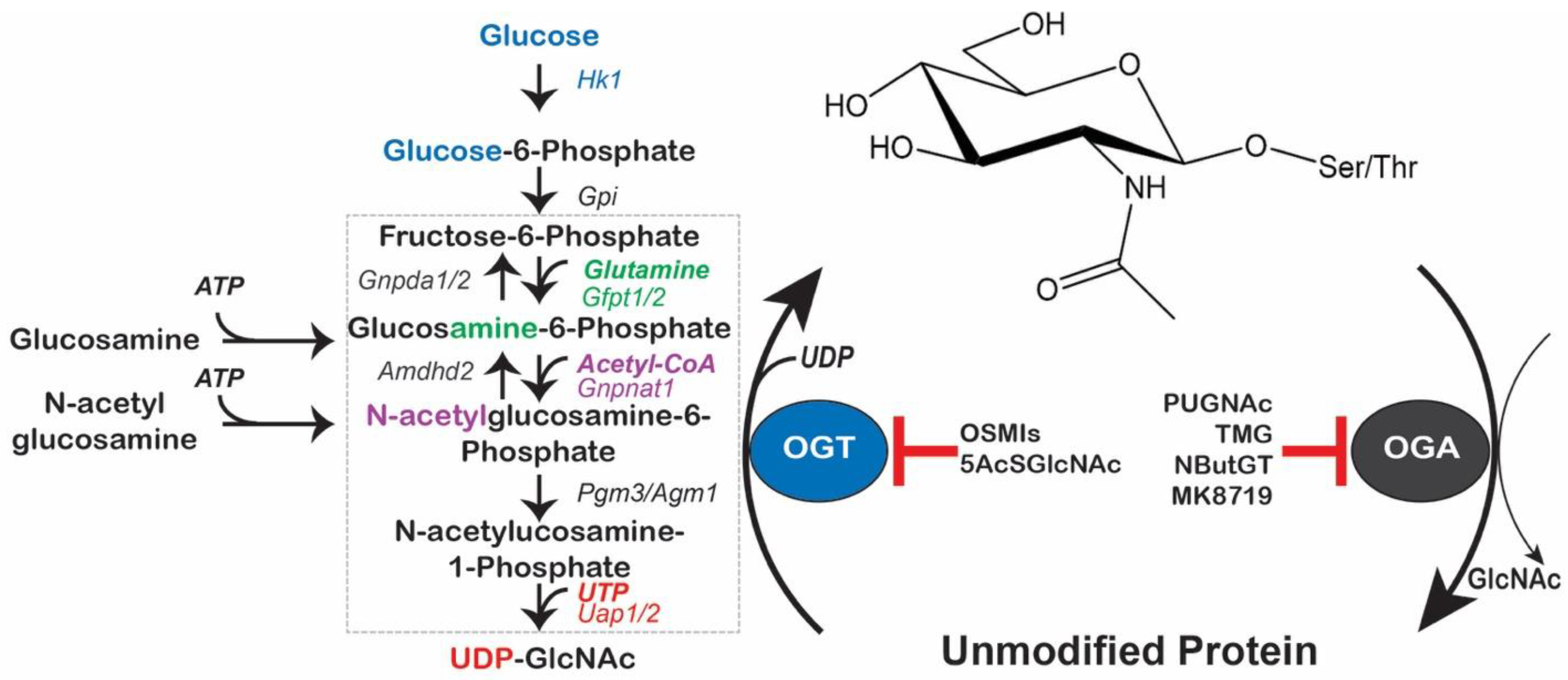{kind=link}
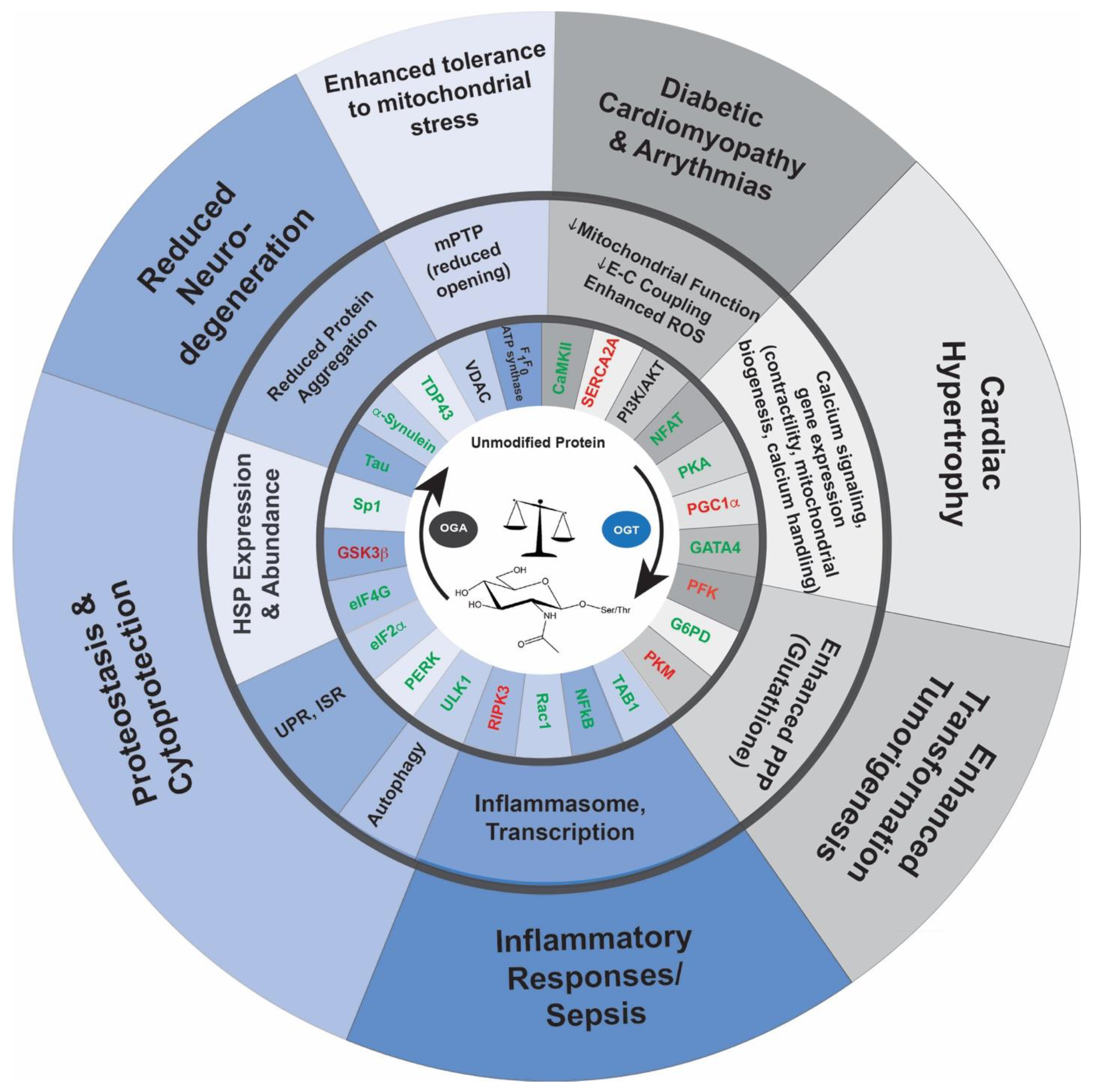{kind=link}
1. Background
2. The O-GlcNAc Cycle
2.1. Coordination of UDP-GlcNAc Production in Health and Disease
2.2. O-GlcNAc-Cycling by the Writer/Eraser: OGT/OGA
3. Impact of O-GlcNAc in Physiological Models of Injury and Pathophysiology
3.1. Cardiac Development, Physiology, Pathophysiology, and Homeostasis
3.2. Pressure Overload Hypertrophy, Myocardial Infarction, and Ischemia Reperfusion Models
3.3. ER Stress Response
3.4. Trauma Hemorrhage
3.5. O-GlcNAc Cycling Resists Proteotoxic Aggregation in Neurodegenerative Diseases
3.5.1. Alzheimer’s Disease
3.5.2. α-Synuclein O-GlcNAcylation Suppresses Aggregation and Prion-like Behavior That Typifies Parkinson’s Disease
3.5.3. O-GlcNAc Modification of Transactive Response DNA Binding Protein 43 Resists Aggregation Observed in Amyotrophic Lateral Sclerosis
4. Other Stress-Responsive Pathways Regulated by O-GlcNAc
4.1. O-GlcNAc Cycling Mediates Pro- and Anti-Inflammatory Signaling
4.1.1. The Role of O-GlcNAcylation in the Activation and Resolution of the Inflammatory Response
4.1.2. Integrated Stress Response (ISR)
4.1.3. O-GlcNAc Cycling Regulates Different Steps in the of Autophagy Pathway
4.1.4. O-GlcNAcylation and the Pentose Phosphate Pathway (PPP)
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hart, G.W. Nutrient Regulation of Signaling and Transcription. J. Biol. Chem. 2019, 294, 2211–2231. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Li, Y.; Hou, C.; Wu, C. O-GlcNAcAtlas: A Database of Experimentally Identified O-GlcNAc Sites and Proteins. Glycobiology 2021, 31, 719–723. [Google Scholar] [CrossRef]
- Wulff-Fuentes, E.; Berendt, R.R.; Massman, L.; Danner, L.; Malard, F.; Vora, J.; Kahsay, R.; Stichelen, S.O.-V. The Human O-GlcNAcome Database and Meta-Analysis. Sci. Data 2020, 8, 25. [Google Scholar] [CrossRef]
- Gewinner, C.; Hart, G.; Zachara, N.; Cole, R.; Beisenherz-Huss, C.; Groner, B. The Coactivator of Transcription CREB-Binding Protein Interacts Preferentially with the Glycosylated Form of Stat5. J. Biol. Chem. 2004, 279, 3563–3572. [Google Scholar] [CrossRef] [Green Version]
- Lamarre-Vincent, N.; Hsieh-Wilson, L.C. Dynamic Glycosylation of the Transcription Factor CREB: A Potential Role in Gene Regulation. J. Am. Chem. Soc. 2003, 125, 6612–6613. [Google Scholar] [CrossRef] [Green Version]
- Deplus, R.; Delatte, B.; Schwinn, M.K.; Defrance, M.; Méndez, J.; Murphy, N.; Dawson, M.A.; Volkmar, M.; Putmans, P.; Calonne, E.; et al. TET2 and TET3 Regulate GlcNAcylation and H3K4 Methylation through OGT and SET1/COMPASS. EMBO J. 2013, 32, 645–655. [Google Scholar] [CrossRef]
- Slawson, C.; Zachara, N.E.; Vosseller, K.; Cheung, W.D.; Lane, M.D.; Hart, G.W. Perturbations in O-Linked β-N-Acetylglucosamine Protein Modification Cause Severe Defects in Mitotic Progression and Cytokinesis. J. Biol. Chem. 2005, 280, 32944–32956. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, T.-W.; Cecioni, S.; Eskandari, R.; Zandberg, W.F.; Vocadlo, D.J. O-GlcNAc Occurs Cotranslationally to Stabilize Nascent Polypeptide Chains. Nat. Chem. Biol. 2015, 11, 319–325. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, T.; Madden, Z.; Yuzwa, S.A.; Murray, K.; Cecioni, S.; Zachara, N.; Vocadlo, D.J. Post-Translational O-GlcNAcylation Is Essential for Nuclear Pore Integrity and Maintenance of the Pore Selectivity Filter. J. Mol. Cell Biol. 2015, 8, 2–16. [Google Scholar] [CrossRef] [Green Version]
- Sümegi, M.; Hunyadi-Gulyás, E.; Medzihradszky, K.F.; Udvardy, A. 26S Proteasome Subunits Are O-Linked N-Acetylglucosamine-Modified in Drosophila Melanogaster. Biochem. Biophys. Res. Commun. 2003, 312, 1284–1289. [Google Scholar] [CrossRef]
- Yoo, T.Y.; Mitchison, T.J. O-GlcNAc Modification of Nuclear Pore Complexes Accelerates Bidirectional Transport. J. Cell Biol. 2021, 220, e202010141. [Google Scholar] [CrossRef]
- Yang, X.; Su, K.; Roos, M.D.; Chang, Q.; Paterson, A.J.; Kudlow, J.E. O-Linkage of N-Acetylglucosamine to Sp1 Activation Domain Inhibits Its Transcriptional Capability. Proc. Natl. Acad. Sci. USA 2001, 98, 6611–6616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toleman, C.A.; Schumacher, M.A.; Yu, S.-H.; Zeng, W.; Cox, N.J.; Smith, T.J.; Soderblom, E.J.; Wands, A.M.; Kohler, J.J.; Boyce, M. Structural Basis of O-GlcNAc Recognition by Mammalian 14-3-3 Proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 5956–5961. [Google Scholar] [CrossRef] [Green Version]
- Dias, W.B.; Cheung, W.D.; Wang, Z.; Hart, G.W. Regulation of Calcium/Calmodulin-Dependent Kinase IV by O-GlcNAc Modification. J. Biol. Chem. 2009, 284, 21327–21337. [Google Scholar] [CrossRef] [Green Version]
- Pathak, S.; Borodkin, V.S.; Albarbarawi, O.; Campbell, D.G.; Ibrahim, A.; van Aalten, D.M. O-GlcNAcylation of TAB1 Modulates TAK1-Mediated Cytokine Release. EMBO J. 2012, 31, 1394–1404. [Google Scholar] [CrossRef] [Green Version]
- Tarrant, M.K.; Rho, H.-S.; Xie, Z.; Jiang, Y.L.; Gross, C.; Culhane, J.C.; Yan, G.; Qian, J.; Ichikawa, Y.; Matsuoka, T.; et al. Regulation of CK2 by Phosphorylation and O-GlcNAcylation Revealed by Semisynthesis. Nat. Chem. Biol. 2012, 8, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Zachara, N.E.; O’Donnell, N.; Cheung, W.D.; Mercer, J.J.; Marth, J.D.; Hart, G.W. Dynamic O-GlcNAc Modification of Nucleocytoplasmic Proteins in Response to Stress. J. Biol. Chem. 2004, 279, 30133–30142. [Google Scholar] [CrossRef] [Green Version]
- Kazemi, Z.; Chang, H.; Haserodt, S.; McKen, C.; Zachara, N.E. O-Linked-N-Acetylglucosamine (O-GlcNAc) Regulates Stress-Induced Heat Shock Protein Expression in a GSK-3 -Dependent Manner. J. Biol. Chem. 2010, 285, 39096–39107. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.P.; Zachara, N.E.; Ngoh, G.A.; Hill, B.G.; Teshima, Y.; Bhatnagar, A.; Hart, G.W.; Marbán, E. Cardioprotection by N-Acetylglucosamine Linkage to Cellular Proteins. Circulation 2008, 117, 1172–1182. [Google Scholar] [CrossRef] [Green Version]
- Karababa, A.; Görg, B.; Schliess, F.; Häussinger, D. O-GlcNAcylation as a Novel Ammonia-Induced Posttranslational Protein Modification in Cultured Rat Astrocytes. Metab. Brain Dis. 2014, 29, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Al-Mukh, H.; Baudoin, L.; Bouaboud, A.; Sanchez-Salgado, J.-L.; Maraqa, N.; Khair, M.; Pagesy, P.; Bismuth, G.; Niedergang, F.; Issad, T. Lipopolysaccharide Induces GFAT2 Expression to Promote O-Linked β-N-Acetylglucosaminylation and Attenuate Inflammation in Macrophages. J. Immunol. 2020, 205, 2499–2510. [Google Scholar] [CrossRef]
- Ngoh, G.A.; Facundo, H.T.; Hamid, T.; Dillmann, W.; Zachara, N.E.; Jones, S.P. Unique Hexosaminidase Reduces Metabolic Survival Signal and Sensitizes Cardiac Myocytes to Hypoxia/Reoxygenation Injury. Circ. Res. 2008, 104, 41–49. [Google Scholar] [CrossRef]
- Zhu, W.Z.; Ledee, D.; Olson, A.K. Temporal Regulation of Protein O-GlcNAc Levels during Pressure-overload Cardiac Hypertrophy. Physiol. Rep. 2021, 9, e14965. [Google Scholar] [CrossRef]
- Zou, L.; Yang, S.; Hu, S.; Chaudry, I.H.; Marchase, R.B.; Chatham, J.C. The Protective Effects of PUGNAc on Cardiac Function after Trauma-Hemorrhage Are Mediated via Increased Protein O-GlcNAc Levels. Shock 2007, 27, 402–408. [Google Scholar] [CrossRef]
- Lee, A.; Miller, D.; Henry, R.; Paruchuri, V.D.P.; O’Meally, R.N.; Boronina, T.; Cole, R.N.; Zachara, N.E. Combined Antibody/Lectin Enrichment Identifies Extensive Changes in the O-GlcNAc Sub-Proteome upon Oxidative Stress. J. Proteome Res. 2016, 15, 4318–4336. [Google Scholar] [CrossRef]
- Haltiwanger, R.S.; Holt, G.D.; Hart, G.W. Enzymatic Addition of O-GlcNAc to Nuclear and Cytoplasmic Proteins. Identification of a Uridine Diphospho-N-Acetylglucosamine:Peptide Beta-N-Acetylglucosaminyltransferase. J. Biol. Chem. 1990, 265, 2563–2568. [Google Scholar] [CrossRef]
- Gao, Y.; Wells, L.; Comer, F.I.; Parker, G.J.; Hart, G.W. Dynamic O-Glycosylation of Nuclear and Cytosolic Proteins: Cloning and Characterization of a Neutral, Cytosolic Beta-N-Acetylglucosaminidase from Human Brain. J. Biol. Chem. 2001, 276, 9838–9845. [Google Scholar] [CrossRef] [Green Version]
- Olson, A.K.; Bouchard, B.; Zhu, W.Z.; Chatham, J.C.; Rosiers, C.D. First Characterization of Glucose Flux through the Hexosamine Biosynthesis Pathway (HBP) in Ex Vivo Mouse Heart. J. Biol. Chem. 2020, 295, 2018–2033. [Google Scholar] [CrossRef] [Green Version]
- Kornfeld, S.; Kornfeld, R.; Neufeld, E.F.; O’Brien, P.J. The Feedback Control of Sugar Nucleotide Biosynthesis in Liver. Proc. Natl. Acad. Sci. USA 1964, 52, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Kornfeld, R. Studies on L-Glutamine D-Fructose 6-Phosphate Amidotransferase. I. Feedback Inhibition by Uridine Diphosphate-N-Acetylglucosamine. J. Biological. Chem. 1967, 242, 3135–3141. [Google Scholar] [CrossRef]
- Eguchi, S.; Oshiro, N.; Miyamoto, T.; Yoshino, K.-I.; Okamoto, S.; Ono, T.; Kikkawa, U.; Yonezawa, K. AMP-Activated Protein Kinase Phosphorylates Glutamine: Fructose-6-Phosphate Amidotransferase 1 at Ser243 to Modulate Its Enzymatic Activity. Genes Cells 2009, 14, 179–189. [Google Scholar] [CrossRef]
- Li, Y.; Roux, C.; Lazereg, S.; LeCaer, J.-P.; Laprévote, O.; Badet, B.; Badet-Denisot, M.-A. Identification of a Novel Serine Phosphorylation Site in Human Glutamine:Fructose-6-Phosphate Amidotransferase Isoform 1. Biochemistry 2007, 46, 13163–13169. [Google Scholar] [CrossRef]
- Hu, Y.; Riesland, L.; Paterson, A.J.; Kudlow, J.E. Phosphorylation of Mouse Glutamine-Fructose-6-Phosphate Amidotransferase 2 (GFAT2) by CAMP-Dependent Protein Kinase Increases the Enzyme Activity. J. Biol. Chem. 2004, 279, 29988–29993. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Aguilar, R.; Grayson, B.E.; Kim, D.-H.; Yalamanchili, S.; Calcagno, M.L.; Woods, S.C.; Seeley, R.J. CNS GNPDA2 Does Not Control Appetite, but Regulates Glucose Homeostasis. Front. Nutr. 2021, 8, 787470. [Google Scholar] [CrossRef]
- Kroef, V.; Ruegenberg, S.; Horn, M.; Allmeroth, K.; Ebert, L.; Bozkus, S.; Miethe, S.; Elling, U.; Schermer, B.; Baumann, U.; et al. GFPT2/GFAT2 and AMDHD2 Act in Tandem to Control the Hexosamine Pathway. eLife 2022, 11, e69223. [Google Scholar] [CrossRef]
- Freeze, H.H.; Boyce, M.; Zachara, N.E.; Hart, G.W.; Schnaar, R.L. Glycosylation Precursors, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2022. [Google Scholar]
