
Cross-Talk and Subset Control of Microglia and Associated Myeloid Cells in Neurological Disorders

 , ,
, ,
Abstract
: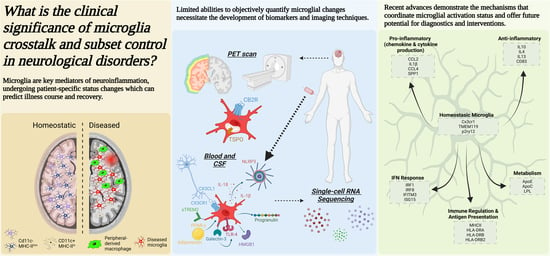
1. Distinct Role for Microglia and Other Myeloid Cells in the Neurological Disorders
- a.
- Overview of the roles of CNS myeloid cells in the pathogenesis of neurological diseases
1.1. CNS Resident Microglia
1.2. Peripheral-Derived Monocytes/Macrophages
1.3. CNS Border-Associated Macrophages
- b.
- New insights into disease-associated CNS myeloid subsets
1.4. CNS Resident Microglial Subsets
1.5. Subsets of Microglia in the Human Brain
1.6. Subsets of Microglia in the Murine Brain
1.7. CNS Border-Associated and Peripheral-Derived Macrophage Subsets
- c.
- Peripheral-derived monocyte engraftment and cross-talk with microglia and BAMs
1.8. Blood–Brain Barrier Integrity and Peripheral-Derived Immune Cell Invasion
1.9. External Factors Influencing CNS Engraftment of Peripheral-Derived Monocytes
1.10. Maintenance of Distinct Identities Following Cell Replacement
- d.
- Divergent neuroinflammatory functions of CNS macrophages in diseases of the brain
2. Clinical Assessment of Microglial Activation and CNS-Infiltrating Macrophages
- a.
- Imaging techniques: Positron emission tomography (PET), diffuse-weighted MRI, and Magnetic Resonance Imaging (MRI)
- b.
- CSF and blood biomarkers to detect microglia activation

{kind=link}
{kind=link}
{kind=link}
| Biomarker Name | Abbreviation | Microglia Connection | Disease State | Source | Direction | Sources |
|---|---|---|---|---|---|---|
| Soluble triggering receptor expressed on myeloid cells 2 | sTREM2 | Expressed in microglia, increases inflammatory cytokines | AD | CSF | Up | [135,145,147] |
| IS | Blood | Up | [149,150,151] | |||
| TBI | CSF | Up | [148] | |||
| MS | CSF | Up | [152,153,154] | |||
| Nod-leucine rich repeat and pyrin containing protein 3 | NLRP3 inflammasome | Activated by microglial cytokines IL-1β and IL-18 | AD | CSF | Up | [41,155] |
| IS | Brain tissue | Up | [156] | |||
| TBI | CSF | Up | [159] | |||
| MS | PBMC | Up | [158] | |||
| Adiponectin | Adiponectin | Modulates microglia through PPAR-γ signaling | AD | CSF/Blood | Down/Up | [161] |
| IS | Blood | Up | [188] | |||
| TBI | Blood | Up | [162] | |||
| MS | Blood | Up | [163] | |||
| High mobility group box protein 1 | HMGB1 | DAMP, expressed in phagocytic microglia | AD | Blood | Up | [165,166] |
| IS | Blood | Up | [167,168] | |||
| TBI | Blood | Up | [169] | |||
| MS | Blood | Up | [170] | |||
| Galectin-3 | Gal-3 | TLR-4 ligand, promotes microglial activation | AD | Blood | Up | [175,176] |
| IS | Blood | Up | [173,177] | |||
| TBI | Blood | Up | [178] | |||
| MS | Blood/AA | Up | [174] | |||
| Fractalkine | CX3CL1 | Expressed in neurons, facilitates neuron-microglia communication | AD | CSF/Blood | Down/- | [136] |
| IS | Blood | Down | [179] | |||
| TBI | Blood/CMD | Mixed | [40,189] | |||
| MS | PBMC | Up | [190] | |||
| Progranulin | PGRN | Expressed in microglia, attenuates neural damage | AD | CSF | Up, sex/age-bias | [183] |
| IS | Blood | Up | [184] | |||
| TBI | Serum | Mixed | [180,191] | |||
| MS | CSF | Up | [185] |
3. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kierdorf, K.; Masuda, T.; Jordao, M.J.C.; Prinz, M. Macrophages at CNS interfaces: Ontogeny and function in health and disease. Nat. Rev. Neurosci. 2019, 20, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Pettas, S.; Karagianni, K.; Kanata, E.; Chatziefstathiou, A.; Christoudia, N.; Xanthopoulos, K.; Sklaviadis, T.; Dafou, D. Profiling Microglia through Single-Cell RNA Sequencing over the Course of Development, Aging, and Disease. Cells 2022, 11, 2383. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Kiyama, K. Non-pathological roles of microglial TREM2/DAP12: TREM2/DAP12 regulates the physiological functions of microglia from development to aging. Neurochem. Int. 2020, 141, 104878. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M.V. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef]
- Oldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 599. [Google Scholar] [CrossRef] [Green Version]
- Derk, J.; Jones, H.E.; Como, C.; Pawlikowski, B.; Siegenthaler, J.A. Living on the Edge of the CNS: Meninges Cell Diversity in Health and Disease. Front. Cell. Neurosci. 2021, 15, 703944. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.; Liu, Y.J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef]
- Cugurra, A.; Mamuladze, T.; Rustenhoven, J.; Dykstra, T.; Beroshvili, G.; Greenberg, Z.J.; Baker, W.; Papadopoulos, Z.; Drieu, A.; Blackburn, S.; et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science 2021, 373, eabf7844. [Google Scholar] [CrossRef]
- Sevenich, L. Brain-Resident Microglia and Blood-Borne Macrophages Orchestrate Central Nervous System Inflammation in Neurodegenerative Disorders and Brain Cancer. Front. Immunol. 2018, 9, 697. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, M.E.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The role of microglia in the healthy brain. J. Neurosci. 2011, 31, 16064–16069. [Google Scholar] [CrossRef] [Green Version]
- Bachiller, S.; Jimenez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [Green Version]
- Andoh, M.; Koyama, R. Comparative Review of Microglia and Monocytes in CNS Phagocytosis. Cells 2021, 10, 255. [Google Scholar] [CrossRef]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Chen, H.R.; Chen, C.W.; Kuo, Y.M.; Chen, B.; Kuan, I.S.; Huang, H.; Lee, J.; Anthony, N.; Kuan, C.Y.; Sun, Y.Y. Monocytes promote acute neuroinflammation and become pathological microglia in neonatal hypoxic-ischemic brain injury. Theranostics 2022, 12, 512–529. [Google Scholar] [CrossRef]
- Chen, J.; Zheng, Z.V.; Lu, G.; Chan, W.Y.; Zhang, Y.; Wong, G.K.C. Microglia activation, classification and microglia-mediated neuroinflammatory modulators in subarachnoid hemorrhage. Neural Regen. Res. 2022, 17, 1404–1411. [Google Scholar]
