
Mitoferrin, Cellular and Mitochondrial Iron Homeostasis

Abstract
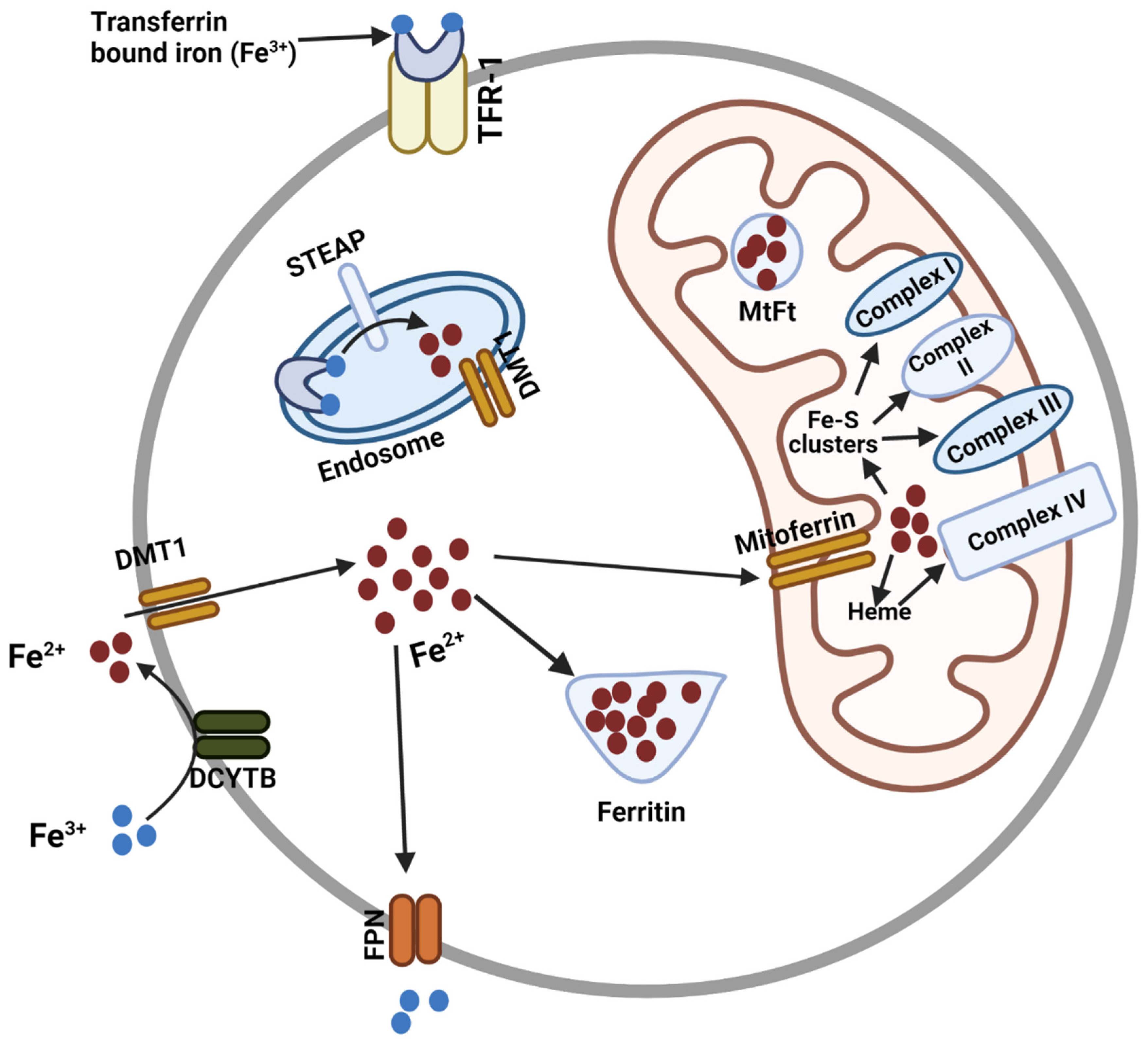
:1. Introduction
2. Discovery of Mitoferrins
3. Mitoferrin Structure and Conservation of Structure in Different Species
4. Regulation of Mitoferrin Expression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors or Regulators of Mitoferrin | Effect in Mitoferrin Expression or Activity | References |
|---|---|---|
| 1. GATA-1 | Displaces GATA-2 from two cis-regulatory elements upstream of mitoferrin-1 to induce mitoferrin-1 transcription during erythroid maturation | [23,31] |
| 2. ENO-1 | Inhibit the transcription of mitoferrin-1 by reducing IRP1 mRNA expression in head and neck cancer cells | [32] |
| 3. ABCB10 | Stabilizes mitoferrin-1 by directly binding to it | [36] |
| 4. ALKBH5 | Regulate the methylation of mitoferrin-2 and enhances RNA stability of mitoferrin-2 in pancreatic ductal carcinoma cells. | [38] |
| 5. miR-7 | Directly target 3’-UTR of mitoferrin-1 and silences it in rhabdomyosarcoma | [41] |
| 6. PINK1 and PARK2 | Regulates the expression of mitoferrin-1 and mitoferrin-2 using ATG5 dependent autophagy pathway | [48] |
| 7. pH | Faster iron transport activity at alkaline pH | [30] |
5. Importance of Mitoferrin in Normal Physiology and Disease Development
| Alteration in Mitoferrin | Associated Diseases | Type of Study (Used Model) | References |
|---|---|---|---|
| Mutation in mitoferrin-1 | Anemia | In vitro | [46] |
| Dysregulated expression of mitoferrin | Myelodysplastic syndrome | In vitro | [62] |
| Loss of mitoferrin-1 | Protoporphyria and hepatotoxicity | Animal (Mouse) | [25] |
| Mitoferrin-1 depletion | Erythropoietic protoporphyria | Patients’ tissue | [47] |
| Downregulation of mitoferrin-1 in hippocampus and peripheral blood | Major depressive disorder (MDD) | Patients’ tissue | [64] |
| Knockout of mitoferrin-1 in neuron | Impairment in spatial learning and memory | Animal (Mouse) | [65] |
| Overexpression of mitoferrin | Rescue of mitochondrial function and improvement in symptoms of Parkinson’s disease | Drosophila melanogaster | [66] |
| Overexpression of mitoferrin | Friedreich’s ataxia | Drosophila melanogaster | [67,68] |
| Downregulation of mitoferrin-1 | Alzheimer’s disease | Caenorhabditis elegans | [69] |
| Increased expression of mitoferrin-1 | Aging in adult skeletal muscle | Patients’ tissue | [70] |
| Knockout of mitoferrin-2 | Reduced fertility | Mouse and Drosophila melanogaster | [71,72] |
| Loss of mitoferrin in hepatocytes | Disruption of liver regeneration | Animal (Mouse) | |
| Knockdown of mitoferrin-1 and mitoferrin-2 | Impaired insulin sensitivity, suppress adipogenic differentiation | In vitro | [73] |
| Increased expression of mitoferrin-2 | Atherosclerosis | Animal (Mouse) | [39] |
| Increased expression of mitoferrin-2 | Huntington’s disease | Animal (Mouse) | [74] |
| Upregulation of mitoferrin-2 | Head and neck cancer | In vitro | [75] |
| Downregulation of mitoferrin-2 | Hepatocellular carcinoma | In vitro | [32] |