- Ryczko, M.C.; Pawling, J.; Chen, R.; Rahman, A.M.A.; Yau, K.; Copeland, J.K.; Zhang, C.; Surendra, A.; Guttman, D.S.; Figeys, D.; et al. Metabolic Reprogramming by Hexosamine Biosynthetic and Golgi N-Glycan Branching Pathways. Sci. Rep. 2016, 6, 23043. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.V.; Deng, Y.; Gao, N.; Pedrozo, Z.; Li, D.L.; Morales, C.R.; Criollo, A.; Luo, X.; Tan, W.; Jiang, N.; et al. Spliced X-Box Binding Protein 1 Couples the Unfolded Protein Response to Hexosamine Biosynthetic Pathway. Cell 2014, 156, 1179–1192. [Google Scholar] [CrossRef] [Green Version]
- Horn, M.; Denzel, S.I.; Srinivasan, B.; Allmeroth, K.; Schiffer, I.; Karthikaisamy, V.; Miethe, S.; Breuer, P.; Antebi, A.; Denzel, M.S. Hexosamine Pathway Activation Improves Protein Homeostasis through the Integrated Stress Response. Iscience 2020, 23, 100887. [Google Scholar] [CrossRef] [Green Version]
- Ruegenberg, S.; Horn, M.; Pichlo, C.; Allmeroth, K.; Baumann, U.; Denzel, M.S. Loss of GFAT-1 Feedback Regulation Activates the Hexosamine Pathway That Modulates Protein Homeostasis. Nat. Commun. 2020, 11, 687. [Google Scholar] [CrossRef] [Green Version]
- Taparra, K.; Wang, H.; Malek, R.; Lafargue, A.; Barbhuiya, M.A.; Wang, X.; Simons, B.W.; Ballew, M.; Nugent, K.; Groves, J.; et al. O-GlcNAcylation Is Required for Mutant KRAS-Induced Lung Tumorigenesis. J. Clin. Investig. 2018, 128, 4924–4937. [Google Scholar] [CrossRef] [PubMed]
- Itkonen, H.M.; Engedal, N.; Babaie, E.; Luhr, M.; Guldvik, I.J.; Minner, S.; Hohloch, J.; Tsourlakis, M.C.; Schlomm, T.; Mills, I.G. UAP1 Is Overexpressed in Prostate Cancer and Is Protective against Inhibitors Of. Nat. Publ. Group 2014, 34, 3744–3750. [Google Scholar] [CrossRef]
- Capotosti, F.; Guernier, S.; Lammers, F.; Waridel, P.; Cai, Y.; Jin, J.; Conaway, J.W.; Conaway, R.C.; Herr, W. O-GlcNAc Transferase Catalyzes Site-Specific Proteolysis of HCF-1. Cell 2011, 144, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daou, S.; Mashtalir, N.; Hammond-Martel, I.; Pak, H.; Yu, H.; Sui, G.; Vogel, J.L.; Kristie, T.M.; Affar, E.B. Crosstalk between O-GlcNAcylation and Proteolytic Cleavage Regulates the Host Cell Factor-1 Maturation Pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 2747–2752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, Z.G.; Potter, S.C.; Joiner, C.M.; Fei, G.Q.; Nabet, B.; Sonnett, M.; Zachara, N.E.; Gray, N.S.; Paulo, J.A.; Walker, S. Mammalian Cell Proliferation Requires Noncatalytic Functions of O-GlcNAc Transferase. Proc. Natl. Acad. Sci. USA 2021, 118, e2016778118. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.J.; Facundo, H.T.; Ngoh, G.A.; Ameen, M.; Brainard, R.E.; Lemma, K.M.; Long, B.W.; Prabhu, S.D.; Xuan, Y.-T.; Jones, S.P. O-Linked β-N-Acetylglucosamine Transferase Is Indispensable in the Failing Heart. Proc. Natl. Acad. Sci. USA 2010, 107, 17797–17802. [Google Scholar] [CrossRef] [Green Version]
- Watson, L.J.; Long, B.W.; DeMartino, A.M.; Brittian, K.R.; Readnower, R.D.; Brainard, R.E.; Cummins, T.D.; Annamalai, L.; Hill, B.G.; Jones, S.P. Cardiomyocyte Ogt Is Essential for Postnatal Viability. AJP Heart Circ. Physiol. 2014, 306, H142–H153. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Li, M.-D.; Yin, R.; Liu, Y.; Yang, Y.; Mitchell-Richards, K.A.; Nam, J.H.; Li, R.; Wang, L.; Iwakiri, Y.; et al. O-GlcNAc Transferase Suppresses Necroptosis and Liver Fibrosis. JCI Insight 2019, 4, e127709. [Google Scholar] [CrossRef] [Green Version]
- Levine, Z.G.; Fan, C.; Melicher, M.S.; Orman, M.; Benjamin, T.; Walker, S. O-GlcNAc Transferase Recognizes Protein Substrates Using an Asparagine Ladder in the Tetratricopeptide Repeat (TPR) Superhelix. J. Am. Chem. Soc. 2018, 140, 3510–3513. [Google Scholar] [CrossRef]
- Joiner, C.M.; Levine, Z.G.; Aonbangkhen, C.; Woo, C.M.; Walker, S. Aspartate Residues Far from the Active Site Drive O-GlcNAc Transferase Substrate Selection. J. Am. Chem. Soc. 2019, 141, 12974–12978. [Google Scholar] [CrossRef]
- Joiner, C.M.; Hammel, F.A.; Janetzko, J.; Walker, S. Protein Substrates Engage the Lumen of O-GlcNAc Transferase’s Tetratricopeptide Repeat Domain in Different Ways. Biochemistry 2021, 60, 847–853. [Google Scholar] [CrossRef]
- Martinez, M.; Renuse, S.; Kreimer, S.; O’Meally, R.; Natov, P.; Madugundu, A.K.; Nirujogi, R.S.; Tahir, R.; Cole, R.; Pandey, A.; et al. Quantitative Proteomics Reveals That the OGT Interactome Is Remodeled in Response to Oxidative Stress. Mol. Cell Proteom. 2021, 20, 100069. [Google Scholar] [CrossRef]
- Cheung, W.D.; Sakabe, K.; Housley, M.P.; Dias, W.B.; Hart, G.W. O-Linked-N-Acetylglucosaminyltransferase Substrate Specificity Is Regulated by Myosin Phosphatase Targeting and Other Interacting Proteins. J. Biol. Chem. 2008, 283, 33935–33941. [Google Scholar] [CrossRef] [Green Version]
- Cheung, W.D.; Hart, G.W. AMP-Activated Protein Kinase and P38 MAPK Activate O-GlcNAcylation of Neuronal Proteins during Glucose Deprivation. J. Biol. Chem. 2008, 283, 13009–13020. [Google Scholar] [CrossRef] [Green Version]
- Groves, J.A.; Maduka, A.O.; O’Meally, R.N.; Cole, R.N.; Zachara, N.E. Fatty Acid Synthase Inhibits the O-GlcNAcase during Oxidative Stress. J. Biol. Chem. 2017, 292, 6493–6511. [Google Scholar] [CrossRef] [Green Version]
- Jensen, R.V.; Zachara, N.E.; Nielsen, P.H.; Kimose, H.H.; Kristiansen, S.B.; Bøtker, H.E. Impact of O-GlcNAc on Cardioprotection by Remote Ischaemic Preconditioning in Non-Diabetic and Diabetic Patients. Cardiovasc. Res. 2013, 97, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Martinez, M.; Sengupta, S.; Lee, A.; Wu, X.; Chaerkady, R.; Chatterjee, A.; O’Meally, R.N.; Cole, R.N.; Pandey, A.; et al. Quantitative Phosphoproteomics Reveals Crosstalk Between Phosphorylation and O-GlcNAc in the DNA Damage Response Pathway. Proteomics 2015, 15, 591–607. [Google Scholar] [CrossRef] [Green Version]
- Jensen, R.V.; Johnsen, J.; Kristiansen, S.B.; Zachara, N.E.; Bøtker, H.E. Ischemic Preconditioning Increases Myocardial O-GlcNAc Glycosylation. Scand. Cardiovasc. J. 2013, 47, 168–174. [Google Scholar] [CrossRef]
- Ryu, I.-H.; Do, S.-I. Denitrosylation of S-Nitrosylated OGT Is Triggered in LPS-Stimulated Innate Immune Response. Biochem. Biophys. Res. Commun. 2011, 408, 52–57. [Google Scholar] [CrossRef]
- Seo, H.G.; Kim, H.B.; Yoon, J.Y.; Kweon, T.H.; Park, Y.S.; Kang, J.; Jung, J.; Son, S.; Yi, E.C.; Lee, T.H.; et al. Mutual Regulation between OGT and XIAP to Control Colon Cancer Cell Growth and Invasion. Cell Death Dis. 2020, 11, 815. [Google Scholar] [CrossRef]
- Kaasik, K.; Kivimäe, S.; Allen, J.J.; Chalkley, R.J.; Huang, Y.; Baer, K.; Kissel, H.; Burlingame, A.L.; Shokat, K.M.; Ptáček, L.J.; et al. Glucose Sensor O-GlcNAcylation Coordinates with Phosphorylation to Regulate Circadian Clock. Cell Metab. 2013, 17, 291–302. [Google Scholar] [CrossRef]
- Rao, F.V.; Dorfmueller, H.C.; Villa, F.; Allwood, M.; Eggleston, I.M.; van Aalten, D.M.F. Structural Insights into the Mechanism and Inhibition of Eukaryotic O-GlcNAc Hydrolysis. EMBO J. 2006, 25, 1569–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimpl, M.; Borodkin, V.S.; Gray, L.J.; van Aalten, D.M.F. Synergy of Peptide and Sugar in O-GlcNAcase Substrate Recognition. Chem. Biol. 2012, 19, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Li, H.; Hu, C.-W.; Jiang, J. Structural Insights into the Substrate Binding Adaptability and Specificity of Human O-GlcNAcase. Nat. Commun. 2017, 8, 666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, J.; Pils, B. Prediction of Structure and Functional Residues for O-GlcNAcase, a Divergent Homologue of Acetyltransferases. FEBS Lett. 2002, 529, 179–182. [Google Scholar] [CrossRef] [Green Version]
- Elsen, N.L.; Patel, S.B.; Ford, R.E.; Hall, D.L.; Hess, F.; Kandula, H.; Kornienko, M.; Reid, J.; Selnick, H.; Shipman, J.M.; et al. Insights into Activity and Inhibition from the Crystal Structure of Human O-GlcNAcase. Nat. Chem. Biol. 2017, 13, 613–615. [Google Scholar] [CrossRef]
- Keembiyehetty, C.N.; Krzeslak, A.; Love, D.C.; Hanover, J.A. A Lipid-Droplet-Targeted O-GlcNAcase Isoform Is a Key Regulator of the Proteasome. J. Cell Sci. 2011, 124, 2851–2860. [Google Scholar] [CrossRef] [Green Version]
- Pagesy, P.; Bouaboud, A.; Feng, Z.; Hulin, P.; Issad, T. Short O-GlcNAcase Is Targeted to the Mitochondria and Regulates Mitochondrial Reactive Oxygen Species Level. Cells 2022, 11, 1827. [Google Scholar] [CrossRef]
- Dontaine, J.; Bouali, A.; Daussin, F.; Bultot, L.; Vertommen, D.; Martin, M.; Rathagirishnan, R.; Cuillerier, A.; Horman, S.; Beauloye, C.; et al. The Intra-Mitochondrial O-GlcNAcylation System Rapidly Modulates OXPHOS Function and ROS Release in the Heart. Commun. Biol. 2022, 5, 349. [Google Scholar] [CrossRef]
- Wells, L.; Gao, Y.; Mahoney, J.A.; Vosseller, K.; Chen, C.; Rosen, A.; Hart, G.W. Dynamic O-Glycosylation of Nuclear and Cytosolic Proteins: Further Characterization of the Nucleocytoplasmic Beta-N-Acetylglucosaminidase, O-GlcNAcase. J. Biol. Chem. 2002, 277, 1755–1761. [Google Scholar] [CrossRef] [Green Version]
- Butkinaree, C.; Cheung, W.D.; Park, S.; Park, K.; Barber, M.; Hart, G.W. Characterization of Beta-N-Acetylglucosaminidase Cleavage by Caspase-3 during Apoptosis. J. Biol. Chem. 2008, 283, 23557–23566. [Google Scholar] [CrossRef]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A Proteome-Wide, Quantitative Survey of in Vivo Ubiquitylation Sites Reveals Widespread Regulatory Roles. Mol. Cell. Proteom. 2011, 10, M111.013284. [Google Scholar] [CrossRef] [Green Version]
- Biarc, J.; Chalkley, R.J.; Burlingame, A.L.; Bradshaw, R.A. The Induction of Serine/Threonine Protein Phosphorylations by a PDGFR/TrkA Chimera in Stably Transfected PC12 Cells. Mol. Cell. Proteom. 2012, 11, 15–30. [Google Scholar] [CrossRef] [Green Version]
- Udeshi, N.D.; Svinkina, T.; Mertins, P.; Kuhn, E.; Mani, D.R.; Qiao, J.W.; Carr, S.A. Refined Preparation and Use of Anti-Diglycine Remnant (K-ε-GG) Antibody Enables Routine Quantification of 10,000s of Ubiquitination Sites in Single Proteomics Experiments*. Mol. Cell. Proteom. 2013, 12, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Muthusamy, S.; DeMartino, A.M.; Watson, L.J.; Brittian, K.R.; Zafir, A.; Dassanayaka, S.; Hong, K.U.; Jones, S.P. MicroRNA-539 Is up-Regulated in Failing Heart, and Suppresses O-GlcNAcase Expression. J. Biol. Chem. 2014, 289, 29665–29676. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Cao, M.; Ruan, X.; Jiang, L.; Lee, S.; Lemanek, A.; Ghassemian, M.; Pizzo, D.P.; Wan, Y.; Qiao, Y.; et al. Cancer-Cell-Secreted MiR-122 Suppresses O-GlcNAcylation to Promote Skeletal Muscle Proteolysis. Nat. Cell Biol. 2022, 24, 793–804. [Google Scholar] [CrossRef]