- Delage, C.; Taib, T.; Mamma, C.; Lerouet, D.; Besson, V.C. Traumatic Brain Injury: An Age-Dependent View of Post-Traumatic Neuroinflammation and Its Treatment. Pharmaceutics 2021, 13, 1624. [Google Scholar] [CrossRef]
- Bellver-Landete, V.; Bretheau, F.; Mailhot, B.; Vallieres, N.; Lessard, M.; Janelle, M.E.; Vernoux, N.; Tremblay, M.E.; Fuehrmann, T.; Shoichet, M.S.; et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat. Commun. 2019, 10, 518. [Google Scholar] [CrossRef] [Green Version]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef] [PubMed]
- De Haas, A.H.; Boddeke, H.W.; Biber, K. Region-specific expression of immunoregulatory proteins on microglia in the healthy CNS. Glia 2008, 56, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Edler, M.K.; Mhatre-Winters, I.; Richardson, J.R. Microglia in Aging and Alzheimer’s Disease: A Comparative Species Review. Cells 2021, 10, 1138. [Google Scholar] [CrossRef] [PubMed]
- Garden, G.A.; Moller, T. Microglia biology in health and disease. J. Neuroimmune Pharmacol. 2006, 1, 127–137. [Google Scholar] [CrossRef]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [Green Version]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef] [Green Version]
- Krukowski, K.; Chou, A.; Feng, X.; Tiret, B.; Paladini, M.S.; Riparip, L.K.; Chaumeil, M.M.; Lemere, C.; Rosi, S. Traumatic Brain Injury in Aged Mice Induces Chronic Microglia Activation, Synapse Loss, and Complement-Dependent Memory Deficits. Int. J. Mol. Sci. 2018, 19, 3753. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Stoica, B.A.; Sabirzhanov, B.; Burns, M.P.; Faden, A.I.; Loane, D.J. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol. Aging 2013, 34, 1397–1411. [Google Scholar] [CrossRef] [Green Version]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275 Pt 3, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Makinde, H.M.; Just, T.B.; Gadhvi, G.T.; Winter, D.R.; Schwulst, S.J. Microglia Adopt Longitudinal Transcriptional Changes After Traumatic Brain Injury. J. Surg. Res. 2020, 246, 113–122. [Google Scholar] [CrossRef]
- Matias, D.; Balca-Silva, J.; da Graca, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Santos, M.F.D.; et al. Microglia/Astrocytes-Glioblastoma Crosstalk: Crucial Molecular Mechanisms and Microenvironmental Factors. Front. Cell. Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef] [Green Version]
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251–277. [Google Scholar] [CrossRef]
- Rodriguez-Gomez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef]
- Sankowski, R.; Bottcher, C.; Masuda, T.; Geirsdottir, L.; Sagar, E.; Sindram, T.; Seredenina, A.; Muhs, C.; Scheiwe, M.J.; Shah, D.H.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.E.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Wlodarczyk, A.; Holtman, I.R.; Krueger, M.; Yogev, N.; Bruttger, J.; Khorooshi, R.; Benmamar-Badel, A.; de Boer-Bergsma, J.J.; Martin, N.A.; Karram, K.; et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 2017, 36, 3292–3308. [Google Scholar] [CrossRef]
- Ziebell, J.M.; Adelson, P.D.; Lifshitz, J. Microglia: Dismantling and rebuilding circuits after acute neurological injury. Metab. Brain Dis. 2015, 30, 393–400. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Begum, G.; Reddy, R.; Yakoub, K.M.; Belli, A.; Davies, D.J.; Di Pietro, V. Differential Expression of Circulating Inflammatory Proteins Following Sport-Related Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 1216. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Lin, Y.; Zhang, B.; Lu, D.; Lu, Z.; Cai, W. Update of inflammasome activation in microglia/macrophage in aging and aging-related disease. CNS Neurosci. Ther. 2019, 25, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Barrett, J.P.; Alvarez-Croda, D.-M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav. Immun. 2016, 58, 291–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, C.D. M1 and M2 Macrophages: Oracles of Health and Disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selenica, M.L.; Alvarez, J.A.; Nash, K.R.; Lee, D.C.; Cao, C.; Lin, X.; Reid, P.; Mouton, P.R.; Morgan, D.; Gordon, M.N. Diverse activation of microglia by chemokine (C-C motif) ligand 2 overexpression in brain. J. Neuroinflamm. 2013, 10, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persidsky, Y.; Ghorpade, A.; Rasmussen, J.; Limoges, J.; Liu, X.J.; Stins, M.; Fiala, M.; Way, D.; Kim, K.S.; Witte, M.H.; et al. Microglial and astrocyte chemokines regulate monocyte migration through the blood-brain barrier in human immunodeficiency virus-1 encephalitis. Am. J. Pathol. 1999, 155, 1599–1611. [Google Scholar] [CrossRef] [Green Version]
- Priego, N.; Valiente, M. The Potential of Astrocytes as Immune Modulators in Brain Tumors. Front. Immunol. 2019, 10, 1314. [Google Scholar] [CrossRef] [Green Version]
- Somebang, K.; Rudolph, J.; Imhof, I.; Li, L.; Niemi, E.C.; Shigenaga, J.; Tran, H.; Gill, T.M.; Lo, I.; Zabel, B.A.; et al. CCR2 deficiency alters activation of microglia subsets in traumatic brain injury. Cell Rep. 2021, 36, 109727. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Kim, C.C.; Ryba, B.E.; Niemi, E.C.; Bando, J.K.; Locksley, R.M.; Liu, J.; Nakamura, M.C.; Seaman, W.E. Traumatic brain injury induces macrophage subsets in the brain. Eur. J. Immunol. 2013, 43, 2010–2022. [Google Scholar] [CrossRef] [Green Version]
- Passlick, B.; Flieger, D.; Ziegler-Heitbrock, H.W. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood 1989, 74, 2527–2534. [Google Scholar] [CrossRef] [Green Version]
- Wolf, A.A.; Yanez, A.; Barman, P.K.; Goodridge, H.S. The Ontogeny of Monocyte Subsets. Front. Immunol. 2019, 10, 1642. [Google Scholar] [CrossRef] [Green Version]
- Hammond, M.D.; Taylor, R.A.; Mullen, M.T.; Ai, Y.; Aguila, H.L.; Mack, M.; Kasner, S.E.; McCullough, L.D.; Sansing, L.H. CCR2+ Ly6C(hi) inflammatory monocyte recruitment exacerbates acute disability following intracerebral hemorrhage. J. Neurosci. 2014, 34, 3901–3909. [Google Scholar] [CrossRef]