| Increased expression of mitoferrin-1 | Pancreatic tumorigenesis | Animal (Mouse) | [48] |
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, M.L.; Lane, D.; Richardson, D. Mitochondrial Mayhem: The Mitochondrion as a Modulator of Iron Metabolism and Its Role in Disease. Antioxid. Redox Signal. 2011, 15, 3003–3019. [Google Scholar] [CrossRef] [PubMed]
- E Lim, J.; Jin, O.; Bennett, C.; Morgan, K.; Wang, F.; Trenor, C.C.; Fleming, M.D.; Andrews, N.C. A mutation in Sec15l1 causes anemia in hemoglobin deficit (hbd) mice. Nat. Genet. 2005, 37, 1270–1273. [Google Scholar] [CrossRef] [PubMed]
- White, R.A.; Boydston, L.A.; Brookshier, T.R.; McNulty, S.G.; Nsumu, N.N.; Brewer, B.P.; Blackmore, K. Iron metabolism mutant hbd mice have a deletion in Sec15l1, which has homology to a yeast gene for vesicle docking. Genomics 2005, 86, 668–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponka, P. Tissue-specific regulation of iron metabolism and heme synthesis: Distinct control mechanisms in erythroid cells. Blood 1997, 89, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Napier, I.; Ponka, P.; Richardson, D.R. Iron trafficking in the mitochondrion: Novel pathways revealed by disease. Blood 2005, 105, 1867–1874. [Google Scholar] [CrossRef]
- Schueck, N.D.; Woontner, M.; Koeller, D.M. The role of the mitochondrion in cellular iron homeostasis. Mitochondrion 2001, 1, 51–60. [Google Scholar] [CrossRef]
- Chen, C.; Paw, B.H. Cellular and mitochondrial iron homeostasis in vertebrates. Biochim. Biophys. Acta 2012, 1823, 1459–1467. [Google Scholar] [CrossRef] [Green Version]
- Andrews, N. Molecular control of iron metabolism. Best Pract. Res. Clin. Haematol. 2005, 18, 159–169. [Google Scholar] [CrossRef]
- Andrews, N.C. Iron Metabolism: Iron Deficiency and Iron Overload. Annu. Rev. Genom. Hum. Genet. 2000, 1, 75–98. [Google Scholar] [CrossRef]
- Kaplan, J. Mechanisms of Cellular Iron Acquisition: Another Iron in the Fire. Cell 2002, 111, 603–606. [Google Scholar] [CrossRef]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Dautry-Varsat, A.; Ciechanover, A.; Lodish, H.F. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 1983, 80, 2258–2262. [Google Scholar] [CrossRef] [Green Version]
- Fleming, M.D.; Romano, M.A.; Su, M.A.; Garrick, L.M.; Garrick, M.D.; Andrews, N.C. Nramp2 is mutated in the anemic Belgrade (b) rat: Evidence of a role for Nramp2 in endosomal iron transport. Proc. Natl. Acad. Sci. USA 1998, 95, 1148–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunshin, H.; MacKenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesenberger, G.; Link, T.A.; von Ahsen, U.; Waldherr, M.; Schweyen, R.J. MRS3 and MRS4, two suppressors of mtRNA splicing defects in yeast, are new members of the mitochondrial carrier family. J. Mol. Biol. 1991, 217, 23–37. [Google Scholar] [CrossRef]
- Waldherr, M.; Ragnini, A.; Jank, B.; Teply, R.; Wiesenberger, G.; Schweyen, R.J. A multitude of suppressors of group II intron-splicing defects in yeast. Curr. Genet. 1993, 24, 301–306. [Google Scholar] [CrossRef]
