
Identifying Candidate Circulating RNA Markers for Coronary Artery Disease by Deep RNA-Sequencing in Human Plasma
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Coronary Heart Disease Cohort Study (CDCS)
2.2. Canterbury Healthy Volunteers Cohort
2.3. Plasma Collection
2.4. Sample Selection
2.5. Extraction of Circulating Cell-Free RNA from Plasma
2.6. Plasma RNA Sequencing
2.7. Bioinformatics Pipeline
3. Results
3.1. Sequencing Quality Control and Patient Characteristics
3.2. Annotated mRNA and lncRNAs Associated with CAD and Progression to HF
3.3. Putative Novel lncRNAs Associated with CAD and Progression to HF
3.4. circRNAs Associated with CAD and Progression to HF
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malakar, A.K.; Choudhury, D.; Halder, B.; Paul, P.; Uddin, A.; Chakraborty, S. A review on coronary artery disease, its risk factors, and therapeutics. J. Cell. Physiol. 2019, 234, 16812–16823. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.L.; Maisel, A.S.; Anand, I.; Bozkurt, B.; de Boer, R.A.; Felker, G.M.; Fonarow, G.C.; Greenberg, B.; Januzzi, J.L., Jr.; Kiernan, M.S.; et al. Role of Biomarkers for the Prevention, Assessment, and Management of Heart Failure: A Scientific Statement from the American Heart Association. Circulation 2017, 135, e1054–e1091. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.P.; Thiele, H.; Barbato, E.; Barthelemy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart. J. 2021, 42, 1289–1367. [Google Scholar] [CrossRef] [PubMed]
- Danesh, J.; Wheeler, J.G.; Hirschfield, G.M.; Eda, S.; Eiriksdottir, G.; Rumley, A.; Lowe, G.D.; Pepys, M.B.; Gudnason, V. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. New. Engl. J. Med. 2004, 350, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Pos, O.; Biro, O.; Szemes, T.; Nagy, B. Circulating cell-free nucleic acids: Characteristics and applications. Eur. J. Hum. Genet. 2018, 26, 937–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Danielson, K.M.; Rubio, R.; Abderazzaq, F.; Das, S.; Wang, Y.E. High Throughput Sequencing of Extracellular RNA from Human Plasma. PLoS ONE 2017, 12, e0164644. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Yao, J.; Wu, D.C.; Nottingham, R.M.; Mohr, S.; Hunicke-Smith, S.; Lambowitz, A.M. High-throughput sequencing of human plasma RNA by using thermostable group II intron reverse transcriptases. RNA 2016, 22, 111–128. [Google Scholar] [CrossRef] [Green Version]
- Savelyeva, A.V.; Kuligina, E.V.; Bariakin, D.N.; Kozlov, V.V.; Ryabchikova, E.I.; Richter, V.A.; Semenov, D.V. Variety of RNAs in Peripheral Blood Cells, Plasma, and Plasma Fractions. Biomed. Res. Int. 2017, 2017, 7404912. [Google Scholar] [CrossRef] [Green Version]
- Cardona-Monzonis, A.; Garcia-Gimenez, J.L.; Mena-Molla, S.; Pareja-Galeano, H.; de la Guia-Galipienso, F.; Lippi, G.; Pallardo, F.V.; Sanchis-Gomar, F. Non-coding RNAs and Coronary Artery Disease. Adv. Exp. Med. Biol. 2020, 1229, 273–285. [Google Scholar] [CrossRef]
- Correia, C.C.M.; Rodrigues, L.F.; de Avila Pelozin, B.R.; Oliveira, E.M.; Fernandes, T. Long Non-Coding RNAs in Cardiovascular Diseases: Potential Function as Biomarkers and Therapeutic Targets of Exercise Training. Noncoding RNA 2021, 7, 65. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Gholipour, M.; Taheri, M. The Emerging Role of Long Non-coding RNAs and Circular RNAs in Coronary Artery Disease. Front. Cardiovasc. Med. 2021, 8, 632393. [Google Scholar] [CrossRef] [PubMed]