- Kowalski, E.A.; Soliman, E.; Kelly, C.; Basso, E.K.G.; Leonard, J.; Pridham, K.J.; Ju, J.; Cash, A.; Hazy, A.; de Jager, C.; et al. Monocyte proinflammatory phenotypic control by ephrin type A receptor 4 mediates neural tissue damage. JCI Insight 2022, 7. [Google Scholar] [CrossRef]
- Kapellos, T.S.; Bonaguro, L.; Gemund, I.; Reusch, N.; Saglam, A.; Hinkley, E.R.; Schultze, J.L. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front. Immunol. 2019, 10, 2035. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.L.; Tai, J.J.; Wong, W.C.; Han, H.; Sem, X.; Yeap, W.H.; Kourilsky, P.; Wong, S.C. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011, 118, e16–e31. [Google Scholar] [CrossRef] [Green Version]
- Hamers, A.A.J.; Dinh, H.Q.; Thomas, G.D.; Marcovecchio, P.; Blatchley, A.; Nakao, C.S.; Kim, C.; McSkimming, C.; Taylor, A.M.; Nguyen, A.T.; et al. Human Monocyte Heterogeneity as Revealed by High-Dimensional Mass Cytometry. Arter. Thromb Vasc. Biol. 2019, 39, 25–36. [Google Scholar] [CrossRef]
- Weinstock, A.; Fisher, E.A. Methods to Study Monocyte and Macrophage Trafficking in Atherosclerosis Progression and Resolution. Methods Mol. Biol. 2019, 1951, 153–165. [Google Scholar]
- Makinde, H.M.; Just, T.B.; Cuda, C.M.; Bertolino, N.; Procissi, D.; Schwulst, S.J. Monocyte depletion attenuates the development of posttraumatic hydrocephalus and preserves white matter integrity after traumatic brain injury. PLoS ONE 2018, 13, e0202722. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Rosen, S.; Weinstein, P.; van Rooijen, N.; Noble-Haeusslein, L.J. Prevention of both neutrophil and monocyte recruitment promotes recovery after spinal cord injury. J. Neurotrauma 2011, 28, 1893–1907. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Zhai, X.; Han, D.; Xiong, X.; Wang, T.; Zeng, X.; He, S.; Liu, R.; Miyata, M.; Xu, B.; et al. CCR2-dependent monocytes/macrophages exacerbate acute brain injury but promote functional recovery after ischemic stroke in mice. Theranostics 2018, 8, 3530–3543. [Google Scholar] [CrossRef]
- Xu, J.; Ganguly, A.; Zhao, J.; Ivey, M.; Lopez, R.; Osterholzer, J.J.; Cho, C.S.; Olszewski, M.A. CCR2 Signaling Promotes Brain Infiltration of Inflammatory Monocytes and Contributes to Neuropathology during Cryptococcal Meningoencephalitis. mBio 2021, 12, e0107621. [Google Scholar] [CrossRef]
- Geng, H.; Chen, L.; Tang, J.; Chen, Y.; Wang, L. The Role of CCL2/CCR2 Axis in Cerebral Ischemia-Reperfusion Injury and Treatment: From Animal Experiments to Clinical Trials. Int. J. Mol. Sci. 2022, 23, 3485. [Google Scholar] [CrossRef] [PubMed]
- Naert, G.; Rivest, S. CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 6208–6220. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Tuo, H.; Wang, S.; Zhao, L. The Roles of Monocyte and Monocyte-Derived Macrophages in Common Brain Disorders. BioMed Res. Int. 2020, 2020, 9396021. [Google Scholar] [CrossRef] [PubMed]
- Makinde, H.M.; Cuda, C.M.; Just, T.B.; Perlman, H.R.; Schwulst, S.J. Nonclassical Monocytes Mediate Secondary Injury, Neurocognitive Outcome, and Neutrophil Infiltration after Traumatic Brain Injury. J. Immunol. 2017, 199, 3583–3591. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.; Han, J.; Wang, Y.; Xu, Y.; Zhang, Y.; Zhu, C. The therapeutic potential of bone marrow-derived macrophages in neurological diseases. CNS Neurosci. Ther. 2022, 1–11. [Google Scholar] [CrossRef]
- Meeker, R.B.; Williams, K.; Killebrew, D.A.; Hudson, L.C. Cell trafficking through the choroid plexus. Cell Adh. Migr. 2012, 6, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Strominger, I.; Elyahu, Y.; Berner, O.; Reckhow, J.; Mittal, K.; Nemirovsky, A.; Monsonego, A. The Choroid Plexus Functions as a Niche for T-Cell Stimulation Within the Central Nervous System. Front. Immunol. 2018, 9, 1066. [Google Scholar] [CrossRef]
- Szmydynger-Chodobska, J.; Strazielle, N.; Zink, B.J.; Ghersi-Egea, J.F.; Chodobski, A. The role of the choroid plexus in neutrophil invasion after traumatic brain injury. J. Cereb. Blood Flow Metab. 2009, 29, 1503–1516. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Mack, J.J.; Guc, E.; Warren, C.M.; Squadrito, M.L.; Kilarski, W.W.; Baer, C.; Freshman, R.D.; McDonald, A.I.; Ziyad, S.; et al. Perivascular Macrophages Limit Permeability. Arter. Thromb. Vasc. Biol. 2016, 36, 2203–2212. [Google Scholar] [CrossRef] [Green Version]
- Venero Galanternik, M.; Castranova, D.; Gore, A.V.; Blewett, N.H.; Jung, H.M.; Stratman, A.N.; Kirby, M.R.; Iben, J.; Miller, M.F.; Kawakami, K.; et al. A novel perivascular cell population in the zebrafish brain. Elife 2017, 6, e24369. [Google Scholar] [CrossRef]
- Axtell, R.C.; Steinman, L. Gaining entry to an uninflamed brain. Nat. Immunol. 2009, 10, 453–455. [Google Scholar] [CrossRef]
- Bodnar, C.N.; Watson, J.B.; Higgins, E.K.; Quan, N.; Bachstetter, A.D. Inflammatory Regulation of CNS Barriers After Traumatic Brain Injury: A Tale Directed by Interleukin-1. Front. Immunol. 2021, 12, 688254. [Google Scholar] [CrossRef]
- Di Marco Barros, R.; Fitzpatrick, Z.; Clatworthy, M. R The gut-meningeal immune axis: Priming brain defense against the most likely invaders. J. Exp. Med. 2022, 219, e20211520. [Google Scholar] [CrossRef]
- Russo, M.V.; Latour, L.L.; McGavern, D.B. Distinct myeloid cell subsets promote meningeal remodeling and vascular repair after mild traumatic brain injury. Nat. Immunol. 2018, 19, 442–452. [Google Scholar] [CrossRef]
- Jordao, M.J.C.; Sankowski, R.; Brendecke, S.M.; Sagar; Locatelli, G.; Tai, Y.-H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019, 363, eaat7554. [Google Scholar]
- Holtman, I.R.; Skola, D.; Glass, C.K. Transcriptional control of microglia phenotypes in health and disease. J. Clin. Investig. 2017, 127, 3220–3229. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Shui, X.; Sun, R.; Wan, L.; Zhang, B.; Xiao, B.; Luo, Z. Microglial Phenotypic Transition: Signaling Pathways and Influencing Modulators Involved in Regulation in Central Nervous System Diseases. Front. Cell. Neurosci. 2021, 15, 736310. [Google Scholar] [CrossRef]