- Park, S.-K.; Zhou, X.; Pendleton, K.E.; Hunter, O.V.; Kohler, J.J.; O’Donnell, K.A.; Conrad, N.K. A Conserved Splicing Silencer Dynamically Regulates O-GlcNAc Transferase Intron Retention and O-GlcNAc Homeostasis. Cell Rep. 2017, 20, 1088–1099. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.-W.; Fei, G.; Paulo, J.A.; Bellaousov, S.; Martin, S.E.S.; Duveau, D.Y.; Thomas, C.J.; Gygi, S.P.; Boutz, P.L.; Walker, S. O-GlcNAc Regulates Gene Expression by Controlling Detained Intron Splicing. Nucleic Acids Res. 2020, 48, 5656–5669. [Google Scholar] [CrossRef] [Green Version]
- Ninomiya, K.; Kataoka, N.; Hagiwara, M. Stress-Responsive Maturation of Clk1/4 Pre-MRNAs Promotes Phosphorylation of SR Splicing Factor. J. Cell Biol. 2011, 195, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Xu, J.; Song, Y.; Xin, C.; Liu, L.; Hou, N.; Teng, Y.; Cheng, X.; Wang, T.; Yu, Z.; et al. PRMT5 Prevents Dilated Cardiomyopathy via Suppression of Protein O-GlcNAcylation. Circ. Res. 2021, 129, 857–871. [Google Scholar] [CrossRef]
- Schimpl, M.; Schüttelkopf, A.W.; Borodkin, V.S.; van Aalten, D.M.F. Human OGA Binds Substrates in a Conserved Peptide Recognition Groove. Biochem. J. 2010, 432, 1–7. [Google Scholar] [CrossRef]
- Stephen, H.M.; Praissman, J.L.; Wells, L. Generation of an Interactome for the Tetratricopeptide Repeat Domain of O-GlcNAc Transferase Indicates a Role for the Enzyme in Intellectual Disability. J. Proteome Res. 2021, 20, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, N.; Zachara, N.E.; Hart, G.W.; Marth, J.D. Ogt-Dependent X-Chromosome-Linked Protein Glycosylation Is a Requisite Modification in Somatic Cell Function and Embryo Viability. Mol. Cell. Biol. 2004, 24, 1680–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafi, R.; Iyer, S.P.; Ellies, L.G.; O’Donnell, N.; Marek, K.W.; Chui, D.; Hart, G.W.; Marth, J.D. The O-GlcNAc Transferase Gene Resides on the X Chromosome and Is Essential for Embryonic Stem Cell Viability and Mouse Ontogeny. Proc. Natl. Acad. Sci. USA 2000, 97, 5735–5739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, Y.; Yu, H.; Wu, T.; Zhang, J.; Evans, S.M.; Chen, J. O-Linked β-N-Acetylglucosamine Transferase Plays an Essential Role in Heart Development through Regulating Angiopoietin-1. PLoS Genet. 2020, 16, e1008730. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Park, S.Y.; Choi, Y.R.; Kang, J.G.; Joo, H.J.; Moon, W.K.; Cho, J.W. Excessive O-GlcNAcylation of Proteins Suppresses Spontaneous Cardiogenesis in ES Cells. FEBS Lett. 2009, 583, 2474–2478. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Woo, J.S.; Joo, H.J.; Moon, W.K. Cardiac Transcription Factor Nkx2.5 Is Downregulated under Excessive O-GlcNAcylation Condition. PLoS ONE 2012, 7, e38053. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.; Kim, T.W.; Yoon, S.; Choi, S.-Y.; Kang, T.-W.; Kim, S.-Y.; Kwon, Y.-W.; Cho, E.-J.; Youn, H.-D. O-GlcNAc Regulates Pluripotency and Reprogramming by Directly Acting on Core Components of the Pluripotency Network. Cell Stem Cell 2012, 11, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Speakman, C.M.; Domke, T.C.E.; Wongpaiboonwattana, W.; Sanders, K.; Mudaliar, M.; van Aalten, D.M.F.; Barton, G.J.; Stavridis, M.P. Elevated O-GlcNAc Levels Activate Epigenetically Repressed Genes and Delay Mouse ESC Differentiation Without Affecting Naïve to Primed Cell Transition. Stem Cells 2014, 32, 2605–2615. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Fan, X.; Shi, Y.; Zhang, C.; Sun, D.-E.; Qin, K.; Qin, W.; Zhou, W.; Chen, X. Next-Generation Unnatural Monosaccharides Reveal That ESRRB O-GlcNAcylation Regulates Pluripotency of Mouse Embryonic Stem Cells. Nat. Commun. 2019, 10, 4065. [Google Scholar] [CrossRef] [Green Version]
- Zafir, A.; Bradley, J.A.; Long, B.W.; Muthusamy, S.; Li, Q.; Hill, B.G.; Wysoczynski, M.; Prabhu, S.D.; Bhatnagar, A.; Bolli, R.; et al. O-GlcNAcylation Negatively Regulates Cardiomyogenic Fate in Adult Mouse Cardiac Mesenchymal Stromal Cells. PLoS ONE 2015, 10, e0142939. [Google Scholar] [CrossRef]
- Umapathi, P.; Mesubi, O.O.; Banerjee, P.S.; Abrol, N.; Wang, Q.; Luczak, E.D.; Wu, Y.; Granger, J.M.; Wei, A.-C.; Gaido, O.E.R.; et al. Excessive O-GlcNAcylation Causes Heart Failure and Sudden Death. Circulation 2021, 143, 1687–1703. [Google Scholar] [CrossRef]
- Ma, J.; Banerjee, P.; Whelan, S.A.; Liu, T.; Wei, A.-C.; Ramirez-Correa, G.; McComb, M.E.; Costello, C.E.; O’Rourke, B.; Murphy, A.; et al. Comparative Proteomics Reveals Dysregulated Mitochondrial O-GlcNAcylation in Diabetic Hearts. J. Proteome Res. 2016, 15, 2254–2264. [Google Scholar] [CrossRef]
- Ma, J.; Liu, T.; Wei, A.-C.; Banerjee, P.; O’Rourke, B.; Hart, G.W. O-GlcNAcomic Profiling Identifies Widespread O-Linked β-N-Acetylglucosamine Modification (O-GlcNAcylation) in Oxidative Phosphorylation System Regulating Cardiac Mitochondrial Function* ♦. J. Biol. Chem. 2015, 290, 29141–29153. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Wen, L.; Mu, Y. O-GlcNAcylation Is Essential for Autophagy in Cardiomyocytes. Oxidative Med. Cell. Longev. 2020, 2020, 5602396. [Google Scholar] [CrossRef]
- Ednie, A.R.; Bennett, E.S. Intracellular O-Linked Glycosylation Directly Regulates Cardiomyocyte L-Type Ca2+ Channel Activity and Excitation–Contraction Coupling. Basic Res. Cardiol. 2020, 115, 59. [Google Scholar] [CrossRef]
- Erickson, J.R.; Pereira, L.; Wang, L.; Han, G.; Ferguson, A.; Dao, K.; Copeland, R.J.; Despa, F.; Hart, G.W.; Ripplinger, C.M.; et al. Diabetic Hyperglycaemia Activates CaMKII and Arrhythmias by O-Linked Glycosylation. Nature 2013, 502, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Hegyi, B.; Fasoli, A.; Ko, C.Y.; Van, B.W.; Alim, C.C.; Shen, E.Y.; Ciccozzi, M.M.; Tapa, S.; Ripplinger, C.M.; Erickson, J.R.; et al. CaMKII Serine 280 O-GlcNAcylation Links Diabetic Hyperglycemia to Proarrhythmia. Circ. Res. 2021, 129, 98–113. [Google Scholar] [CrossRef]
- Zhu-Mauldin, X.; Marsh, S.A.; Zou, L.; Marchase, R.B.; Chatham, J.C. Modification of STIM1 by O-Linked N-Acetylglucosamine (O-GlcNAc) Attenuates Store-Operated Calcium Entry in Neonatal Cardiomyocytes*. J. Biol. Chem. 2012, 287, 39094–39106. [Google Scholar] [CrossRef] [Green Version]
- Nomura, A.; Yokoe, S.; Tomoda, K.; Nakagawa, T.; Martin-Romero, F.J.; Asahi, M. Fluctuation in O-GlcNAcylation Inactivates STIM1 to Reduce Store-Operated Calcium Ion Entry via down-Regulation of Ser621 Phosphorylation. J. Biological. Chem. 2020, 295, 17071–17082. [Google Scholar] [CrossRef]
- Keembiyehetty, C.; Love, D.C.; Harwood, K.R.; Gavrilova, O.; Comly, M.E.; Hanover, J.A. Conditional Knock-out Reveals a Requirement for O-Linked N-Acetylglucosaminase (O-GlcNAcase) in Metabolic Homeostasis. J. Biol. Chem. 2015, 290, 7097–7113. [Google Scholar] [CrossRef]
- Yang, Y.R.; Song, M.; Lee, H.; Jeon, Y.; Choi, E.-J.; Jang, H.-J.; Moon, H.Y.; Byun, H.-Y.; Kim, E.-K.; Kim, D.H.; et al. O-GlcNAcase Is Essential for Embryonic Development and Maintenance of Genomic Stability. Aging Cell 2012, 11, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muha, V.; Authier, F.; Szoke-Kovacs, Z.; Johnson, S.; Gallagher, J.; McNeilly, A.; McCrimmon, R.J.; Teboul, L.; van Aalten, D.M.F. Loss of O-GlcNAcase Catalytic Activity Leads to Defects in Mouse Embryogenesis. J. Biological. Chem. 2021, 296, 100439. [Google Scholar] [CrossRef] [PubMed]
- Dassanayaka, S.; Brittian, K.R.; Long, B.W.; Higgins, L.A.; Bradley, J.A.; Audam, T.N.; Jurkovic, A.; Gumpert, A.M.; Harrison, L.T.; Hartyánszky, I.; et al. Cardiomyocyte Oga Haploinsufficiency Increases O-GlcNAcylation but Hastens Ventricular Dysfunction Following Myocardial Infarction. PLoS ONE 2020, 15, e0242250. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.S.; Ma, J.; Hart, G.W. Diabetes-Associated Dysregulation of O-GlcNAcylation in Rat Cardiac Mitochondria. Proc. Natl. Acad. Sci. USA 2015, 112, 6050–6055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Suarez, J.; Fricovsky, E.; Wang, H.; Scott, B.T.; Trauger, S.A.; Han, W.; Hu, Y.; Oyeleye, M.O.; Dillmann, W.H. Increased Enzymatic O-GlcNAcylation of Mitochondrial Proteins Impairs Mitochondrial Function in Cardiac Myocytes Exposed to High Glucose. J. Biol. Chem. 2009, 284, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Prakoso, D.; Lim, S.Y.; Erickson, J.R.; Wallace, R.S.; Lees, J.G.; Tate, M.; Kiriazis, H.; Donner, D.G.; Henstridge, D.C.; Davey, J.R.; et al. Fine-Tuning the Cardiac O-GlcNAcylation Regulatory Enzymes Governs the Functional and Structural Phenotype of the Diabetic Heart. Cardiovasc. Res. 2021, 118, cvab043. [Google Scholar] [CrossRef]
- Ramirez-Correa, G.A.; Ma, J.; Slawson, C.; Zeidan, Q.; Lugo-Fagundo, N.S.; Xu, M.; Shen, X.; Gao, W.D.; Caceres, V.; Chakir, K.; et al. Removal of Abnormal Myofilament O-GlcNAcylation Restores Ca2+ Sensitivity in Diabetic Cardiac Muscle. Diabetes 2015, 64, 3573–3587. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Liao, Z.; Lu, X.; Katschinski, D.M.; Mercola, M.; Chen, J.; Brown, J.H.; Molkentin, J.D.; Bossuyt, J.; Bers, D.M. Hyperglycemia Acutely Increases Cytosolic Reactive Oxygen Species via O-Linked GlcNAcylation and CaMKII Activation in Mouse Ventricular Myocytes. Circ. Res. 2020, 126, e80–e96. [Google Scholar] [CrossRef]
- Mesubi, O.O.; Rokita, A.G.; Abrol, N.; Wu, Y.; Chen, B.; Wang, Q.; Granger, J.M.; Tucker-Bartley, A.; Luczak, E.D.; Murphy, K.R.; et al. Oxidized CaMKII and O-GlcNAcylation Cause Increased Atrial Fibrillation in Diabetic Mice by Distinct Mechanisms. J. Clin. Investig. 2021, 131, e95747. [Google Scholar] [CrossRef]
- Lunde, I.G.; Aronsen, J.M.; Kvaløy, H.; Qvigstad, E.; Sjaastad, I.; Tønnessen, T.; Christensen, G.; Grønning-Wang, L.M.; Carlson, C.R. Cardiac O-GlcNAc Signaling Is Increased in Hypertrophy and Heart Failure. Physiol. Genom. 2012, 44, 162–172. [Google Scholar] [CrossRef]
- Facundo, H.T.; Brainard, R.E.; Watson, L.J.; Ngoh, G.A.; Hamid, T.; Prabhu, S.D.; Jones, S.P. O-GlcNAc Signaling Is Essential for NFAT-Mediated Transcriptional Reprogramming during Cardiomyocyte Hypertrophy. AJP Heart Circ. Physiol. 2012, 302, H2122–H2130. [Google Scholar] [CrossRef] [Green Version]