- Rutherford, J.C.; Jaron, S.; Winge, D.R. Aft1p and Aft2p Mediate Iron-responsive Gene Expression in Yeast through Related Promoter Elements. J. Biol. Chem. 2003, 278, 27636–27643. [Google Scholar] [CrossRef] [Green Version]
- Foury, F.; Roganti, T. Deletion of the Mitochondrial Carrier Genes MRS3 andMRS4 Suppresses Mitochondrial Iron Accumulation in a Yeast Frataxin-deficient Strain. J. Biol. Chem. 2002, 277, 24475–24483. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Kaplan, J. A Mitochondrial-Vacuolar Signaling Pathway in Yeast That Affects Iron and Copper Metabolism. J. Biol. Chem. 2004, 279, 33653–33661. [Google Scholar] [CrossRef]
- Mühlenhoff, U.; Stadler, J.A.; Richhardt, N.; Seubert, A.; Eickhorst, T.; Schweyen, R.J.; Lill, R.; Wiesenberger, G. A Specific Role of the Yeast Mitochondrial Carriers Mrs3/4p in Mitochondrial Iron Acquisition under Iron-limiting Conditions. J. Biol. Chem. 2003, 278, 40612–40620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lyver, E.R.; Knight, S.A.; Pain, D.; Lesuisse, E.; Dancis, A. Mrs3p, Mrs4p, and Frataxin Provide Iron for Fe-S Cluster Synthesis in Mitochondria. J. Biol. Chem. 2006, 281, 22493–22502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of Mitochondrial Iron Import through Differential Turnover of Mitoferrin 1 and Mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troadec, M.-B.; Warner, D.; Wallace, J.; Thomas, K.; Spangrude, G.J.; Phillips, J.; Khalimonchuk, O.; Paw, B.; Ward, D.M.; Kaplan, J. Targeted deletion of the mouse Mitoferrin1 gene: From anemia to protoporphyria. Blood 2011, 117, 5494–5502. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- McGuffin, L.J.; Bryson, K.; Jones, D.T.J.B. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [Green Version]
- Brazzolotto, X.; Pierrel, F.; Pelosi, L. Three conserved histidine residues contribute to mitochondrial iron transport through mitoferrins. Biochem. J. 2014, 460, 79–92. [Google Scholar] [CrossRef]
- Shawki, A.; Knight, P.B.; Maliken, B.D.; Niespodzany, E.J.; Mackenzie, B. Chapter Five–H+-Coupled Divalent Metal-Ion Transporter-1: Functional Properties, Physiological Roles and Therapeutics, in Current Topics in Membranes; Bevensee, M.O., Ed.; Academic Press: Cambridge, MA, USA, 2012; pp. 169–214. [Google Scholar]
- Christenson, E.; Gallegos, A.S.; Banerjee, A. In vitro reconstitution, functional dissection, and mutational analysis of metal ion transport by mitoferrin-1. J. Biol. Chem. 2018, 293, 3819–3828. [Google Scholar] [CrossRef] [Green Version]
- Amigo, J.D.; Yu, M.; Troadec, M.-B.; Gwynn, B.; Cooney, J.D.; Lambert, A.J.; Chi, N.C.; Weiss, M.J.; Peters, L.L.; Kaplan, J.; et al. Identification of Distal cis-Regulatory Elements at Mouse Mitoferrin Loci Using Zebrafish Transgenesis. Mol. Cell. Biol. 2011, 31, 1344–1356. [Google Scholar] [CrossRef]