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS ONE 2011, 6, e26317. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Xu, Z.; Xiong, S.; Sun, F.; Qin, G.; Hu, G.; Wang, J.; Zhao, L.; Liang, Y.X.; Wu, T.; et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat. Neurosci. 2018, 21, 530–540. [Google Scholar] [CrossRef]
- Zhan, L.; Krabbe, G.; Du, F.; Jones, I.; Reichert, M.C.; Telpoukhovskaia, M.; Kodama, L.; Wang, C.; Cho, S.H.; Sayed, F.; et al. Proximal recolonization by self-renewing microglia re-establishes microglial homeostasis in the adult mouse brain. PLoS Biol. 2019, 17, e3000134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boza-Serrano, A.; Ruiz, R.; Sanchez-Varo, R.; Garcia-Revilla, J.; Yang, Y.; Jimenez-Ferrer, I.; Paulus, A.; Wennstrom, M.; Vilalta, A.; Allendorf, D.; et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 2019, 138, 251–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, L.; Fan, L.; Kodama, L.; Sohn, P.D.; Wong, M.Y.; Mousa, G.A.; Zhou, Y.; Li, Y.; Gan, L. A MAC2-positive progenitor-like microglial population is resistant to CSF1R inhibition in adult mouse brain. Elife 2020, 9, e51796. [Google Scholar] [CrossRef] [PubMed]
- Immig, K.; Gericke, M.; Menzel, F.; Merz, F.; Krueger, M.; Schiefenhovel, F.; Losche, A.; Jager, K.; Hanisch, U.K.; Biber, K.; et al. CD11c-positive cells from brain, spleen, lung, and liver exhibit site-specific immune phenotypes and plastically adapt to new environments. Glia 2015, 63, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Jiang, Y.; Li, J.; Wang, Y.; Tian, Y.; Guo, Q.; Cheng, Z. DUSP1 Promotes Microglial Polarization toward M2 Phenotype in the Medial Prefrontal Cortex of Neuropathic Pain Rats via Inhibition of MAPK Pathway. ACS Chem. Neurosci. 2021, 12, 966–978. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Esaulova, E.; Cantoni, C.; Shchukina, I.; Zaitsev, K.; Bucelli, R.C.; Wu, G.F.; Artyomov, M.N.; Cross, A.H.; Edelson, B.T. Single-cell RNA-seq analysis of human CSF microglia and myeloid cells in neuroinflammation. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e732. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Bottcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar]
- Olah, M.; Menon, V.; Habib, N.; Taga, M.F.; Ma, Y.; Yung, C.J.; Cimpean, M.; Khairallah, A.; Coronas-Samano, G.; Sankowski, R.; et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 2020, 11, 6129. [Google Scholar] [CrossRef]
- Jinno, S.; Fleischer, F.; Eckel, S.; Schmidt, V.; Kosaka, T. Spatial arrangement of microglia in the mouse hippocampus: A stereological study in comparison with astrocytes. Glia 2007, 55, 1334–1347. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Vela, J.M.; Dalmau, I.; Gonzalez, B.; Castellano, B. Morphology and distribution of microglial cells in the young and adult mouse cerebellum. J. Comp. Neurol. 1995, 361, 602–616. [Google Scholar] [CrossRef]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 24. [Google Scholar] [CrossRef]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Gerganova, G.; Riddell, A.; Miller, A.A. CNS border-associated macrophages in the homeostatic and ischaemic brain. Pharmacol. Ther. 2022, 240, 108220. [Google Scholar] [CrossRef]
- Yanez, A.; Coetzee, S.G.; Olsson, A.; Muench, D.E.; Berman, B.P.; Hazelett, D.J.; Salomonis, N.; Grimes, H.L.; Goodridge, H.S. Granulocyte-Monocyte Progenitors and Monocyte-Dendritic Cell Progenitors Independently Produce Functionally Distinct Monocytes. Immunity 2017, 47, 890–902.e4. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.C.; Nakamura, M.C.; Hsieh, C.L. Brain trauma elicits non-canonical macrophage activation states. J. Neuroinflamm. 2016, 13, 117. [Google Scholar] [CrossRef] [Green Version]
- Mildner, A.; Schmidt, H.; Nitsche, M.; Merkler, D.; Hanisch, U.K.; Mack, M.; Heikenwalder, M.; Bruck, W.; Priller, J.; Prinz, M.; et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 2007, 10, 1544–1553. [Google Scholar] [CrossRef]
- Hickey, W.F.; Vass, K.; Lassmann, H. Bone marrow-derived elements in the central nervous system: An immunohistochemical and ultrastructural survey of rat chimeras. J. Neuropathol. Exp. Neurol. 1992, 51, 246–256. [Google Scholar] [CrossRef]
- Priller, J.; Flugel, A.; Wehner, T.; Boentert, M.; Haas, C.A.; Prinz, M.; Fernandez-Klett, F.; Prass, K.; Bechmann, I.; de Boer, B.A.; et al. Targeting gene-modified hematopoietic cells to the central nervous system: Use of green fluorescent protein uncovers microglial engraftment. Nat. Med. 2001, 7, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.F.; Goods, B.A.; Askenase, M.H.; Beatty, H.E.; Osherov, A.; DeLong, J.H.; Hammond, M.D.; Massey, J.; Landreneau, M.; Love, J.C.; et al. Divergent Functions of Tissue-Resident and Blood-Derived Macrophages in the Hemorrhagic Brain. Stroke 2021, 52, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, D.; Fumagalli, S.; Schafer, M.K.; Pedragosa, J.; Ngassam, L.D.C.; Wilhelmi, V.; Winterberg, S.; Planas, A.M.; Weihe, E.; de Simoni, M.G. Protein Expression of the Microglial Marker Tmem119 Decreases in Association With Morphological Changes and Location in a Mouse Model of Traumatic Brain Injury. Front. Cell. Neurosci. 2022, 16, 820127. [Google Scholar] [CrossRef] [PubMed]
- Van Wageningen, T.A.; Vlaar, E.; Kooij, G.; Jongenelen, C.A.M.; Geurts, J.J.G.; van Dam, A.M. Regulation of microglial TMEM119 and P2RY12 immunoreactivity in multiple sclerosis white and grey matter lesions is dependent on their inflammatory environment. Acta Neuropathol. Commun. 2019, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Vankriekelsvenne, E.; Chrzanowski, U.; Manzhula, K.; Greiner, T.; Wree, A.; Hawlitschka, A.; Llovera, G.; Zhan, J.; Joost, S.; Schmitz, C.; et al. Transmembrane protein 119 is neither a specific nor a reliable marker for microglia. Glia 2022, 70, 1170–1190. [Google Scholar] [CrossRef]