- Dassanayaka, S.; Brainard, R.E.; Watson, L.J.; Long, B.W.; Brittian, K.R.; DeMartino, A.M.; Aird, A.L.; Gumpert, A.M.; Audam, T.N.; Kilfoil, P.J.; et al. Cardiomyocyte Ogt Limits Ventricular Dysfunction in Mice Following Pressure Overload without Affecting Hypertrophy. Basic Res. Cardiol. 2017, 112, 23. [Google Scholar] [CrossRef] [Green Version]
- Ledee, D.; Smith, L.; Bruce, M.; Kajimoto, M.; Isern, N.; Portman, M.A.; Olson, A.K. C-Myc Alters Substrate Utilization and O-GlcNAc Protein Posttranslational Modifications without Altering Cardiac Function during Early Aortic Constriction. PLoS ONE 2015, 10, e0135262. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; El-Nachef, D.; Yang, X.; Ledee, D.; Olson, A.K. O-GlcNAc Transferase Promotes Compensated Cardiac Function and Protein Kinase A O-GlcNAcylation During Early and Established Pathological Hypertrophy From Pressure Overload. J. Am. Heart Assoc. 2019, 8, e011260. [Google Scholar] [CrossRef]
- Gélinas, R.; Mailleux, F.; Dontaine, J.; Bultot, L.; Demeulder, B.; Ginion, A.; Daskalopoulos, E.P.; Esfahani, H.; Dubois-Deruy, E.; Lauzier, B.; et al. AMPK Activation Counteracts Cardiac Hypertrophy by Reducing O-GlcNAcylation. Nat. Commun. 2018, 9, 374. [Google Scholar] [CrossRef] [Green Version]
- Nabeebaccus, A.A.; Verma, S.; Zoccarato, A.; Emanuelli, G.; Santos, C.X.C.; Streckfuss-Bömeke, K.; Shah, A.M. Cardiomyocyte Protein O-GlcNAcylation Is Regulated by GFAT1 Not GFAT2. Biochem. Biophys. Res. Commun. 2021, 583, 121–127. [Google Scholar] [CrossRef]
- Tran, D.H.; May, H.I.; Li, Q.; Luo, X.; Huang, J.; Zhang, G.; Niewold, E.; Wang, X.; Gillette, T.G.; Deng, Y.; et al. Chronic Activation of Hexosamine Biosynthesis in the Heart Triggers Pathological Cardiac Remodeling. Nat. Commun. 2020, 11, 1771. [Google Scholar] [CrossRef] [Green Version]
- Ishikita, A.; Matsushima, S.; Ikeda, S.; Okabe, K.; Nishimura, R.; Tadokoro, T.; Enzan, N.; Yamamoto, T.; Sada, M.; Tsutsui, Y.; et al. GFAT2 Mediates Cardiac Hypertrophy through HBP-O-GlcNAcylation-Akt Pathway. Iscience 2021, 24, 103517. [Google Scholar] [CrossRef]
- Brainard, R.E.; Facundo, H.T. Cardiac Hypertrophy Drives PGC-1α Suppression Associated with Enhanced O-Glycosylation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2021, 1867, 166080. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Belliard, A.; Mulder, P.; Bouvet, M.; Smet-Nocca, C.; Janel, S.; Lafont, F.; Beseme, O.; Amouyel, P.; Richard, V.; et al. Interplay between Troponin T Phosphorylation and O-N-Acetylglucosaminylation in Ischaemic Heart Failure. Cardiovasc. Res. 2015, 107, 56–65. [Google Scholar] [CrossRef]
- Laczy, B.; Marsh, S.A.; Brocks, C.A.; Wittmann, I.; Chatham, J.C. Inhibition of O-GlcNAcase in Perfused Rat Hearts by NAG-Thiazolines at the Time of Reperfusion Is Cardioprotective in an O-GlcNAc-Dependent Manner. AJP Heart Circ. Physiol. 2010, 299, H1715–H1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fülöp, N.; Zhang, Z.; Marchase, R.B.; Chatham, J.C. Glucosamine Cardioprotection in Perfused Rat Hearts Associated with Increased O-Linked N-Acetylglucosamine Protein Modification and Altered P38 Activation. AJP Heart Circ. Physiol. 2007, 292, H2227–H2236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; PANG, Y.; Chang, T.; Bounelis, P.; Chatham, J.; Marchase, R. Increased Hexosamine Biosynthesis and Protein O-GlcNAc Levels Associated with Myocardial Protection against Calcium Paradox and Ischemia. J. Mol. Cell. Cardiol. 2006, 40, 303–312. [Google Scholar] [CrossRef]
- Liu, J.; Marchase, R.B.; Chatham, J.C. Glutamine-Induced Protection of Isolated Rat Heart from Ischemia/Reperfusion Injury Is Mediated via the Hexosamine Biosynthesis Pathway and Increased Protein O-GlcNAc Levels. J. Mol. Cell. Cardiol. 2007, 42, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Marchase, R.B.; Chatham, J.C. Increased O-GlcNAc Levels during Reperfusion Lead to Improved Functional Recovery and Reduced Calpain Proteolysis. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1391–H1399. [Google Scholar] [CrossRef] [Green Version]
- Ngoh, G.A.; Watson, L.J.; Facundo, H.T.; Dillmann, W.; Jones, S.P. Non-Canonical Glycosyltransferase Modulates Post-Hypoxic Cardiac Myocyte Death and Mitochondrial Permeability Transition. J. Mol. Cell. Cardiol. 2008, 45, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of Mitochondrial ATP Synthase Form the Permeability Transition Pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [Green Version]
- Ngoh, G.A.; Hamid, T.; Prabhu, S.D.; Jones, S.P. O-GlcNAc Signaling Attenuates ER Stress-Induced Cardiomyocyte Death. AJP Heart Circ. Physiol. 2009, 297, H1711–H1719. [Google Scholar] [CrossRef] [Green Version]
- Latorre-Muro, P.; O’Malley, K.E.; Bennett, C.F.; Perry, E.A.; Balsa, E.; Tavares, C.D.J.; Jedrychowski, M.; Gygi, S.P.; Puigserver, P. A Cold-Stress-Inducible PERK/OGT Axis Controls TOM70-Assisted Mitochondrial Protein Import and Cristae Formation. Cell Metab. 2021, 33, 598–614.e7. [Google Scholar] [CrossRef]
- Zeidan, Q.; Wang, Z.; Maio, A.D.; Hart, G.W. O-GlcNAc Cycling Enzymes Associate with the Translational Machinery and Modify Core Ribosomal Proteins. Mol. Biol. Cell 2010, 21, 1922–1936. [Google Scholar] [CrossRef]
- Li, X.; Zhu, Q.; Shi, X.; Cheng, Y.; Li, X.; Xu, H.; Duan, X.; Hsieh-Wilson, L.C.; Chu, J.; Pelletier, J.; et al. O-GlcNAcylation of Core Components of the Translation Initiation Machinery Regulates Protein Synthesis. Proc. Natl. Acad. Sci. USA 2019, 116, 7857–7866. [Google Scholar] [CrossRef] [Green Version]
- Jang, I.; Kim, H.B.; Seo, H.; Kim, J.Y.; Choi, H.; Yoo, J.S.; Kim, J.; Cho, J.W. O-GlcNAcylation of EIF2α Regulates the Phospho-EIF2α-Mediated ER Stress Response. Biochim. Biophys. Acta 2015, 1853, 1860–1869. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.-H.; Chang, H.-I. O-Linked N-Acetylglucosamine Suppresses Thermal Aggregation of Sp1. FEBS Lett. 2006, 580, 4645–4652. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shu, X.E.; Qian, S.-B. O-GlcNAc Modification of EIF4GI Acts as a Translational Switch in Heat Shock Response. Nat. Publ. Group 2018, 14, 909–916. [Google Scholar] [CrossRef]
- Alejandro, E.U.; Bozadjieva, N.; Kumusoglu, D.; Abdulhamid, S.; Levine, H.; Haataja, L.; Vadrevu, S.; Satin, L.S.; Arvan, P.; Bernal-Mizrachi, E. Disruption of O-Linked N-Acetylglucosamine Signaling Induces ER Stress and β Cell Failure. Cell Rep. 2015, 13, 2527–2538. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Collins, H.E.; Young, M.E.; Zhang, J.; Wende, A.R.; Darley-Usmar, V.M.; Chatham, J.C. The Identification of a Novel Calcium-Dependent Link Between NAD+ and Glucose Deprivation-Induced Increases in Protein O-GlcNAcylation and ER Stress. Front. Mol. Biosci. 2021, 8, 780865. [Google Scholar] [CrossRef]
- Taylor, R.P.; Parker, G.J.; Hazel, M.W.; Soesanto, Y.; Fuller, W.; Yazzie, M.J.; McClain, D.A. Glucose Deprivation Stimulates O-GlcNAc Modification of Proteins through Up-Regulation of O-Linked N-Acetylglucosaminyltransferase*. J. Biol. Chem. 2008, 283, 6050–6057. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.P.; Mihailescu, M.L.; Yang, C.; Barber, A.J.; Kimball, S.R.; Jefferson, L.S.; Dennis, M.D. The Translational Repressor 4E-BP1 Contributes to Diabetes-Induced Visual Dysfunction. Investig. Opthalmology Vis. Sci. 2016, 57, 1327–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, M.D.; Shenberger, J.S.; Stanley, B.A.; Kimball, S.R.; Jefferson, L.S. Hyperglycemia Mediates a Shift from Cap-Dependent to Cap-Independent Translation Via a 4E-BP1–Dependent Mechanism. Diabetes 2013, 62, 2204–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierschke, S.K.; Miller, W.P.; Favate, J.S.; Shah, P.; Kawasawa, Y.I.; Salzberg, A.C.; Kimball, S.R.; Jefferson, L.S.; Dennis, M.D. O-GlcNAcylation Alters the Selection of MRNAs for Translation and Promotes 4E-BP1–Dependent Mitochondrial Dysfunction in the Retina. J. Biol. Chem. 2019, 294, 5508–5520. [Google Scholar] [CrossRef]
- Yang, S.; Zou, L.-Y.; Bounelis, P.; Chaudry, I.; Chatham, J.C.; Marchase, R.B. Glucosamine Administration during Resuscitation Improves Organ Function after Trauma Hemorrhage. Shock 2006, 25, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Nöt, L.G.; Marchase, R.B.; Fülöp, N.; Brocks, C.A.; Chatham, J.C. Glucosamine Administration Improves Survival Rate after Severe Hemorrhagic Shock Combined With Trauma in Rats. Shock 2007, 28, 345–352. [Google Scholar] [CrossRef]
- Zou, L.; Yang, S.; Champattanachai, V.; Hu, S.; Chaudry, I.H.; Marchase, R.B.; Chatham, J.C. Glucosamine Improves Cardiac Function Following Trauma-Hemorrhage by Increased Protein O-GlcNAcylation and Attenuation of NF- B Signaling. AJP Heart Circ. Physiol. 2008, 296, H515–H523. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, P.; Clark, P.M.; Mason, D.E.; Peters, E.C.; Hsieh-Wilson, L.C.; Baltimore, D. Activation of the Transcriptional Function of the NF-ΚB Protein c-Rel by O-GlcNAc Glycosylation. Sci. Signal. 2013, 6, ra75. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Chalkley, R.J.; Vosseller, K. Hyper-O-GlcNAcylation Activates Nuclear Factor κ-Light-Chain-Enhancer of Activated B Cells (NF-ΚB) Signaling through Interplay with Phosphorylation and Acetylation. J. Biological. Chem. 2017, 292, 9150–9163. [Google Scholar] [CrossRef] [Green Version]
- Nöt, L.G.; Brocks, C.A.; Vámhidy, L.; Marchase, R.B.; Chatham, J.C. Increased O-Linked β-N-Acetylglucosamine Levels on Proteins Improves Survival, Reduces Inflammation and Organ Damage 24 Hours after Trauma-Hemorrhage in Rats. Crit. Care Med. 2010, 38, 562–571. [Google Scholar] [CrossRef]
- Ferron, M.; Cadiet, J.; Persello, A.; Prat, V.; Denis, M.; Erraud, A.; Aillerie, V.; Mevel, M.; Bigot, E.; Chatham, J.C.; et al. O-GlcNAc Stimulation: A New Metabolic Approach to Treat Septic Shock. Sci. Rep. 2019, 9, 18751. [Google Scholar] [CrossRef] [Green Version]
- Denis, M.; Dupas, T.; Persello, A.; Dontaine, J.; Bultot, L.; Betus, C.; Pelé, T.; Dhot, J.; Erraud, A.; Maillard, A.; et al. An O-GlcNAcylomic Approach Reveals ACLY as a Potential Target in Sepsis in the Young Rat. Int. J. Mol. Sci. 2021, 22, 9236. [Google Scholar] [CrossRef]
- Dupas, T.; Persello, A.; Blangy-Letheule, A.; Denis, M.; Erraud, A.; Aillerie, V.; Leroux, A.A.; Rivière, M.; Lebreton, J.; Tessier, A.; et al. Beneficial Effects of O-GlcNAc Stimulation in a Young Rat Model of Sepsis: Beyond Modulation of Gene Expression. Int. J. Mol. Sci. 2022, 23, 6430. [Google Scholar] [CrossRef]