- Zhang, T.; Sun, L.; Hao, Y.; Suo, C.; Shen, S.; Wei, H.; Ma, W.; Zhang, P.; Wang, T.; Gu, X.; et al. ENO1 suppresses cancer cell ferroptosis by degrading the mRNA of iron regulatory protein 1. Nat. Cancer 2021, 3, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Kuang, Y.; Yuan, Y.; Yu, B. Mitochondrion-mediated iron accumulation promotes carcinogenesis and Warburg effect through reactive oxygen species in osteosarcoma. Cancer Cell Int. 2020, 20, 399. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, X.; Li, D.; Shao, Z.; Cao, H.; Zhang, Y.; Trompouki, E.; Bowman, T.V.; Zon, L.I.; Yuan, G.-C.; et al. Dynamic Control of Enhancer Repertoires Drives Lineage and Stage-Specific Transcription during Hematopoiesis. Dev. Cell 2016, 36, 9–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyde, B.B.; Liesa, M.; A Elorza, A.; Qiu, W.; E Haigh, S.; Richey, L.; Mikkola, H.K.; Schlaeger, T.M.; Shirihai, O.S. The mitochondrial transporter ABC-me (ABCB10), a downstream target of GATA-1, is essential for erythropoiesis in vivo. Cell Death Differ. 2012, 19, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Paradkar, P.N.; Li, L.; Pierce, E.L.; Langer, N.B.; Takahashi-Makise, N.; Hyde, B.B.; Shirihai, O.S.; Ward, D.M.; Kaplan, J.; et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc. Natl. Acad. Sci. USA 2009, 106, 16263–16268. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Dailey, H.A.; Paw, B.H. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 2010, 116, 628–630. [Google Scholar] [CrossRef]
- Huang, R.; Yang, L.; Zhang, Z.; Liu, X.; Fei, Y.; Tong, W.-M.; Niu, Y.; Liang, Z. RNA m6A Demethylase ALKBH5 Protects Against Pancreatic Ductal Adenocarcinoma via Targeting Regulators of Iron Metabolism. Front. Cell Dev. Biol. 2021, 9, 724282. [Google Scholar] [CrossRef]
- Wang, D.; Ye, P.; Kong, C.; Chao, Y.; Yu, W.; Jiang, X.; Luo, J.; Gu, Y.; Chen, S.-L. Mitoferrin 2 deficiency prevents mitochondrial iron overload-induced endothelial injury and alleviates atherosclerosis. Exp. Cell Res. 2021, 402, 112552. [Google Scholar] [CrossRef]
- Bijkerk, R.; Esguerra, J.L.; Ellenbroek, J.H.; Au, Y.W.; Hanegraaf, M.A.; de Koning, E.J.; Eliasson, L.; van Zonneveld, A.J. In Vivo Silencing of MicroRNA-132 Reduces Blood Glucose and Improves Insulin Secretion. Nucleic Acid Ther. 2019, 29, 67–72. [Google Scholar] [CrossRef]
- Yang, L.; Kong, D.; He, M.; Gong, J.; Nie, Y.; Tai, S.; Teng, C.-B. MiR-7 mediates mitochondrial impairment to trigger apoptosis and necroptosis in Rhabdomyosarcoma. Biochim. Biophys. Acta 2020, 1867, 118826. [Google Scholar] [CrossRef]
- Lenkala, D.; LaCroix, B.; Gamazon, E.; Geeleher, P.; Im, H.K.; Huang, R.S. The impact of microRNA expression on cellular proliferation. Qual. Life Res. 2014, 133, 931–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maio, N.; Kim, K.S.; Holmes-Hampton, G.; Singh, A.; Rouault, T.A. Dimeric ferrochelatase bridges ABCB7 and ABCB10 homodimers in an architecturally defined molecular complex required for heme biosynthesis. Haematologica 2019, 104, 1756–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Jia, H.; Zhang, Q.; Wan, Y.; Zhou, Y.; Jia, Q.; Zhang, W.; Yuan, W.; Cheng, T.; Zhu, X.; et al. Assessment of hematopoietic failure due to Rpl11 deficiency in a zebrafish model of Diamond-Blackfan anemia by deep sequencing. BMC Genom. 