- Young, K.F.; Gardner, R.; Sariana, V.; Whitman, S.A.; Bartlett, M.J.; Falk, T.; Morrison, H.W. Can quantifying morphology and TMEM119 expression distinguish between microglia and infiltrating macrophages after ischemic stroke and reperfusion in male and female mice? J. Neuroinflamm. 2021, 18, 58. [Google Scholar] [CrossRef]
- Su, N.; Marz, S.; Plagemann, T.; Cao, J.; Schnittler, H.J.; Eter, N.; Heiduschka, P. Occurrence of Transmembrane Protein 119 in the Retina is Not Restricted to the Microglia: An Immunohistochemical Study. Transl. Vis. Sci. Technol. 2019, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Soliman, E.; Mills, J.; Ju, J.; Kaloss, A.M.; Basso, E.K.G.; Groot, N.; Kelly, C.; Kowalski, E.A.; Elhassanny, M.; Chen, M.; et al. Conditional Deletion of EphA4 on Cx3cr1-Expressing Microglia Fails to Influence Histopathological Outcome and Blood Brain Barrier Disruption Following Brain Injury. Front. Mol. Neurosci. 2021, 14, 747770. [Google Scholar] [CrossRef]
- Grassivaro, F.; Menon, R.; Acquaviva, M.; Ottoboni, L.; Ruffini, F.; Bergamaschi, A.; Muzio, L.; Farina, C.; Martino, G. Convergence between Microglia and Peripheral Macrophages Phenotype during Development and Neuroinflammation. J. Neurosci. 2020, 40, 784–795. [Google Scholar] [CrossRef]
- Miyata, S. New aspects in fenestrated capillary and tissue dynamics in the sensory circumventricular organs of adult brains. Front. Neurosci. 2015, 9, 390. [Google Scholar] [CrossRef] [Green Version]
- Ousman, S.S.; Kubes, P. Immune surveillance in the central nervous system. Nat. Neurosci. 2012, 15, 1096–1101. [Google Scholar] [CrossRef]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The blood-brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Qiu, Y.M.; Zhang, C.L.; Chen, A.Q.; Wang, H.L.; Zhou, Y.F.; Li, Y.N.; Hu, B. Immune Cells in the BBB Disruption After Acute Ischemic Stroke: Targets for Immune Therapy? Front. Immunol. 2021, 12, 678744. [Google Scholar] [CrossRef]
- Cronk, J.C.; Filiano, A.J.; Louveau, A.; Marin, I.; Marsh, R.; Ji, E.; Goldman, D.H.; Smirnov, I.; Geraci, N.; Acton, S.; et al. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J. Exp. Med. 2018, 215, 1627–1647. [Google Scholar] [CrossRef] [Green Version]
- Rua, R.; Lee, J.Y.; Silva, A.B.; Swafford, I.S.; Maric, D.; Johnson, K.R.; McGavern, D.B. Infection drives meningeal engraftment by inflammatory monocytes that impairs CNS immunity. Nat. Immunol. 2019, 20, 407–419. [Google Scholar] [CrossRef]
- Lund, H.; Pieber, M.; Parsa, R.; Han, J.; Grommisch, D.; Ewing, E.; Kular, L.; Needhamsen, M.; Espinosa, A.; Nilsson, E.; et al. Competitive repopulation of an empty microglial niche yields functionally distinct subsets of microglia-like cells. Nat. Commun. 2018, 9, 4845. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Rao, Y.; Huang, Y.; Zhou, T.; Feng, R.; Xiong, S.; Yuan, T.F.; Qin, S.; Lu, Y.; Zhou, X.; et al. Efficient Strategies for Microglia Replacement in the Central Nervous System. Cell Rep. 2020, 32, 108041. [Google Scholar] [CrossRef]
- Herisson, F.; Frodermann, V.; Courties, G.; Rohde, D.; Sun, Y.; Vandoorne, K.; Wojtkiewicz, G.R.; Masson, G.S.; Vinegoni, C.; Kim, J.; et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat. Neurosci. 2018, 21, 1209–1217. [Google Scholar] [CrossRef]
- Cai, R.; Pan, C.; Ghasemigharagoz, A.; Todorov, M.I.; Forstera, B.; Zhao, S.; Bhatia, H.S.; Parra-Damas, A.; Mrowka, L.; Theodorou, D.; et al. Panoptic imaging of transparent mice reveals whole-body neuronal projections and skull-meninges connections. Nat. Neurosci. 2019, 22, 317–327. [Google Scholar] [CrossRef]
- Yao, H.; Price, T.T.; Cantelli, G.; Ngo, B.; Warner, M.J.; Olivere, L.; Ridge, S.M.; Jablonski, E.M.; Therrien, J.; Tannheimer, S.; et al. Leukaemia hijacks a neural mechanism to invade the central nervous system. Nature 2018, 560, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, I.; Priller, J.; Kovac, A.; Bontert, M.; Wehner, T.; Klett, F.F.; Bohsung, J.; Stuschke, M.; Dirnagl, U.; Nitsch, R. Immune surveillance of mouse brain perivascular spaces by blood-borne macrophages. Eur. J. Neurosci. 2001, 14, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Hickey, W.F.; Kimura, H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science 1988, 239, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Iwasaki, A. Monocytes Inadequately Fill In for Meningeal Macrophages. Trends Immunol. 2019, 40, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Ruedl, C.; Karjalainen, K. Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity 2015, 43, 382–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Saito, T.; Saido, T.C.; Barron, A.M.; Ruedl, C. Microglia and CD206+ border-associated mouse macrophages maintain their embryonic origin during Alzheimer’s disease. Elife 2021, 10, e71879. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, A.; Lelios, I.; Yu, X.; Vrohlings, M.; Krakoski, N.R.; Gautier, E.L.; Nishinakamura, R.; Becher, B.; Greter, M. Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 2016, 17, 1397–1406. [Google Scholar]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Koso, H.; Tsuhako, A.; Lai, C.Y.; Baba, Y.; Otsu, M.; Ueno, K.; Nagasaki, M.; Suzuki, Y.; Watanabe, S. Conditional rod photoreceptor ablation reveals Sall1 as a microglial marker and regulator of microglial morphology in the retina. Glia 2016, 64, 2005–2024. [Google Scholar] [CrossRef]
- Lloyd-Burton, S.M.; York, E.M.; Anwar, M.A.; Vincent, A.J.; Roskams, A.J. SPARC regulates microgliosis and functional recovery following cortical ischemia. J. Neurosci. 2013, 33, 4468–4481. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.W.; McGeachy, M.J.; Bayir, H.; Clark, R.S.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [Green Version]
- Nakada, M.; Hayashi, Y.; Hamada, J. Role of Eph/ephrin tyrosine kinase in malignant glioma. Neuro. Oncol. 2011, 13, 1163–1170. [Google Scholar] [CrossRef] [Green Version]