- Liao, L.; Cheng, D.; Wang, J.; Duong, D.M.; Losik, T.G.; Gearing, M.; Rees, H.D.; Lah, J.J.; Levey, A.I.; Peng, J. Proteomic Characterization of Postmortem Amyloid Plaques Isolated by Laser Capture Microdissection*. J. Biol. Chem. 2004, 279, 37061–37068. [Google Scholar] [CrossRef]
- Woerner, A.C.; Frottin, F.; Hornburg, D.; Feng, L.R.; Meissner, F.; Patra, M.; Tatzelt, J.; Mann, M.; Winklhofer, K.F.; Hartl, F.U.; et al. Cytoplasmic Protein Aggregates Interfere with Nucleocytoplasmic Transport of Protein and RNA. Science 2016, 351, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, H.; Guo, Q.; Bader, J.; Frottin, F.; Farny, D.; Kleinberger, G.; Haass, C.; Mann, M.; Hartl, F.U.; Baumeister, W.; et al. Gel-like Inclusions of C-terminal Fragments of TDP-43 Sequester Stalled Proteasomes in Neurons. EMBO Rep. 2022, 23, e53890. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.C.; Jensen, E.H.; Rexach, J.E.; Vinters, H.V.; Hsieh-Wilson, L.C. Loss of O-GlcNAc Glycosylation in Forebrain Excitatory Neurons Induces Neurodegeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 15120–15125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Ha, H.-J.; Chung, E.S.; Baek, S.H.; Cho, Y.; Kim, H.K.; Han, J.; Sul, J.H.; Lee, J.; Kim, E.; et al. O-GlcNAcylation Ameliorates the Pathological Manifestations of Alzheimer’s Disease by Inhibiting Necroptosis. Sci. Adv. 2021, 7, eabd3207. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.J.; Coleman, P.D. Reduction of O-Linked N-Acetylglucosamine-Modified Assembly Protein-3 in Alzheimer’s Disease. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 2399–2411. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.-X. O-GlcNAcylation Regulates Phosphorylation of Tau: A Mechanism Involved in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Yang, F.; Petyuk, V.A.; Shukla, A.K.; Monroe, M.E.; Gritsenko, M.A.; Rodland, K.D.; Smith, R.D.; Qian, W.-J.; Gong, C.-X.; et al. Quantitative Proteomics Identifies Altered O-GlcNAcylation of Structural, Synaptic and Memory-Associated Proteins in Alzheimer’s Disease. J. Pathol. 2017, 243, 78–88. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.S.; Johnson, G.V.; Cole, R.N.; Dong, D.L.; Lee, M.; Hart, G.W. The Microtubule-Associated Protein Tau Is Extensively Modified with O-Linked N-Acetylglucosamine. J. Biol. Chem. 1996, 271, 28741–28744. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Shi, J.; Tanimukai, H.; Gu, J.; Gu, J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.-X. Reduced O-GlcNAcylation Links Lower Brain Glucose Metabolism and Tau Pathology in Alzheimer’s Disease. Brain 2009, 132, 1820–1832. [Google Scholar] [CrossRef] [Green Version]
- Yuzwa, S.A.; Yadav, A.K.; Skorobogatko, Y.; Clark, T.; Vosseller, K.; Vocadlo, D.J. Mapping O-GlcNAc Modification Sites on Tau and Generation of a Site-Specific O-GlcNAc Tau Antibody. Amino Acids 2011, 40, 857–868. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Shan, X.; Macauley, M.S.; Clark, T.; Skorobogatko, Y.; Vosseller, K.; Vocadlo, D.J. Increasing O-GlcNAc Slows Neurodegeneration and Stabilizes Tau against Aggregation. Nat. Chem. Biol. 2012, 8, 393–399. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Cheung, A.H.; Okon, M.; McIntosh, L.P.; Vocadlo, D.J. O-GlcNAc Modification of Tau Directly Inhibits Its Aggregation without Perturbing the Conformational Properties of Tau Monomers. J. Mol. Biol. 2014, 426, 1736–1752. [Google Scholar] [CrossRef]
- Cantrelle, F.-X.; Loyens, A.; Trivelli, X.; Reimann, O.; Despres, C.; Gandhi, N.S.; Hackenberger, C.P.R.; Landrieu, I.; Smet-Nocca, C. Phosphorylation and O-GlcNAcylation of the PHF-1 Epitope of Tau Protein Induce Local Conformational Changes of the C-Terminus and Modulate Tau Self-Assembly Into Fibrillar Aggregates. Front. Mol. Neurosci. 2021, 14, 661368. [Google Scholar] [CrossRef]
- Borghgraef, P.; Menuet, C.; Theunis, C.; Louis, J.V.; Devijver, H.; Maurin, H.; Smet-Nocca, C.; Lippens, G.; Hilaire, G.; Gijsen, H.; et al. Increasing Brain Protein O-GlcNAc-Ylation Mitigates Breathing Defects and Mortality of Tau.P301L Mice. PLoS ONE 2013, 8, e84442. [Google Scholar] [CrossRef] [Green Version]
- Hastings, N.B.; Wang, X.; Song, L.; Butts, B.D.; Grotz, D.; Hargreaves, R.; Hess, J.F.; Hong, K.-L.K.; Huang, C.R.-R.; Hyde, L.; et al. Inhibition of O-GlcNAcase Leads to Elevation of O-GlcNAc Tau and Reduction of Tauopathy and Cerebrospinal Fluid Tau in RTg4510 Mice. Mol. Neurodegener. 2017, 12, 39. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, W.; Marcus, J.; Pearson, M.; Song, L.; Smith, K.; Terracina, G.; Lee, J.; Hong, K.-L.K.; Lu, S.X.; et al. MK-8719, a Novel and Selective O-GlcNAcase Inhibitor That Reduces the Formation of Pathological Tau and Ameliorates Neurodegeneration in a Mouse Model of Tauopathy. J. Pharmacol. Exp. Ther. 2020, 374, 252–263. [Google Scholar] [CrossRef]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau Post-Translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol. 2021, 11, 595532. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Macauley, M.S.; Heinonen, J.E.; Shan, X.; Dennis, R.J.; He, Y.; Whitworth, G.E.; Stubbs, K.A.; McEachern, E.J.; Davies, G.J.; et al. A Potent Mechanism-Inspired O-GlcNAcase Inhibitor That Blocks Phosphorylation of Tau in Vivo. Nat. Chem. Biol. 2008, 4, 483–490. [Google Scholar] [CrossRef]
- Graham, D.L.; Gray, A.J.; Joyce, J.A.; Yu, D.; O’Moore, J.; Carlson, G.A.; Shearman, M.S.; Dellovade, T.L.; Hering, H. Increased O-GlcNAcylation Reduces Pathological Tau without Affecting Its Normal Phosphorylation in a Mouse Model of Tauopathy. Neuropharmacology 2014, 79, 307–313. [Google Scholar] [CrossRef]
- Jiménez, J.S. Macromolecular Structures and Proteins Interacting with the Microtubule Associated Tau Protein. Neuroscience 2022, in press. [CrossRef]
- Tracy, T.E.; Madero-Pérez, J.; Swaney, D.L.; Chang, T.S.; Moritz, M.; Konrad, C.; Ward, M.E.; Stevenson, E.; Hüttenhain, R.; Kauwe, G.; et al. Tau Interactome Maps Synaptic and Mitochondrial Processes Associated with Neurodegeneration. Cell 2022, 185, 712–728.e14. [Google Scholar] [CrossRef] [PubMed]
- Sui, D.; Liu, M.; Kuo, M.-H. In Vitro Aggregation Assays Using Hyperphosphorylated Tau Protein. J. Vis. Exp. 2015, 95, e51537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorantla, N.V.; Chinnathambi, S. Autophagic Pathways to Clear the Tau Aggregates in Alzheimer’s Disease. Cell Mol. Neurobiol. 2021, 41, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Caballero, B.; Bourdenx, M.; Luengo, E.; Diaz, A.; Sohn, P.D.; Chen, X.; Wang, C.; Juste, Y.R.; Wegmann, S.; Patel, B.; et al. Acetylated Tau Inhibits Chaperone-Mediated Autophagy and Promotes Tau Pathology Propagation in Mice. Nat. Commun. 2021, 12, 2238. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Liang, Q.; Li, L.; Hu, Z.; Wu, F.; Zhang, P.; Ma, Y.; Zhao, B.; Kovács, A.L.; Zhang, Z.; et al. O-GlcNAc-Modification of SNAP-29 Regulates Autophagosome Maturation. Nat. Cell Biol. 2014, 16, 1215–1226. [Google Scholar] [CrossRef]
- Ruan, H.-B.; Ma, Y.; Torres, S.; Zhang, B.; Feriod, C.; Heck, R.M.; Qian, K.; Fu, M.; Li, X.; Nathanson, M.H.; et al. Calcium-Dependent O-GlcNAc Signaling Drives Liver Autophagy in Adaptation to Starvation. Genes Dev. 2017, 31, 1655–1665. [Google Scholar] [CrossRef] [Green Version]
- Luu, L.; Ciccotosto, G.D.; Cappai, R. The Alzheimer’s Disease Amyloid Precursor Protein and Its Neuritogenic Actions. Curr. Alzheimer Res. 2021, 18, 772–786. [Google Scholar] [CrossRef]
- Higgins, L.S.; Murphy, G.M.; Forno, L.S.; Catalano, R.; Cordell, B. P3 Beta-Amyloid Peptide Has a Unique and Potentially Pathogenic Immunohistochemical Profile in Alzheimer’s Disease Brain. Am. J. Pathol. 1996, 149, 585–596. [Google Scholar]
- Kuhn, A.J.; Abrams, B.S.; Knowlton, S.; Raskatov, J.A. Alzheimer’s Disease “Non-Amyloidogenic” P3 Peptide Revisited: A Case for Amyloid-α. ACS Chem. Neurosci. 2020, 11, 1539–1544. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Shan, X.; Jones, B.A.; Zhao, G.; Woodward, M.L.; Li, X.; Zhu, Y.; McEachern, E.J.; Silverman, M.A.; Watson, N.V.; et al. Pharmacological Inhibition of O-GlcNAcase (OGA) Prevents Cognitive Decline and Amyloid Plaque Formation in Bigenic Tau/APP Mutant Mice. Mol. Neurodegener. 2014, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Nam, D.W.; Park, S.Y.; Song, H.; Hong, H.S.; Boo, J.H.; Jung, E.S.; Kim, Y.; Baek, J.Y.; Kim, K.S.; et al. O-Linked β-N-Acetylglucosaminidase Inhibitor Attenuates β-Amyloid Plaque and Rescues Memory Impairment. Neurobiol. Aging 2013, 34, 275–285. [Google Scholar] [CrossRef]
- Jacobsen, K.T.; Iverfeldt, K. O-GlcNAcylation Increases Non-Amyloidogenic Processing of the Amyloid-β Precursor Protein (APP). Biochem. Biophys. Res. Commun. 2011, 404, 882–886. [Google Scholar] [CrossRef]
- Chun, Y.S.; Kwon, O.-H.; Chung, S. O-GlcNAcylation of Amyloid-β Precursor Protein at Threonine 576 Residue Regulates Trafficking and Processing. Biochem. Biophys. Res. Commun. 2017, 490, 486–491. [Google Scholar] [CrossRef]
- Chun, Y.S.; Park, Y.; Oh, H.G.; Kim, T.-W.; Yang, H.O.; Park, M.K.; Chung, S. O-GlcNAcylation Promotes Non-Amyloidogenic Processing of Amyloid-β Protein Precursor via Inhibition of Endocytosis from the Plasma Membrane. J. Alzheimer’s Dis. 2015, 44, 261–275. [Google Scholar] [CrossRef]
- Griffith, L.S.; Mathes, M.; Schmitz, B. Beta-Amyloid Precursor Protein Is Modified with O-Linked N-Acetylglucosamine. J. Neurosci. Res. 1995, 41, 270–278. [Google Scholar] [CrossRef]
- Runwal, G.; Edwards, R.H. The Membrane Interactions of Synuclein: Physiology and Pathology. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 465–485. [Google Scholar] [CrossRef]
- de Boni, L.; Watson, A.H.; Zaccagnini, L.; Wallis, A.; Zhelcheska, K.; Kim, N.; Sanderson, J.; Jiang, H.; Martin, E.; Cantlon, A.; et al. Brain Region-Specific Susceptibility of Lewy Body Pathology in Synucleinopathies Is Governed by α-Synuclein Conformations. Acta Neuropathol. 2022, 143, 453–469. [Google Scholar] [CrossRef]
- Dettmer, U.; Newman, A.J.; von Saucken, V.E.; Bartels, T.; Selkoe, D. KTKEGV Repeat Motifs Are Key Mediators of Normal α-Synuclein Tetramerization: Their Mutation Causes Excess Monomers and Neurotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 9596–9601. [Google Scholar] [CrossRef] [Green Version]