2013, 14, 896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karayel, O.; Xu, P.; Bludau, I.; Bhoopalan, S.V.; Yao, Y.; Rita, F.C.A.; Santos, A.; A Schulman, B.; Alpi, A.F.; Weiss, M.J.; et al. Integrative proteomics reveals principles of dynamic phosphosignaling networks in human erythropoiesis. Mol. Syst. Biol. 2020, 16, e9813. [Google Scholar] [CrossRef] [PubMed]
- A Sekeres, M.; Heuer, A.H.; Saunthararajah, Y.; Barnard, J.; Tiu, R.V.; Visconte, V.; Avishai, N.; Mahfouz, R.Z.; Tabarroki, A.; Cowen, J.; et al. Distinct iron architecture in SF3B1-mutant myelodysplastic syndrome patients is linked to an SLC25A37 splice variant with a retained intron. Leukemia 2014, 29, 188–195. [Google Scholar] [CrossRef]
- Wang, Y.; Langer, N.B.; Shaw, G.C.; Yang, G.; Li, L.; Kaplan, J.; Paw, B.H.; Bloomer, J.R. Abnormal mitoferrin-1 expression in patients with erythropoietic protoporphyria. Exp. Hematol. 2011, 39, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhang, Y.; Cheng, X.; Yuan, H.; Zhu, S.; Liu, J.; Wen, Q.; Xie, Y.; Liu, J.; Kroemer, G.; et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev. Cell 2018, 46, 441–455.e8. [Google Scholar] [CrossRef] [Green Version]
- Key, J.; Sen, N.; Arsović, A.; Krämer, S.; Hülse, R.; Khan, N.; Meierhofer, D.; Gispert, S.; Koepf, G.; Auburger, G. Systematic Surveys of Iron Homeostasis Mechanisms Reveal Ferritin Superfamily and Nucleotide Surveillance Regulation to be Modified by PINK1 Absence. Cells 2020, 9, 2229. [Google Scholar] [CrossRef]
- Bozzo, F.; Mirra, A.; Carrì, M. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: New perspectives. Neurosci. Lett. 2017, 636, 3–8. [Google Scholar] [CrossRef]
- Cardoso, S.; Carvalho, C.; Correia, S.C.; Seiça, R.M.; Moreira, P.I. Alzheimer’s disease: From mitochondrial perturbations to mitochondrial medicine. Brain Pathol. 2016, 26, 632–647. [Google Scholar] [CrossRef]
- Requejo-Aguilar, R.; Bolaños, J.P. Mitochondrial control of cell bioenergetics in Parkinson’s disease. Free Radic. Biol. Med. 2016, 100, 123–137. [Google Scholar] [CrossRef] [Green Version]
- Hadzhieva, M.; Kirches, E.; Mawrin, C. Review: Iron metabolism and the role of iron in neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2014, 40, 240–257. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.G.; Kalkwarf, D.R. Iron, radiation, and cancer. Environ. Health Perspect. 1990, 87, 291–300. [Google Scholar] [CrossRef]
- Leach, J.K.; Van Tuyle, G.; Lin, P.S.; Schmidt-Ullrich, R.; Mikkelsen, R.B. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001, 61, 3894–3901. [Google Scholar] [PubMed]
- Kazan, H.H.; Urfali-Mamatoglu, C.; Gunduz, U. Iron metabolism and drug resistance in cancer. BioMetals 2017, 30, 629–641. [Google Scholar] [CrossRef]
- Morales, M.; Xue, X. Targeting iron metabolism in cancer therapy. Theranostics 2021, 11, 8412–8429. [Google Scholar] [CrossRef] [PubMed]