- Nakada, M.; Nakada, S.; Demuth, T.; Tran, N.L.; Hoelzinger, D.B.; Berens, M.E. Molecular targets of glioma invasion. Cell. Mol. Life Sci. 2007, 64, 458–478. [Google Scholar] [CrossRef]
- Wang, L.F.; Fokas, E.; Bieker, M.; Rose, F.; Rexin, P.; Zhu, Y.; Pagenstecher, A.; Engenhart-Cabillic, R.; An, H.X. Increased expression of EphA2 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. Oncol. Rep. 2008, 19, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef]
- Perea, J.R.; Lleó, A.; Alcolea, D.; Fortea, J.; Ávila, J.; Bolós, M. Decreased CX3CL1 Levels in the Cerebrospinal Fluid of Patients With Alzheimer’s Disease. Front. Neurosci. 2018, 12, 609. [Google Scholar] [CrossRef] [Green Version]
- Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11, 2091. [Google Scholar] [CrossRef]
- Beuker, C.; Schafflick, D.; Strecker, J.K.; Heming, M.; Li, X.; Wolbert, J.; Schmidt-Pogoda, A.; Thomas, C.; Kuhlmann, T.; Aranda-Pardos, I.; et al. Stroke induces disease-specific myeloid cells in the brain parenchyma and pia. Nat. Commun. 2022, 13, 945. [Google Scholar] [CrossRef]
- Shah, F.; Hume, S.P.; Pike, V.W.; Ashworth, S.; McDermott, J. Synthesis of the enantiomers of [N-methyl-11C]PK 11195 and comparison of their behaviours as radioligands for PK binding sites in rats. Nucl. Med. Biol. 1994, 21, 573–581. [Google Scholar] [CrossRef]
- Parbo, P.; Ismail, R.; Hansen, K.V.; Amidi, A.; Mårup, F.H.; Gottrup, H.; Brændgaard, H.; Eriksson, B.O.; Eskildsen, S.F.; Lund, T.E.; et al. Brain inflammation accompanies amyloid in the majority of mild cognitive impairment cases due to Alzheimer’s disease. Brain 2017, 140, 2002–2011. [Google Scholar] [CrossRef]
- Venneti, S.; Wagner, A.; Wang, G.; Slagel, S.; Chen, X.; Lopresti, B.; Mathis, C.; Wiley, C. The high affinity peripheral benzodiazepine receptor ligand DAA1106 binds specifically to microglia in a rat model of traumatic brain injury: Implications for PET imaging. Exp. Neurol. 2007, 207, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, N.; Mandhair, H.; Smyth, E.; Dakin, S.G.; Kiriakidis, S.; Wells, L.; Owen, D.; Sabokbar, A.; Taylor, P. The macrophage marker translocator protein (TSPO) is down-regulated on pro-inflammatory ‘M1’ human macrophages. PLoS ONE 2017, 12, e0185767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Hernandez, R.; Cerdá, A.C.; Carpena, A.T.; Drakesmith, M.; Koller, K.; Jones, D.K.; Canals, S.; de Santis, S. Mapping microglia and astrocyte activation in vivo using diffusion MRI. Sci. Adv. 2022, 8, eabq2923. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.; Bendszus, M. Imaging of inflammation in the peripheral and central nervous system by magnetic resonance imaging. Neuroscience 2009, 158, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Khrestian, M.; Dyne, E.; Shao, Y.; Pillai, J.A.; Rao, S.M.; Bemiller, S.M.; Lamb, B.; Fernandez, H.H.; Leverenz, J.B. Soluble TREM2 and biomarkers of central and peripheral inflammation in neurodegenerative disease. J. Neuroimmunol. 2018, 319, 19–27. [Google Scholar] [CrossRef]
- Zhong, L.; Chen, X.-F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Calvet, M.; Morenas-Rodríguez, E.; Kleinberger, G.; Schlepckow, K.; Caballero, M.Á.A.; Franzmeier, N.; Capell, A.; Fellerer, K.; Nuscher, B.; Eren, E.; et al. Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol. Neurodegener. 2019, 14, 1. [Google Scholar] [CrossRef] [Green Version]
- Alosco, M.L.; Tripodis, Y.; Fritts, N.G.; Heslegrave, A.; Baugh, C.M.; Conneely, S.; Mariani, M.; Martin, B.M.; Frank, S.; Mez, J.; et al. Cerebrospinal fluid tau, Aβ, and sTREM2 in Former National Football League Players: Modeling the relationship between repetitive head impacts, microglial activation, and neurodegeneration. Alzheimer Dement. 2018, 14, 1159–1170. [Google Scholar] [CrossRef]
- Rauchmann, B.-S.; Schneider-Axmann, T.; Alexopoulos, P.; Perneczky, R. CSF soluble TREM2 as a measure of immune response along the Alzheimer’s disease continuum. Neurobiol. Aging 2019, 74, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.S.; Lee, E.-H.; Park, H.-H.; Jin, J.-H.; Choi, H.; Lee, K.-Y.; Lee, Y.J.; Lee, J.-H.; de Oliveira, F.M.S.; Kim, H.Y.; et al. Early increment of soluble triggering receptor expressed on myeloid cells 2 in plasma might be a predictor of poor outcome after ischemic stroke. J. Clin. Neurosci. 2020, 73, 215–218. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, Y.; Lu, Y.; Fang, C.; Zhang, Q.; Zhang, J.; Ju, Z.; Zhang, Y.; Xu, T.; Zhong, C. The association between plasma soluble triggering receptor expressed on myeloid cells 2 and cognitive impairment after acute ischemic stroke. J. Affect. Disord. 2022, 299, 287–293. [Google Scholar] [CrossRef]
- Ohrfelt, A.; Axelsson, M.; Malmestrom, C.; Novakova, L.; Heslegrave, A.; Blennow, K.; Lycke, J.; Zetterberg, H. Soluble TREM-2 in cerebrospinal fluid from patients with multiple sclerosis treated with natalizumab or mitoxantrone. Mult. Scler. 2016, 22, 1587–1595. [Google Scholar] [CrossRef]
- Ioannides, Z.A.; Csurhes, P.A.; Swayne, A.; Foubert, P.; Aftab, B.T.; Pender, M.P. Correlations between macrophage/microglial activation marker sTREM-2 and measures of T-cell activation, neuroaxonal damage and disease severity in multiple sclerosis. Mult. Scler. J. Exp. Transl. Clin. 2021, 7, 20552173211019772. [Google Scholar] [CrossRef]
- Dong, M.H.; Zhou, L.Q.; Tang, Y.; Chen, M.; Xiao, J.; Shang, K.; Deng, G.; Qin, C.; Tian, D.S. CSF sTREM2 in neurological diseases: A two-sample Mendelian randomization study. J. Neuroinflamm. 2022, 19, 79. [Google Scholar] [CrossRef]
- Feng, Y.-S.; Tan, Z.-X.; Wu, L.-Y.; Dong, F.; Zhang, F. The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101192. [Google Scholar] [CrossRef]
- Gao, L.; Dong, Q.; Song, Z.; Shen, F.; Shi, J.; Li, Y. NLRP3 inflammasome: A promising target in ischemic stroke. Inflamm. Res. 2017, 66, 17–24. [Google Scholar] [CrossRef]