- Imberdis, T.; Fanning, S.; Newman, A.; Ramalingam, N.; Dettmer, U. Alpha-Synuclein, Methods and Protocols. Methods Mol. Biol. 2019, 1948, 77–91. [Google Scholar] [CrossRef]
- Benskey, M.J.; Perez, R.G.; Manfredsson, F.P. The Contribution of Alpha Synuclein to Neuronal Survival and Function—Implications for Parkinson’s Disease. J. Neurochem. 2016, 137, 331–359. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.; Lee, J.; Kim, B.; Moon, H.; Jeong, H.; Lee, K.; Song, W.J.; Hur, J.K.; Oh, Y. Role of Post-Translational Modifications on the Alpha-Synuclein Aggregation-Related Pathogenesis of Parkinson’s Disease. BMB Rep. 2022, 55, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Balana, A.T.; Pratt, M.R. Mechanistic Roles for Altered O-GlcNAcylation in Neurodegenerative Disorders. Biochem. J. 2021, 478, 2733–2758. [Google Scholar] [CrossRef] [PubMed]
- Wani, W.Y.; Ouyang, X.; Benavides, G.A.; Redmann, M.; Cofield, S.S.; Shacka, J.J.; Chatham, J.C.; Darley-Usmar, V.; Zhang, J. O-GlcNAc Regulation of Autophagy and α-Synuclein Homeostasis; Implications for Parkinson’s Disease. Mol. Brain 2017, 10, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marotta, N.P.; Lin, Y.H.; Lewis, Y.E.; Ambroso, M.R.; Zaro, B.W.; Roth, M.T.; Arnold, D.B.; Langen, R.; Pratt, M.R. O-GlcNAc Modification Blocks the Aggregation and Toxicity of the Protein α-Synuclein Associated with Parkinson’s Disease. Nat. Chem. 2015, 7, 913–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, P.M.; Galesic, A.; Balana, A.T.; Mahul-Mellier, A.-L.; Navarro, M.X.; Leon, C.A.D.; Lashuel, H.A.; Pratt, M.R. α-Synuclein O-GlcNAcylation Alters Aggregation and Toxicity, Revealing Certain Residues as Potential Inhibitors of Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2019, 116, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, Y.E.; Galesic, A.; Levine, P.M.; Leon, C.A.D.; Lamiri, N.; Brennan, C.K.; Pratt, M.R. O-GlcNAcylation of α-Synuclein at Serine 87 Reduces Aggregation without Affecting Membrane Binding. ACS Chem. Biol. 2017, 12, 1020–1027. [Google Scholar] [CrossRef] [Green Version]
- Tavassoly, O.; Yue, J.; Vocadlo, D.J. Pharmacological Inhibition and Knockdown of O-GlcNAcase Reduces Cellular Internalization of A-synuclein Preformed Fibrils. FEBS J. 2021, 288, 452–470. [Google Scholar] [CrossRef]
- Ihse, E.; Yamakado, H.; van Wijk, X.M.; Lawrence, R.; Esko, J.D.; Masliah, E. Cellular Internalization of Alpha-Synuclein Aggregates by Cell Surface Heparan Sulfate Depends on Aggregate Conformation and Cell Type. Sci. Rep. 2017, 7, 9008. [Google Scholar] [CrossRef] [Green Version]
- Aulić, S.; Masperone, L.; Narkiewicz, J.; Isopi, E.; Bistaffa, E.; Ambrosetti, E.; Pastore, B.; Cecco, E.D.; Scaini, D.; Zago, P.; et al. α-Synuclein Amyloids Hijack Prion Protein to Gain Cell Entry, Facilitate Cell-to-Cell Spreading and Block Prion Replication. Sci. Rep. 2017, 7, 10050. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.-Q.; Jia, C.; Lim, Y.-J.; Feng, G.; Xu, E.; Long, H.; Kimura, Y.; Tao, Y.; Zhao, C.; et al. Mechanistic Basis for Receptor-Mediated Pathological α-Synuclein Fibril Cell-to-Cell Transmission in Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2011196118. [Google Scholar] [CrossRef]
- Scarlino, S.; Domi, T.; Pozzi, L.; Romano, A.; Pipitone, G.B.; Falzone, Y.M.; Mosca, L.; Penco, S.; Lunetta, C.; Sansone, V.; et al. Burden of Rare Variants in ALS and Axonal Hereditary Neuropathy Genes Influence Survival in ALS: Insights from a Next Generation Sequencing Study of an Italian ALS Cohort. Int. J. Mol. Sci. 2020, 21, 3346. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef]
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z.; Guo, L.; Shorter, J.; Cruz, S.D.; Cleveland, D.W. Cytoplasmic TDP-43 De-Mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 2019, 102, 339–357.e7. [Google Scholar] [CrossRef] [Green Version]
- Maraschi, A.; Gumina, V.; Dragotto, J.; Colombrita, C.; Mompeán, M.; Buratti, E.; Silani, V.; Feligioni, M.; Ratti, A. SUMOylation Regulates TDP-43 Splicing Activity and Nucleocytoplasmic Distribution. Mol. Neurobiol. 2021, 58, 5682–5702. [Google Scholar] [CrossRef]
- Buratti, E. TDP-43 Post-Translational Modifications in Health and Disease. Expert Opin. Ther. Targets 2018, 22, 279–293. [Google Scholar] [CrossRef]
- da Silva, L.A.G.; Simonetti, F.; Hutten, S.; Riemenschneider, H.; Sternburg, E.L.; Pietrek, L.M.; Gebel, J.; Dötsch, V.; Edbauer, D.; Hummer, G.; et al. Disease-linked TDP-43 Hyperphosphorylation Suppresses TDP-43 Condensation and Aggregation. EMBO J. 2022, 41, e108443. [Google Scholar] [CrossRef]
- Zhao, M.; Yao, X.; Wei, P.; Zhao, C.; Cheng, M.; Zhang, D.; Xue, W.; He, W.; Xue, W.; Zuo, X.; et al. O-GlcNAcylation of TDP-43 Suppresses Proteinopathies and Promotes TDP-43’s MRNA Splicing Activity. EMBO Rep. 2021, 22, e51649. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxidative Med. Cell. Longev. 2020, 2020, 5021694. [Google Scholar] [CrossRef]
- Ratti, A.; Gumina, V.; Lenzi, P.; Bossolasco, P.; Fulceri, F.; Volpe, C.; Bardelli, D.; Pregnolato, F.; Maraschi, A.; Fornai, F.; et al. Chronic Stress Induces Formation of Stress Granules and Pathological TDP-43 Aggregates in Human ALS Fibroblasts and IPSC-Motoneurons. Neurobiol. Dis. 2020, 145, 105051. [Google Scholar] [CrossRef]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 Aggregation Induced by Oxidative Stress Causes Global Mitochondrial Imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef]
- Romano, N.; Catalani, A.; Lattante, S.; Belardo, A.; Proietti, S.; Bertini, L.; Silvestri, F.; Catalani, E.; Cervia, D.; Zolla, L.; et al. ALS Skin Fibroblasts Reveal Oxidative Stress and ERK1/2-Mediated Cytoplasmic Localization of TDP-43. Cell Signal. 2020, 70, 109591. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Castanedo-Vázquez, D.; Gil-Bea, F.; Tapia, O.; Arozamena, J.; Durán-Vían, C.; Sedano, M.J.; Berciano, M.T.; de Munain, A.L.; Lafarga, M. ALS-Derived Fibroblasts Exhibit Reduced Proliferation Rate, Cytoplasmic TDP-43 Aggregation and a Higher Susceptibility to DNA Damage. J. Neurol. 2020, 267, 1291–1299. [Google Scholar] [CrossRef]
- Shan, X.; Vocadlo, D.J.; Krieger, C. Reduced Protein O-Glycosylation in the Nervous System of the Mutant SOD1 Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2012, 516, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-L.; Su, F.-Y.; Tsai, L.-K.; Huang, C.-C.; Ko, Y.-L.; Su, L.-W.; Chen, K.-Y.; Shih, H.-M.; Hu, C.-M.; Lee, W.-H. NPGPx-Mediated Adaptation to Oxidative Stress Protects Motor Neurons from Degeneration in Aging by Directly Modulating O-GlcNAcase. Cell Rep. 2019, 29, 2134–2143.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comer, F.I.; Vosseller, K.; Wells, L.; Accavitti, M.A.; Hart, G.W. Characterization of a Mouse Monoclonal Antibody Specific for O-Linked N-Acetylglucosamine. Anal. Biochem. 2001, 293, 169–177. [Google Scholar] [CrossRef]
- Teo, C.F.; Ingale, S.; Wolfert, M.A.; Elsayed, G.A.; Nöt, L.G.; Chatham, J.C.; Wells, L.; Boons, G.-J. Glycopeptide-Specific Monoclonal Antibodies Suggest New Roles for O-GlcNAc. Nat. Chem. Biol. 2010, 6, 338–343. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Q.; Zhang, N.; Zhang, K.; Dou, T.; Cao, Y.; Liu, Y.; Li, K.; Hao, X.; Xie, X.; et al. Proteomic Profiling and Genome-Wide Mapping of O-GlcNAc Chromatin-Associated Proteins Reveal an O-GlcNAc-Regulated Genotoxic Stress Response. Nat. Commun. 2020, 11, 5898. [Google Scholar] [CrossRef]
- Zachara, N.E.; Molina, H.; Wong, K.Y.; Pandey, A.; Hart, G.W. The Dynamic Stress-Induced “O-GlcNAc-Ome” Highlights Functions for O-GlcNAc in Regulating DNA Damage/Repair and Other Cellular Pathways. Amino Acids 2010, 40, 793–808. [Google Scholar] [CrossRef] [Green Version]
- Heap, G.A.; Heel, D.A. van The Genetics of Chronic Inflammatory Diseases. Hum. Mol. Genet. 2009, 18, R101–R106. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Inflammation and Metabolic Disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Schmid-Schönbein, G.W. Analysis of Inflammation. Biomed. Eng. 2006, 8, 93–151. [Google Scholar] [CrossRef]
- Xu, Y.-R.; Lei, C.-Q. TAK1-TABs Complex: A Central Signalosome in Inflammatory Responses. Front. Immunol. 2021, 11, 608976. [Google Scholar] [CrossRef]
- Shinohara, H.; Yasuda, T.; Kurosaki, T. TAK1 Adaptor Proteins, TAB2 and TAB3, Link the Signalosome to B-cell Receptor-induced IKK Activation. FEBS Lett. 2016, 590, 3264–3269. [Google Scholar] [CrossRef] [Green Version]
- Ninomiya-Tsuji, J.; Kishimoto, K.; Hiyama, A.; Inoue, J.; Cao, Z.; Matsumoto, K. The Kinase TAK1 Can Activate the NIK-IκB as Well as the MAP Kinase Cascade in the IL-1 Signalling Pathway. Nature 1999, 398, 252–256. [Google Scholar] [CrossRef]
- Fan, Y.; Yu, Y.; Shi, Y.; Sun, W.; Xie, M.; Ge, N.; Mao, R.; Chang, A.; Xu, G.; Schneider, M.D.; et al. Lysine 63-Linked Polyubiquitination of TAK1 at Lysine 158 Is Required for Tumor Necrosis Factor α- and Interleukin-1β-Induced IKK/NF-ΚB and JNK/AP-1 Activation*. J. Biol. Chem. 2010, 285, 5347–5360. [Google Scholar] [CrossRef] [Green Version]
- Tao, T.; He, Z.; Shao, Z.; Lu, H. TAB3 O-GlcNAcylation Promotes Metastasis of Triple Negative Breast Cancer. Oncotarget 2016, 7, 22807–22818. [Google Scholar] [CrossRef] [Green Version]
- Woo, C.M.; Lund, P.J.; Huang, A.C.; Davis, M.M.; Bertozzi, C.R.; Pitteri, S.J. Mapping and Quantification of Over 2000 O-Linked Glycopeptides in Activated Human T Cells with Isotope-Targeted Glycoproteomics (Isotag). Mol. Cell Proteom. 2018, 17, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.H.; Park, S.Y.; Nam, H.W.; Kim, D.H.; Kang, J.G.; Kang, E.S.; Kim, Y.S.; Lee, H.C.; Kim, K.S.; Cho, J.W. NFκB Activation Is Associated with Its O-GlcNAcylation State under Hyperglycemic Conditions. Proc. Natl. Acad. Sci. USA 2008, 105, 17345–17350. [Google Scholar] [CrossRef] [Green Version]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Loss of P53 Enhances Catalytic Activity of IKKβ through O-Linked β-N-Acetyl Glucosamine Modification. Proc. Natl. Acad. Sci. USA 2009, 106, 3431–3436. [Google Scholar] [CrossRef] [Green Version]
- Higashimoto, T.; Chan, N.; Lee, Y.-K.; Zandi, E. Regulation of IκB Kinase Complex by Phosphorylation of γ-Binding Domain of IκB Kinase β by Polo-like Kinase 1*. J. Biol. Chem. 2008, 283, 35354–35367. [Google Scholar] [CrossRef] [Green Version]