- Bashir, K.; Ishimaru, Y.; Shimo, H.; Nagasaka, S.; Fujimoto, M.; Takanashi, H.; Tsutsumi, N.; An, G.; Nakanishi, H.; Nishizawa, N.K. The rice mitochondrial iron transporter is essential for plant growth. Nat. Commun. 2011, 2, 322. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Connolly, E.L. Mitochondrial iron transport and homeostasis in plants. Front. Plant Sci. 2013, 4, 348. [Google Scholar] [CrossRef] [Green Version]
- Vigani, G.; Bashir, K.; Ishimaru, Y.; Lehmann, M.; Casiraghi, F.M.; Nakanishi, H.; Seki, M.; Geigenberger, P.; Zocchi, G.; Nishizawa, N.K. Knocking down mitochondrial iron transporter (MIT) reprograms primary and secondary metabolism in rice plants. J. Exp. Bot. 2015, 67, 1357–1368. [Google Scholar] [CrossRef] [Green Version]
- Azzam, E.I.; Jay-Gerin, J.-P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef]
- Dolatshad, H.; Pellagatti, A.; Fernandez-Mercado, M.; Yip, B.H.; Malcovati, L.; Attwood, M.; Przychodzen, B.; Sahgal, N.; Kanapin, A.; Lockstone, H.; et al. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia 2015, 29, 1798. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, S.; Yildiz, S.; Korucu, T.; Gundogan, B.; Sumbul, Z.E.; Korkmaz, H.H.; Atmaca, M. Frequency of anemia in chronic psychiatry patients. Neuropsychiatr. Dis. Treat. 2015, ume 11, 2737–2741. [Google Scholar] [CrossRef] [Green Version]
- Huo, Y.-X.; Huang, L.; Zhang, D.-F.; Yao, Y.-G.; Fang, Y.-R.; Zhang, C.; Luo, X.-J. Identification of SLC25A37 as a major depressive disorder risk gene. J. Psychiatr. Res. 2016, 83, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, L.; Endres, T.; Scholz, J.; Kirches, E.; Ward, D.M.; Lessmann, V.; Borucki, K.; Mawrin, C. Mitoferrin-1 is required for brain energy metabolism and hippocampus-dependent memory. Neurosci. Lett. 2019, 713, 134521. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Xu, J.; Huang, Y.; Zhai, Y.; Ma, Z.; Zhou, B.; Cao, Z. Elevating bioavailable iron levels in mitochondria suppresses the defective phenotypes caused by PINK1 loss-of-function in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2020, 532, 285–291. [Google Scholar] [CrossRef]
- Navarro, J.A.; Botella, J.A.; Metzendorf, C.; Lind, M.I.; Schneuwly, S. Mitoferrin modulates iron toxicity in a Drosophila model of Friedreich’s ataxia. Free Radic. Biol. Med. 2015, 85, 71–82. [Google Scholar] [CrossRef]
- Edenharter, O.; Clement, J.; Schneuwly, S.; Navarro, J.A. Overexpression of Drosophila frataxin triggers cell death in an iron-dependent manner. J. Neurogenet. 2017, 31, 189–202. [Google Scholar] [CrossRef]
- Huang, J.; Chen, S.; Hu, L.; Niu, H.; Sun, Q.; Li, W.; Tan, G.; Li, J.; Jin, L.; Lyu, J.; et al. Mitoferrin-1 is Involved in the Progression of Alzheimer’s Disease Through Targeting Mitochondrial Iron Metabolism in a Caenorhabditis elegans Model of Alzheimer’s Disease. Neuroscience 2018, 385, 90–101. [Google Scholar] [CrossRef]