- Zhang, D.; Yan, H.; Hu, Y.; Zhuang, Z.; Yu, Z.; Hang, C. Increased Expression of NLRP3 Inflammasome in Wall of Ruptured and Unruptured Human Cerebral Aneurysms: Preliminary Results. J. Stroke Cerebrovasc. Dis. 2015, 24, 972–979. [Google Scholar] [CrossRef]
- Malhotra, S.; Costa, C.; Eixarch, H.; Keller, C.W.; Amman, L.; Martinez-Banaclocha, H.; Midaglia, L.; Sarro, E.; Machin-Diaz, I.; Villar, L.M.; et al. NLRP3 inflammasome as prognostic factor and therapeutic target in primary progressive multiple sclerosis patients. Brain 2020, 143, 1414–1430. [Google Scholar] [CrossRef]
- Wallisch, J.S.; Simon, D.W.; Bayır, H.; Bell, M.J.; Kochanek, P.M.; Clark, R.S.B. Cerebrospinal Fluid NLRP3 is Increased After Severe Traumatic Brain Injury in Infants and Children. Neurocritical Care 2017, 27, 44–50. [Google Scholar] [CrossRef]
- Song, J.; Choi, S.-M.; Kim, B.C. Adiponectin Regulates the Polarization and Function of Microglia via PPAR-γ Signaling Under Amyloid β Toxicity. Front. Cell. Neurosci. 2017, 11, 64. [Google Scholar] [CrossRef] [Green Version]
- Waragai, M.; Adame, A.; Trinh, I.; Sekiyama, K.; Takamatsu, Y.; Une, K.; Masliah, E.; Hashimoto, M. Possible Involvement of Adiponectin, the Anti-Diabetes Molecule, in the Pathogenesis of Alzheimer’s Disease. J. Alzheimer Dis. 2016, 52, 1453–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.-J.; Yang, S.-B.; Lv, Q.-W.; Zhang, G.-H.; Zhou, J.; Guo, M.; Huang, H.-B.; Li, Z.; Yang, C.-S. High plasma adiponectin levels in patients with severe traumatic brain injury. Clin. Chim. Acta 2014, 427, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Signoriello, E.; Lus, G.; Polito, R.; Casertano, S.; Scudiero, O.; Coletta, M.; Monaco, M.L.; Rossi, F.; Nigro, E.; Daniele, A. Adiponectin profile at baseline is correlated to progression and severity of multiple sclerosis. Eur. J. Neurol. 2019, 26, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Ghaith, H.S.; Nawar, A.A.; Gabra, M.D.; Abdelrahman, M.E.; Nafady, M.H.; Bahbah, E.I.; Ebada, M.A.; Ashraf, G.M.; Negida, A.; Barreto, G.E. A Literature Review of Traumatic Brain Injury Biomarkers. Mol. Neurobiol. 2022, 59, 4141–4158. [Google Scholar] [CrossRef] [PubMed]
- Festoff, B.W.; Sajja, R.K.; van Dreden, P.; Cucullo, L. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J. Neuroinflamm. 2016, 13, 194. [Google Scholar] [CrossRef]
- Jeong, J.-h.; Lee, D.H.; Song, J. HMGB1 signaling pathway in diabetes-related dementia: Blood-brain barrier breakdown, brain insulin resistance, and Aβ accumulation. Biomed. Pharmacother. 2022, 150, 112933. [Google Scholar] [CrossRef]
- Le, K.; Mo, S.; Lu, X.; Ali, A.I.; Yu, D.; Guo, Y. Association of circulating blood HMGB1 levels with ischemic stroke: A systematic review and meta-analysis. Neurol. Res. 2018, 40, 907–916. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, Y.; Zeng, D.; Zhou, W.; Hong, X. Prognostic value of plasma HMGB1 in ischemic stroke patients with cerebral ischemia-reperfusion injury after intravenous thrombolysis. J. Stroke Cerebrovasc. Dis. 2020, 29, 105055. [Google Scholar] [CrossRef]
- Wang, K.-Y.; Yu, G.-F.; Zhang, Z.-Y.; Huang, Q.; Dong, X.-Q. Plasma high-mobility group box 1 levels and prediction of outcome in patients with traumatic brain injury. Clin. Chim. Acta 2012, 413, 1737–1741. [Google Scholar] [CrossRef]
- Bucova, M.; Majernikova, B.; Durmanova, V.; Cudrakova, D.; Gmitterova, K.; Lisa, I.; Klimova, E.; Kluckova, K.; Buc, M. HMGB1 as a potential new marker of disease activity in patients with multiple sclerosis. Neurol. Sci. 2020, 41, 599–604. [Google Scholar] [CrossRef]
- Burguillos, M.A.; Svensson, M.; Schulte, T.; Boza-Serrano, A.; Garcia-Quintanilla, A.; Kavanagh, E.; Santiago, M.; Viceconte, N.; Oliva-Martin, M.J.; Osman, A.M.; et al. Microglia-Secreted Galectin-3 Acts as a Toll-like Receptor 4 Ligand and Contributes to Microglial Activation. Cell Rep. 2015, 10, 1626–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frunza, O.; Frangogiannis, N.G. Inflammatory Biomarkers in Post-infarction Heart Failure and Cardiac Remodeling, In Inflammation in Heart Failure; Elsevier: Amsterdam, The Netherlands, 2015; pp. 105–116. [Google Scholar]
- Zhuang, J.-J.; Zhou, L.; Zheng, Y.-H.; Ding, Y.-S. The serum galectin-3 levels are associated with the severity and prognosis of ischemic stroke. Aging 2021, 13, 7454–7464. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, H.; Shimizu, F.; Kitagawa, T.; Yamanaka, N.; Akada, J.; Kuramitsu, Y.; Sano, Y.; Takeshita, Y.; Maeda, T.; Abe, M.; et al. Identification of galectin-3 as a possible antibody target for secondary progressive multiple sclerosis. Mult. Scler. 2017, 23, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, S.; Lin, F.; Chu, W.; Yue, S. Elevated Galectin-3 Levels in the Serum of Patients With Alzheimer’s Disease. Am. J. Alzheimer Dis. Other Dement. 2015, 30, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Yazar, T.; Olgun Yazar, H.; Cihan, M. Evaluation of serum galectin-3 levels at Alzheimer patients by stages: A preliminary report. Acta Neurol. Belg. 2021, 121, 949–954. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, K.; Ma, Y.; Li, S.; Xu, Y. Serum Galectin-3 as a Potential Predictive Biomarker Is Associated with Poststroke Cognitive Impairment. Oxidative Med. Cell. Longev. 2021, 2021, 5827812. [Google Scholar] [CrossRef]
- Shen, Y.-F.; Yu, W.-H.; Dong, X.-Q.; Du, Q.; Yang, D.-B.; Wu, G.-Q.; Zhang, Z.-Y.; Wang, H.; Jiang, L. The change of plasma galectin-3 concentrations after traumatic brain injury. Clin. Chim. Acta 2016, 456, 75–80. [Google Scholar] [CrossRef]
- Grosse, G.M.; ATryc, B.; Dirks, M.; Schuppner, R.; Pflugrad, H.; Lichtinghagen, R.; Weissenborn, K.; Worthmann, H. The temporal dynamics of plasma fractalkine levels in ischemic stroke: Association with clinical severity and outcome. J. Neuroinflamm. 2014, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Menzel, L.; Kleber, L.; Friedrich, C.; Hummel, R.; Dangel, L.; Winter, J.; Schmitz, K.; Tegeder, I.; Schäfer, M.K.E. Progranulin protects against exaggerated axonal injury and astrogliosis following traumatic brain injury: Progranulin in Traumatic Brain Injury. Glia 2017, 65, 278–292. [Google Scholar] [CrossRef]