- Schomer-Miller, B.; Higashimoto, T.; Lee, Y.-K.; Zandi, E. Regulation of IκB Kinase (IKK) Complex by IKKγ-Dependent Phosphorylation of the T-Loop and C Terminus of IKKβ*. J. Biol. Chem. 2006, 281, 15268–15276. [Google Scholar] [CrossRef] [PubMed]
- Kneass, Z.T.; Marchase, R.B. Neutrophils Exhibit Rapid Agonist-Induced Increases in Protein-Associated O-GlcNAc*. J. Biol. Chem. 2004, 279, 45759–45765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kneass, Z.T.; Marchase, R.B. Protein O-GlcNAc Modulates Motility-Associated Signaling Intermediates in Neutrophils*. J. Biol. Chem. 2005, 280, 14579–14585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Gong, W.; Wang, H.; Li, T.; Attri, K.S.; Lewis, R.E.; Kalil, A.C.; Bhinderwala, F.; Powers, R.; Yin, G.; et al. O-GlcNAc Transferase Suppresses Inflammation and Necroptosis by Targeting Receptor-Interacting Serine/Threonine-Protein Kinase 3. Immunity 2019, 50, 576–590.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speir, M.; Lawlor, K.E. RIP-Roaring Inflammation: RIPK1 and RIPK3 Driven NLRP3 Inflammasome Activation and Autoinflammatory Disease. Semin. Cell Dev. Biol. 2020, 109, 114–124. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent Advances in the Mechanisms of NLRP3 Inflammasome Activation and Its Inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Li, X.; Attri, K.S.; Liu, C.; Li, L.; Herring, L.E.; Asara, J.M.; Lei, Y.L.; Singh, P.K.; Gao, C.; et al. O-GlcNAc Transferase Links Glucose Metabolism to MAVS-Mediated Antiviral Innate Immunity. Cell Host Microbe 2018, 24, 791–803.e6. [Google Scholar] [CrossRef] [Green Version]
- Shikhman, A.R.; Kuhn, K.; Alaaeddine, N.; Lotz, M. N-Acetylglucosamine Prevents IL-1β-Mediated Activation of Human Chondrocytes. J. Immunol. 2001, 166, 5155–5160. [Google Scholar] [CrossRef] [Green Version]
- Largo, R.; Alvarez-Soria, M.A.; Díez-Ortego, I.; Calvo, E.; Sánchez-Pernaute, O.; Egido, J.; Herrero-Beaumont, G. Glucosamine Inhibits IL-1β-Induced NFκB Activation in Human Osteoarthritic Chondrocytes. Osteoarthr. Cartil. 2003, 11, 290–298. [Google Scholar] [CrossRef] [Green Version]
- d’Abusco, A.S.; Calamia, V.; Cicione, C.; Grigolo, B.; Politi, L.; Scandurra, R. Glucosamine Affects Intracellular Signalling through Inhibition of Mitogen-Activated Protein Kinase Phosphorylation in Human Chondrocytes. Arthritis Res. Ther. 2007, 9, R104. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.P.; Kim, H.G.; Han, E.H.; Choi, J.H.; Park, B.H.; Jung, K.H.; Shin, Y.C.; Jeong, H.G. N-Acetylglucosamine Suppress Collagenases Activation in Ultraviolet B-Irradiated Human Dermal Fibroblasts: Involvement of Calcium Ions and Mitogen-Activated Protein Kinases. J. Dermatol. Sci. 2011, 63, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Z.; Li, L.; Gong, W.; Lazenby, A.J.; Swanson, B.J.; Herring, L.E.; Asara, J.M.; Singer, J.D.; Wen, H. Myeloid-Derived Cullin 3 Promotes STAT3 Phosphorylation by Inhibiting OGT Expression and Protects against Intestinal Inflammation. J. Exp. Med. 2017, 214, 1093–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandhwal, M.; Behl, T.; Singh, S.; Sharma, N.; Arora, S.; Bhatia, S.; Al-Harrasi, A.; Sachdeva, M.; Bungau, S. Role of Matrix Metalloproteinase in Wound Healing. Am. J. Transl. Res. 2021, 14, 4391–4405. [Google Scholar]
- Gorter, R.; Baron, W. Matrix Metalloproteinases Shape the Oligodendrocyte (Niche) during Development and upon Demyelination. Neurosci. Lett. 2020, 729, 134980. [Google Scholar] [CrossRef]
- Huang, J.-B.; Clark, A.J.; Petty, H.R. The Hexosamine Biosynthesis Pathway Negatively Regulates IL-2 Production by Jurkat T Cells. Cell Immunol. 2007, 245, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Xing, D.; Gong, K.; Feng, W.; Nozell, S.E.; Chen, Y.-F.; Chatham, J.C.; Oparil, S. O-GlcNAc Modification of NFκB P65 Inhibits TNF-α-Induced Inflammatory Mediator Expression in Rat Aortic Smooth Muscle Cells. PLoS ONE 2011, 6, e24021. [Google Scholar] [CrossRef]
- Someya, A.; Ikegami, T.; Sakamoto, K.; Nagaoka, I. Glucosamine Downregulates the IL-1β-Induced Expression of Proinflammatory Cytokine Genes in Human Synovial MH7A Cells by O-GlcNAc Modification-Dependent and -Independent Mechanisms. PLoS ONE 2016, 11, e0165158. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Meoz, R.F.; Jiang, J.; Lazarus, M.B.; Orman, M.; Janetzko, J.; Fan, C.; Duveau, D.Y.; Tan, Z.-W.; Thomas, C.J.; Walker, S. A Small Molecule That Inhibits OGT Activity in Cells. ACS Chem. Biol. 2015, 10, 1392–1397. [Google Scholar] [CrossRef] [Green Version]
- Zebrucka, K.P.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The Integrated Stress Response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, T.; Ramaglia, V.; Abdel-Nour, M.; Bianchi, A.A.; Tsalikis, J.; Chau, H.N.; Kalia, S.K.; Kalia, L.V.; Chen, J.-J.; Arnoult, D.; et al. The EIF2α Kinase HRI Triggers the Autophagic Clearance of Cytosolic Protein Aggregates. J. Biol. Chem. 2021, 296, 100050. [Google Scholar] [CrossRef]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The EIF2α Kinases: Their Structures and Functions. Cell Mol. Life Sci. 2012, 70, 3493–3511. [Google Scholar] [CrossRef]
- Dar, A.C.; Dever, T.E.; Sicheri, F. Higher-Order Substrate Recognition of EIF2α by the RNA-Dependent Protein Kinase PKR. Cell 2005, 122, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; McGrath, B.C.; Reinert, J.; Olsen, D.S.; Lei, L.; Gill, S.; Wek, S.A.; Vattem, K.M.; Wek, R.C.; Kimball, S.R.; et al. The GCN2 EIF2alpha Kinase Is Required for Adaptation to Amino Acid Deprivation in Mice. Mol. Cell Biol. 2002, 22, 6681–6688. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation Involving Upstream ORFs Regulates ATF4 MRNA Translation in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [Green Version]
- Palam, L.R.; Baird, T.D.; Wek, R.C. Phosphorylation of EIF2 Facilitates Ribosomal Bypass of an Inhibitory Upstream ORF to Enhance CHOP Translation. J. Biol. Chem. 2011, 286, 10939–10949. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.-Y.; Wek, S.A.; McGrath, B.C.; Lu, D.; Hai, T.; Harding, H.P.; Wang, X.; Ron, D.; Cavener, D.R.; Wek, R.C. Activating Transcription Factor 3 Is Integral to the Eukaryotic Initiation Factor 2 Kinase Stress Response. Mol. Cell Biol. 2004, 24, 1365–1377. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, S.; Oertlin, C.; Szczepanowska, K.; Kukat, A.; Senft, K.; Lucas, C.; Brodesser, S.; Hatzoglou, M.; Larsson, O.; Topisirovic, I.; et al. Adaptation to Mitochondrial Stress Requires CHOP-Directed Tuning of ISR. Sci. Adv. 2020, 7, eabf0971. [Google Scholar] [CrossRef]
- Dey, S.; Baird, T.D.; Zhou, D.; Palam, L.R.; Spandau, D.F.; Wek, R.C. Both Transcriptional Regulation and Translational Control of ATF4 Are Central to the Integrated Stress Response. J. Biol. Chem. 2010, 285, 33165–33174. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.-Y.; Jiang, L.; Wek, R.C. The Eukaryotic Initiation Factor-2 Kinase Pathway Facilitates Differential GADD45a Expression in Response to Environmental Stress*. J. Biol. Chem. 2007, 282, 3755–3765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, S.; Baumeister, P.; Yang, S.; Abcouwer, S.F.; Lee, A.S. Induction of Grp78/BiP by Translational Block: Activation of the Grp78 Promoter by ATF4 through and Upstream ATF/CRE Site Independent of the Endoplasmic Reticulum Stress Elements. J. Biological. Chem. 2003, 278, 37375–37385. [Google Scholar] [CrossRef]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP Induces Activating Transcription Factor 5 (ATF5) to Trigger Apoptosis in Response to Perturbations in Protein Homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef] [PubMed]
- Hahne, H.; Gholami, A.M.; Kuster, B. Discovery of O-GlcNAc-Modified Proteins in Published Large-Scale Proteome Data. Mol. Cell. Proteom. 2012, 11, 843–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Zhu, T.; Yu, K.; Shi, M.; Wang, X.; Wang, L.; Huang, T.; Li, W.; Liu, Y.; Zhang, J. Elevation of O-GlcNAc and GFAT Expression by Nicotine Exposure Promotes Epithelial-mesenchymal Transition and Invasion in Breast Cancer Cells. Cell Death Dis. 2019, 10, 343. [Google Scholar] [CrossRef] [Green Version]
- Chaveroux, C.; Sarcinelli, C.; Barbet, V.; Belfeki, S.; Barthelaix, A.; Ferraro-Peyret, C.; Lebecque, S.; Renno, T.; Bruhat, A.; Fafournoux, P.; et al. Nutrient Shortage Triggers the Hexosamine Biosynthetic Pathway via the GCN2-ATF4 Signalling Pathway. Sci. Rep. 2016, 6, 27278. [Google Scholar] [CrossRef] [Green Version]
- Nabeebaccus, A.A.; Zoccarato, A.; Hafstad, A.D.; Santos, C.X.; Aasum, E.; Brewer, A.C.; Zhang, M.; Beretta, M.; Yin, X.; West, J.A.; et al. Nox4 Reprograms Cardiac Substrate Metabolism via Protein O-GlcNAcylation to Enhance Stress Adaptation. JCI Insight 2017, 2, e96184. [Google Scholar] [CrossRef] [Green Version]
- Jousse, C.; Oyadomari, S.; Novoa, I.; Lu, P.; Zhang, Y.; Harding, H.P.; Ron, D. Inhibition of a Constitutive Translation Initiation Factor 2α Phosphatase, CReP, Promotes Survival of Stressed Cells. J. Cell Biol. 2003, 163, 767–775. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback Inhibition of the Unfolded Protein Response by GADD34-Mediated Dephosphorylation of EIF2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Wells, L.; Kreppel, L.K.; Comer, F.I.; Wadzinski, B.E.; Hart, G.W. O-GlcNAc Transferase Is in a Functional Complex with Protein Phosphatase 1 Catalytic Subunits. J. Biol. Chem. 2004, 279, 38466–38470. [Google Scholar] [CrossRef] [Green Version]
- Slawson, C.; Lakshmanan, T.; Knapp, S.; Hart, G.W. A Mitotic GlcNAcylation/Phosphorylation Signaling Complex Alters the Posttranslational State of the Cytoskeletal Protein Vimentin. Mol. Biol. Cell 2008, 19, 4130–4140. [Google Scholar] [CrossRef] [Green Version]
- van Huizen, R.; Martindale, J.L.; Gorospe, M.; Holbrook, N.J. P58IPK, a Novel Endoplasmic Reticulum Stress-Inducible Protein and Potential Negative Regulator of EIF2α Signaling*. J. Biol. Chem. 2003, 278, 15558–15564. [Google Scholar] [CrossRef]
- Ray, M.K.; Datta, B.; Chakraborty, A.; Chattopadhyay, A.; Meza-Keuthen, S.; Gupta, N.K. The Eukaryotic Initiation Factor 2-Associated 67-KDa Polypeptide (P67) Plays a Critical Role in Regulation of Protein Synthesis Initiation in Animal Cells. Proc. Natl. Acad. Sci. USA 1992, 89, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chang, Y.H. Evidence That the Human Homologue of a Rat Initiation Factor-2 Associated Protein (P67) Is a Methionine Aminopeptidase. Biochem. Biophys. Res. Commun. 1996, 227, 152–159. [Google Scholar] [CrossRef]