- Picca, A.; Saini, S.K.; Mankowski, R.T.; Kamenov, G.; Anton, S.D.; Manini, T.M.; Buford, T.W.; Wohlgemuth, S.E.; Xiao, R.; Calvani, R.; et al. Altered Expression of Mitoferrin and Frataxin, Larger Labile Iron Pool and Greater Mitochondrial DNA Damage in the Skeletal Muscle of Older Adults. Cells 2020, 9, 2579. [Google Scholar] [CrossRef]
- Seguin, A.; Jia, X.; Earl, A.M.; Li, L.; Wallace, J.; Qiu, A.; Bradley, T.; Shrestha, R.; Troadec, M.-B.; Hockin, M.; et al. The mitochondrial metal transporters mitoferrin1 and mitoferrin2 are required for liver regeneration and cell proliferation in mice. J. Biol. Chem. 2020, 295, 11002–11020. [Google Scholar] [CrossRef]
- Metzendorf, C.; I Lind, M. Drosophila mitoferrin is essential for male fertility: Evidence for a role of mitochondrial iron metabolism during spermatogenesis. BMC Dev. Biol. 2010, 10, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-C.; Wu, Y.-T.; Wei, Y.-H. Depletion of mitoferrins leads to mitochondrial dysfunction and impairment of adipogenic differentiation in 3T3-L1 preadipocytes. Free Radic. Res. 2015, 49, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Fox, J.; Thyagarajan, B.; Fox, J.H. Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free Radic. Biol. Med. 2018, 120, 317–329. [Google Scholar] [CrossRef]
- Hung, H.-I.; Schwartz, J.M.; Maldonado, E.N.; Lemasters, J.J.; Nieminen, A.-L. Mitoferrin-2-dependent Mitochondrial Iron Uptake Sensitizes Human Head and Neck Squamous Carcinoma Cells to Photodynamic Therapy. J. Biol. Chem. 2013, 288, 677–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Chen, X.; Zou, H.; Chen, X.; Liu, Y.; Zhao, S. The roles of mitoferrin-2 in the process of arsenic trioxide-induced cell damage in human gliomas. Eur. J. Med. Res. 2014, 19, 49. [Google Scholar] [CrossRef] [Green Version]
- Tomita, K.; Fukumoto, M.; Itoh, K.; Kuwahara, Y.; Igarashi, K.; Nagasawa, T.; Suzuki, M.; Kurimasa, A.; Sato, T. MiR-7-5p is a key factor that controls radioresistance via intracellular Fe2+ content in clinically relevant radioresistant cells. Biochem. Biophys. Res. Commun. 2019, 518, 712–718. [Google Scholar] [CrossRef]




| Gene | mRNA Seq | Protein Sequence | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Name | Chromosomal Location | Ensembl Accession No. | Transcripts | Ensembl Transcript ID | Exon Count | Coding Exons | Uniprot Accession No. | Isoform | Protein Length (aa) |
| Mitoferrin-1 | 8p21.2 | ENSG00000147454 | 12 | ENST00000519973.6 | 9 | 4 | Q9NYZ2 | 3 | 338 |
| Mitoferrin-2 | 10q24.2 | ENSG00000155287 | 4 | ENST00000370495.6 | 11 | 4 | Q96A46-1 | 3 | 364 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.Y.; Oliva, C.R.; Flor, S.; Griguer, C.E. Mitoferrin, Cellular and Mitochondrial Iron Homeostasis. Cells 2022, 11, 3464. https://doi.org/10.3390/cells11213464
Ali MY, Oliva CR, Flor S, Griguer CE. Mitoferrin, Cellular and Mitochondrial Iron Homeostasis. Cells. 2022; 11(21):3464. https://doi.org/10.3390/cells11213464
Chicago/Turabian StyleAli, Md Yousuf, Claudia R. Oliva, Susanne Flor, and Corinne E. Griguer. 2022. "Mitoferrin, Cellular and Mitochondrial Iron Homeostasis" Cells 11, no. 21: 3464. https://doi.org/10.3390/cells11213464