- Petkau, T.L.; Kosior, N.; de Asis, K.; Connolly, C.; Leavitt, B.R. Selective depletion of microglial progranulin in mice is not sufficient to cause neuronal ceroid lipofuscinosis or neuroinflammation. J. Neuroinflamm. 2017, 14, 225. [Google Scholar] [CrossRef]
- Townley, R.A.; Boeve, B.F.; Benarroch, E.E. Progranulin: Functions and neurologic correlations. Neurology 2018, 90, 118–125. [Google Scholar] [CrossRef]
- Piscopo, P.; Rivabene, R.; Galimberti, D.; Crestini, A.; Talarico, G.; Vanacore, N.; Scarpini, E.; Bruno, G.; Confaloni, A. Gender Effects on Plasma PGRN Levels in Patients with Alzheimer’s Disease: A Preliminary Study. J. Alzheimer Dis. 2013, 35, 313–318. [Google Scholar] [CrossRef]
- Xie, S.; Lu, L.; Liu, L.; Bi, G.; Zheng, L. Progranulin and short-term outcome in patients with acute ischaemic stroke. Eur. J. Neurol. 2016, 23, 648–655. [Google Scholar] [CrossRef]
- Pawlitzki, M.; Sweeney-Reed, C.M.; Bittner, D.; Lux, A.; Vielhaber, S.; Schreiber, S.; Paul, F.; Neumann, J. CSF-Progranulin and Neurofilament Light Chain Levels in Patients With Radiologically Isolated Syndrome-Sign of Inflammation. Front. Neurol. 2018, 9, 1075. [Google Scholar] [CrossRef] [Green Version]
- Winston, C.N.; Sarsoza, F.; Spencer, B.; Rissman, R.A. Characterizing blood-based, microglial derived exosomes (MDEs) as biomarkers for Alzheimer’s disease. Alzheimer Dement. 2021, 17 (Suppl. S5), e055371. [Google Scholar] [CrossRef]
- Shaffer, J.L.; Petrella, J.R.; Sheldon, F.C.; Choudhury, K.R.; Calhoun, V.D.; Coleman, R.E.; Doraiswamy, P.M. Alzheimer’s Disease Neuroimaging Initiative. Predicting Cognitive Decline in Subjects at Risk for Alzheimer Disease by Using Combined Cerebrospinal Fluid, MR Imaging, and PET Biomarkers. Radiology 2013, 266, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, B.; Wang, Y.; Maimaitili, A.; Qin, H.; Dangmurenjiafu, G.; Wang, S. The association between serum adiponectin and 3-month outcome after ischemic stroke. Cardiovasc. Diabetol. 2019, 18, 105. [Google Scholar] [CrossRef] [Green Version]
- Dyhrfort, P.; Shen, Q.; Clausen, F.; Thulin, M.; Enblad, P.; Kamali-Moghaddam, M.; Lewén, A.; Hillered, L. Monitoring of Protein Biomarkers of Inflammation in Human Traumatic Brain Injury Using Microdialysis and Proximity Extension Assay Technology in Neurointensive Care. J. Neurotrauma 2019, 36, 2872–2885. [Google Scholar] [CrossRef] [Green Version]
- Stojković, L.; Stanković, A.; Životić, I.; Dinčić, E.; Alavantić, D.; Živković, M. Gene expression of chemokines CX3CL1 and CXCL16 and their receptors, CX3CR1 and CXCR6, in peripheral blood mononuclear cells of patients with relapsing-remitting multiple sclerosis—A pilot study. Vojnosanit. Pregl. 2020, 77, 967–973. [Google Scholar] [CrossRef] [Green Version]
- Olczak, M.; Poniatowski, Ł.; Siwińska, A.; Kwiatkowska, M.; Chutorański, D.; Wierzba-Bobrowicz, T. Elevated serum and urine levels of progranulin (PGRN) as a predictor of microglia activation in the early phase of traumatic brain injury: A further link with the development of neurodegenerative diseases. Folia Neuropathol. 2021, 59, 81–90. [Google Scholar] [CrossRef] [PubMed]


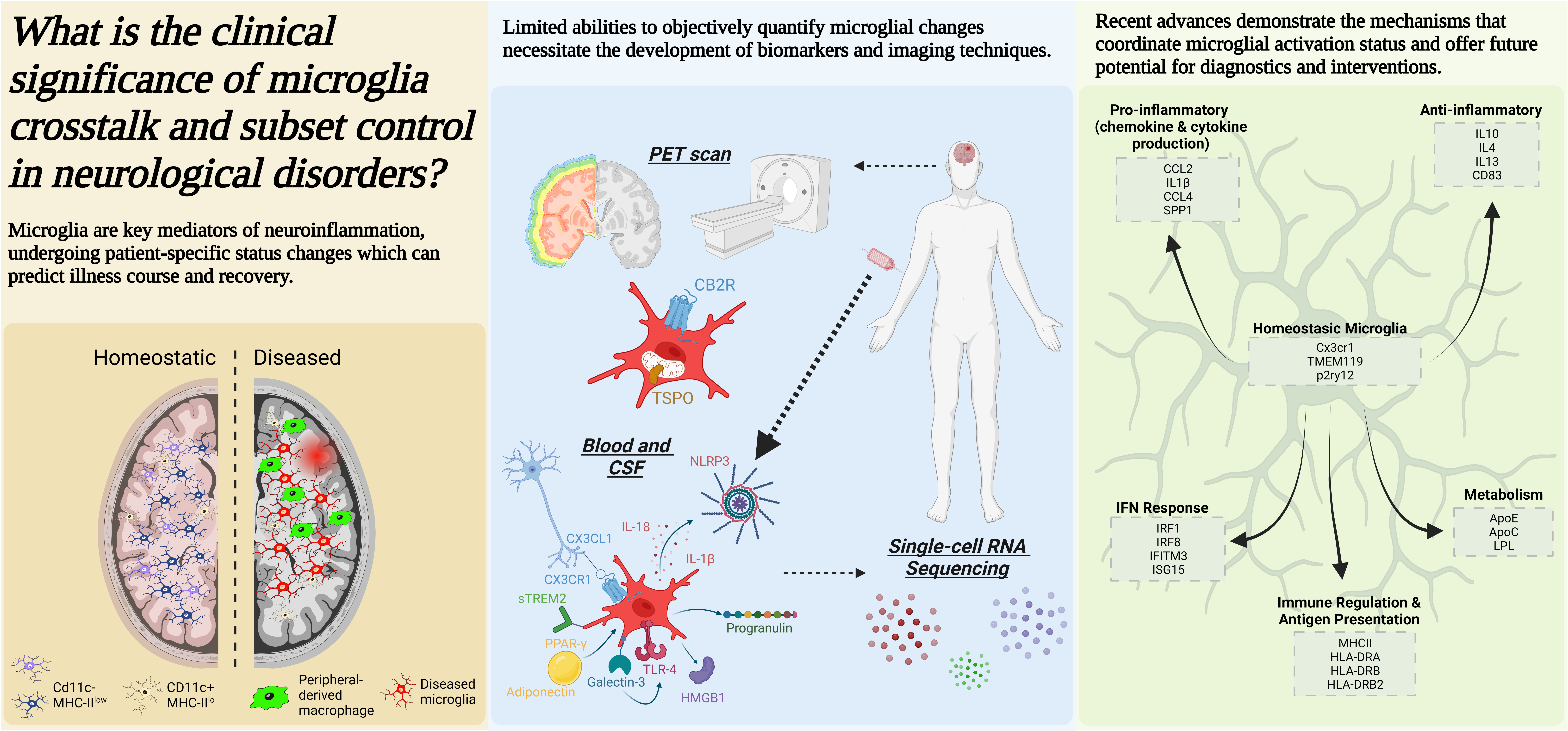
| Study | Human Tissue | Techniques | Subsets/Clusters | Enriched Genes | Function |
|---|---|---|---|---|---|
| [35] | Healthy tissue Temporal lobe (Grey & white matter) | scRNAseq Time-of-flight mass cytometry Cross-data comparison | 9 clusters (C1–C9) | ||
| C1 | CCL2, IL1B | Chemokine and cytokine inflammatory genes | |||
| C2 (WM) | MHC-II, HLA-DRA, CD74 IFI44L | Chemokine and cytokine inflammatory genes | |||
| C3 (GM) | CX3CR1, TMEM119 | Homeostasis c | |||
| C5 (WM) | MHC-II,CCL2 IL1B | Homeostasis | |||
| C8 (GM) | CCL2, IL1B | Chemokine and cytokine inflammatory genes | |||
| C6–C7 (WM) | MHC-II, SPP1, APOE (>50 y) and LPL | Integrin-receptor-binding protein and metabolism genes | |||
| [89] | Healthy tissue MS tissue | scRNA -seq (Cel-Seq2 protocol) | 4 clusters (C1–C4) | ||
| C1–C2 | CST3, P2RY13 | Microglia activation and homeostasis | |||
| C4 | CCL4, CCL2, EGR2, EGR3 | Cytokine inflammatory genes and zinc finger transcription factors | |||
| 7 clusters (C2–C8) | |||||
| C2 | APOE, MAFB, CTSD, APOC1, GPNMB, ANXA2, LGALS1 | Microglia activation | |||
| C3 | APOE, MAFB, CD74, HLA-DRA, HLA-DRB1 HLA-DPB1 | Microglia Activation & Immunoregulation | |||
| C4 | CCL2, CCL4, EGR4 | Cytokines and zinc finger TF | |||
| C5–C7 | TMEM119, P2RY12 | Homeostasis | |||
| C8 | APOE, MAFB, SPP1, PADI2, LPL | Microglial Activation & Demyelination | |||
| [90] | TLE, MCI, AD | scRNA-seq | 9 clusters (C1–C9) | ||
| C4 (MS, AD) | IRF1, IRF8, IFITM3, ISG15+ | IFN-response | |||
| C5 (TLE) | CREB, ATF, IL10, IL4, IL13, CD83 | TF & anti-inflammatory | |||
| C6 (TLE) | IL10, IL4, IL13, CD83+ | Anti-inflammatory | |||
| C7 (TLE, MCI; reduced in AD) | APOE, TREM2, CD74hi | Microglia activation and Antigen presentation | |||
| C9 | E2F1, CBFB, NRF1 | Cell cycle |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mills, J.; Ladner, L.; Soliman, E.; Leonard, J.; Morton, P.D.; Theus, M.H. Cross-Talk and Subset Control of Microglia and Associated Myeloid Cells in Neurological Disorders. Cells 2022, 11, 3364. https://doi.org/10.3390/cells11213364
Mills J, Ladner L, Soliman E, Leonard J, Morton PD, Theus MH. Cross-Talk and Subset Control of Microglia and Associated Myeloid Cells in Neurological Disorders. Cells. 2022; 11(21):3364. https://doi.org/10.3390/cells11213364
Chicago/Turabian StyleMills, Jatia, Liliana Ladner, Eman Soliman, John Leonard, Paul D. Morton, and Michelle H. Theus. 2022. "Cross-Talk and Subset Control of Microglia and Associated Myeloid Cells in Neurological Disorders" Cells 11, no. 21: 3364. https://doi.org/10.3390/cells11213364