- Datta, B.; Ray, M.K.; Chakrabarti, D.; Wylie, D.E.; Gupta, N.K. Glycosylation of Eukaryotic Peptide Chain Initiation Factor 2 (EIF-2)-Associated 67-KDa Polypeptide (P67) and Its Possible Role in the Inhibition of EIF-2 Kinase-Catalyzed Phosphorylation of the EIF-2 α-Subunit*. J. Biol. Chem. 1989, 264, 20620–20624. [Google Scholar] [CrossRef]
- Datta, B.; Datta, R.; Mukherjee, S.; Zhang, Z. Increased Phosphorylation of Eukaryotic Initiation Factor 2α at the G2/M Boundary in Human Osteosarcoma Cells Correlates with Deglycosylation of P67 and a Decreased Rate of Protein Synthesis. Exp. Cell Res. 1999, 250, 223–230. [Google Scholar] [CrossRef]
- Datta, R.; Choudhury, P.; Ghosh, A.; Datta, B. A Glycosylation Site, 60SGTS63, of P67 Is Required for Its Ability To Regulate the Phosphorylation and Activity of Eukaryotic Initiation Factor 2α. Biochemistry 2003, 42, 5453–5460. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy Pathway: Cellular and Molecular Mechanisms. Autophagy 2017, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Hung, Y.-H.; Chen, L.M.-W.; Yang, J.-Y.; Yang, W.Y. Spatiotemporally Controlled Induction of Autophagy-Mediated Lysosome Turnover. Nat. Commun. 2013, 4, 2111. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, C.; Meyer, H. Detection and Clearance of Damaged Lysosomes by the Endo-Lysosomal Damage Response and Lysophagy. Curr. Biol. 2017, 27, R1330–R1341. [Google Scholar] [CrossRef] [Green Version]
- Wyant, G.A.; Abu-Remaileh, M.; Wolfson, R.L.; Chen, W.W.; Freinkman, E.; Danai, L.V.; Heiden, M.G.V.; Sabatini, D.M. MTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 2017, 171, 642–654.e12. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Lee, Y.; Pak, J.W.; Kim, H.; Choi, H.; Kim, J.; Roth, J.; Cho, J.W. O-GlcNAc Modification Is Essential for the Regulation of Autophagy in Drosophila Melanogaster. Cell Mol. Life Sci. 2015, 72, 3173–3183. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The Mammalian ULK1 Complex and Autophagy Initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Audesse, A.J.; Dhakal, S.; Hassell, L.-A.; Gardell, Z.; Nemtsova, Y.; Webb, A.E. FOXO3 Directly Regulates an Autophagy Network to Functionally Regulate Proteostasis in Adult Neural Stem Cells. PLoS Genet. 2019, 15, e1008097. [Google Scholar] [CrossRef]
- Malta, C.D.; Cinque, L.; Settembre, C. Transcriptional Regulation of Autophagy: Mechanisms and Diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.-R.; Wang, K.; Whisenhunt, T.R.; Huang, P.; Zhu, X.; Kudlow, J.E.; Paterson, A.J. O-GlcNAcylation Enhances FOXO4 Transcriptional Regulation in Response to Stress. FEBS Lett. 2010, 584, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Housley, M.P.; Udeshi, N.D.; Rodgers, J.T.; Shabanowitz, J.; Puigserver, P.; Hunt, D.F.; Hart, G.W. A PGC-1alpha-O-GlcNAc Transferase Complex Regulates FoxO Transcription Factor Activity in Response to Glucose. J. Biol. Chem. 2009, 284, 5148–5157. [Google Scholar] [CrossRef] [Green Version]
- Housley, M.P.; Rodgers, J.T.; Udeshi, N.D.; Kelly, T.J.; Shabanowitz, J.; Hunt, D.F.; Puigserver, P.; Hart, G.W. O-GlcNAc Regulates FoxO Activation in Response to Glucose. J. Biol. Chem. 2008, 283, 16283–16292. [Google Scholar] [CrossRef] [Green Version]
- Yoon, I.; Nam, M.; Kim, H.K.; Moon, H.-S.; Kim, S.; Jang, J.; Song, J.A.; Jeong, S.J.; Kim, S.B.; Cho, S.; et al. Glucose-Dependent Control of Leucine Metabolism by Leucyl-TRNA Synthetase 1. Science 2020, 367, 205–210. [Google Scholar] [CrossRef]
- Kim, K.; Yoo, H.C.; Kim, B.G.; Kim, S.; Sung, Y.; Yoon, I.; Yu, Y.C.; Park, S.J.; Kim, J.H.; Myung, K.; et al. O-GlcNAc Modification of Leucyl-TRNA Synthetase 1 Integrates Leucine and Glucose Availability to Regulate MTORC1 and the Metabolic Fate of Leucine. Nat. Commun. 2022, 13, 2904. [Google Scholar] [CrossRef]
- Jin, L.; Yuan, F.; Dai, G.; Yao, Q.; Xiang, H.; Wang, L.; Xue, B.; Shan, Y.; Liu, X. Blockage of O-Linked GlcNAcylation Induces AMPK-Dependent Autophagy in Bladder Cancer Cells. Cell Mol. Biol. Lett. 2020, 25, 17. [Google Scholar] [CrossRef]
- Shi, Y.; Yan, S.; Shao, G.-C.; Wang, J.; Jian, Y.-P.; Liu, B.; Yuan, Y.; Qin, K.; Nai, S.; Huang, X.; et al. O-GlcNAcylation Stabilizes the Autophagy-Initiating Kinase ULK1 by Inhibiting Chaperone-Mediated Autophagy upon HPV Infection. J. Biol. Chem. 2022, 298, 102341. [Google Scholar] [CrossRef]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 Affects Autophagosome Fusion and Links Chaperone-Mediated Autophagy to Macroautophagy. Nat. Commun. 2018, 9, 3492. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Lak, B.; Li, J.; Jokitalo, E.; Wang, Y. GRASP55 Senses Glucose Deprivation through O-GlcNAcylation to Promote Autophagosome-Lysosome Fusion. Dev. Cell 2018, 45, 245–261.e6. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Liu, P.; Jiang, T.; Ambrose, A.J.; Luo, G.; de la Vega, M.R.; Cholanians, A.B.; Wong, P.K.; Chapman, E.; Zhang, D.D. Increased O-GlcNAcylation of SNAP29 Drives Arsenic-Induced Autophagic Dysfunction. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, G.; Prencipe, L.; Iodice, L.; Beznoussenko, G.; Savarese, M.; Marra, P.; Tullio, G.D.; Martire, G.; Matteis, M.A.D.; Bonatti, S. GRASP65 and GRASP55 Sequentially Promote the Transport of C-Terminal Valine-Bearing Cargos to and through the Golgi Complex*. J. Biol. Chem. 2009, 284, 34849–34860. [Google Scholar] [CrossRef] [Green Version]
- Pothukuchi, P.; Agliarulo, I.; Pirozzi, M.; Rizzo, R.; Russo, D.; Turacchio, G.; Nüchel, J.; Yang, J.; Gehin, C.; Capolupo, L.; et al. GRASP55 Regulates Intra-Golgi Localization of Glycosylation Enzymes to Control Glycosphingolipid Biosynthesis. EMBO J. 2021, 40, e107766. [Google Scholar] [CrossRef] [PubMed]
- Nüchel, J.; Tauber, M.; Nolte, J.L.; Mörgelin, M.; Türk, C.; Eckes, B.; Demetriades, C.; Plomann, M. An MTORC1-GRASP55 Signaling Axis Controls Unconventional Secretion to Reshape the Extracellular Proteome upon Stress. Mol. Cell 2021, 81, 3275–3293.e12. [Google Scholar] [CrossRef] [PubMed]
- Sou, Y.; Waguri, S.; Iwata, J.; Ueno, T.; Fujimura, T.; Hara, T.; Sawada, N.; Yamada, A.; Mizushima, N.; Uchiyama, Y.; et al. The Atg8 Conjugation System Is Indispensable for Proper Development of Autophagic Isolation Membranes in Mice. Mol. Biol. Cell 2008, 19, 4762–4775. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a Mammalian Homologue of Yeast Apg8p, Is Localized in Autophagosome Membranes after Processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Fracchiolla, D.; Sawa-Makarska, J.; Zens, B.; de Ruiter, A.; Zaffagnini, G.; Brezovich, A.; Romanov, J.; Runggatscher, K.; Kraft, C.; Zagrovic, B.; et al. Mechanism of Cargo-Directed Atg8 Conjugation during Selective Autophagy. eLife 2016, 5, e18544. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.K.; Park, N.Y.; Park, S.J.; Kim, B.-G.; Shin, J.H.; Jo, D.S.; Bae, D.-J.; Suh, Y.-A.; Chang, J.H.; Lee, E.K.; et al. O-GlcNAcylation of ATG4B Positively Regulates Autophagy by Increasing Its Hydroxylase Activity. Oncotarget 2016, 7, 57186–57196. [Google Scholar] [CrossRef] [Green Version]
- Shaw, J.; Yurkova, N.; Zhang, T.; Gang, H.; Aguilar, F.; Weidman, D.; Scramstad, C.; Weisman, H.; Kirshenbaum, L.A. Antagonism of E2F-1 Regulated Bnip3 Transcription by NF-ΚB Is Essential for Basal Cell Survival. Proc. Natl. Acad. Sci. USA 2008, 105, 20734–20739. [Google Scholar] [CrossRef] [Green Version]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, Å.B. Response to Myocardial Ischemia/Reperfusion Injury Involves Bnip3 and Autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Tracy, K.; Dibling, B.C.; Spike, B.T.; Knabb, J.R.; Schumacker, P.; Macleod, K.F. BNIP3 Is an RB/E2F Target Gene Required for Hypoxia-Induced Autophagy. Mol. Cell Biol. 2007, 27, 6229–6242. [Google Scholar] [CrossRef] [Green Version]
- Rao, X.; Duan, X.; Mao, W.; Li, X.; Li, Z.; Li, Q.; Zheng, Z.; Xu, H.; Chen, M.; Wang, P.G.; et al. O-GlcNAcylation of G6PD Promotes the Pentose Phosphate Pathway and Tumor Growth. Nat. Commun. 2015, 6, 8468. [Google Scholar] [CrossRef] [Green Version]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A.; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 Glycosylation Regulates Cell Growth and Metabolism. Science 2012, 337, 975–980. [Google Scholar] [CrossRef] [Green Version]
- Mullarky, E.; Cantley, L.C. Diverting Glycolysis to Combat Oxidative Stress. In Innovative Medicine; Springer: Tokyo, Japan, 2015; pp. 3–23. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Jin, X.; Zhang, D.; Li, D.; Hao, F.; Feng, Y.; Gu, S.; Meng, F.; Tian, M.; et al. O-GlcNAcylation Destabilizes the Active Tetrameric PKM2 to Promote the Warburg Effect. Proc. Natl. Acad. Sci. USA 2017, 114, 13732–13737. [Google Scholar] [CrossRef] [Green Version]
- Nie, H.; Ju, H.; Fan, J.; Shi, X.; Cheng, Y.; Cang, X.; Zheng, Z.; Duan, X.; Yi, W. O-GlcNAcylation of PGK1 Coordinates Glycolysis and TCA Cycle to Promote Tumor Growth. Nat. Commun. 2020, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Burt, R.A.; Dejanovic, B.; Peckham, H.J.; Lee, K.A.; Li, X.; Ounadjela, J.R.; Rao, A.; Malaker, S.A.; Carr, S.A.; Myers, S.A. Novel Antibodies for the Simple and Efficient Enrichment of Native O-GlcNAc Modified Peptides. Mol. Cell Proteom. 2021, 20, 100167. [Google Scholar] [CrossRef]
- Gorelik, A.; Bartual, S.G.; Borodkin, V.S.; Varghese, J.; Ferenbach, A.T.; van Aalten, D.M.F. Genetic Recoding to Dissect the Roles of Site-Specific Protein O-GlcNAcylation. Nat. Struct. Mol. Biol. 2019, 26, 1071–1077. [Google Scholar] [CrossRef]
- Shan, H.; Sun, J.; Shi, M.; Liu, X.; Shi, Z.; Yu, W.; Gu, Y. Generation and Characterization of a Site-Specific Antibody for SIRT1 O-GlcNAcylated at Serine 549. Glycobiology 2018, 28, 482–487. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahie, K.M.M.; Papanicolaou, K.N.; Zachara, N.E. Integration of O-GlcNAc into Stress Response Pathways. Cells 2022, 11, 3509. https://doi.org/10.3390/cells11213509
Fahie KMM, Papanicolaou KN, Zachara NE. Integration of O-GlcNAc into Stress Response Pathways. Cells. 2022; 11(21):3509. https://doi.org/10.3390/cells11213509
Chicago/Turabian StyleFahie, Kamau M. M., Kyriakos N. Papanicolaou, and Natasha E. Zachara. 2022. "Integration of O-GlcNAc into Stress Response Pathways" Cells 11, no. 21: 3509. https://doi.org/10.3390/cells11213509