- Viereck, J.; Thum, T. Circulating Noncoding RNAs as Biomarkers of Cardiovascular Disease and Injury. Circ. Res. 2017, 120, 381–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, Z.; Pearson, J.; Schmeier, S.; Cameron, V.; Pilbrow, A. Insights into circular RNAs: Their biogenesis, detection, and emerging role in cardiovascular disease. RNA Biol. 2021, 18, 2055–2072. [Google Scholar] [CrossRef] [PubMed]
- Zareba, L.; Fitas, A.; Wolska, M.; Junger, E.; Eyileten, C.; Wicik, Z.; De Rosa, S.; Siller-Matula, J.M.; Postula, M. MicroRNAs and Long Noncoding RNAs in Coronary Artery Disease: New and Potential Therapeutic Targets. Cardiol. Clin. 2020, 38, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Everaert, C.; Helsmoortel, H.; Decock, A.; Hulstaert, E.; Van Paemel, R.; Verniers, K.; Nuytens, J.; Anckaert, J.; Nijs, N.; Tulkens, J.; et al. Performance assessment of total RNA sequencing of human biofluids and extracellular vesicles. Sci. Rep. 2019, 9, 17574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvanin, A.; Dostert, G.; Ayadi, L.; Marchand, V.; Velot, E.; Motorin, Y. Diversity and heterogeneity of extracellular RNA in human plasma. Biochimie 2019, 164, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Xiong, G.; Jiang, X.J.; Song, T. The overexpression of lncRNA H19 as a diagnostic marker for coronary artery disease. Rev. Assoc. Med. Bras. 2019, 65, 110–117. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, W.; Long, Q.Q.; Zhang, J.; Li, Y.F.; Liu, D.C.; Yan, J.J.; Yang, Z.J.; Wang, L.S. Increased plasma levels of lncRNA H19 and LIPCAR are associated with increased risk of coronary artery disease in a Chinese population. Sci. Rep. 2017, 7, 7491. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Cai, Y.; Wu, G.; Chen, X.; Liu, Y.; Wang, X.; Yu, J.; Li, C.; Chen, X.; Jose, P.A.; et al. Plasma long non-coding RNA, CoroMarker, a novel biomarker for diagnosis of coronary artery disease. Clin. Sci. 2015, 129, 675–685. [Google Scholar] [CrossRef]
- Ballantyne, M.D.; Pinel, K.; Dakin, R.; Vesey, A.T.; Diver, L.; Mackenzie, R.; Garcia, R.; Welsh, P.; Sattar, N.; Hamilton, G.; et al. Smooth Muscle Enriched Long Noncoding RNA (SMILR) Regulates Cell Proliferation. Circulation 2016, 133, 2050–2065. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, Y.; Guo, S.; Yao, R.; Wu, L.; Xiao, L.; Wang, Z.; Liu, Y.; Zhang, Y. Circulating Long Noncoding RNA HOTAIR is an Essential Mediator of Acute Myocardial Infarction. Cell. Physiol. Biochem. 2017, 44, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamy, R.; Bauters, C.; Volkmann, I.; Maury, F.; Fetisch, J.; Holzmann, A.; Lemesle, G.; de Groote, P.; Pinet, F.; Thum, T. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ. Res. 2014, 114, 1569–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prickett, T.C.; Doughty, R.N.; Troughton, R.W.; Frampton, C.M.; Whalley, G.A.; Ellis, C.J.; Espiner, E.A.; Richards, A.M. C-Type Natriuretic Peptides in Coronary Disease. Clin. Chem. 2017, 63, 316–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, K.L.; Frampton, C.M.; Pilbrow, A.P.; Troughton, R.W.; Doughty, R.N.; Whalley, G.A.; Ellis, C.J.; Skelton, L.; Thomson, J.; Yandle, T.G.; et al. Genomic risk variants at 1p13.3, 1q41, and 3q22.3 are associated with subsequent cardiovascular outcomes in healthy controls and in established coronary artery disease. Circ. Cardiovasc. Genet. 2011, 4, 636–646. [Google Scholar] [CrossRef]
- Ho, D.E.; Imai, K.; King, G.; Stuart, E.A. Matching as nonparametric preprocessing for reducing model dependence in parametric causal inference. Political Anal. 2007, 15, 199–236. [Google Scholar] [CrossRef] [Green Version]
- R Core Team R. A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. Software. 2020. Available online: https://www.R-project.org/ (accessed on 1 August 2017).
- Li, X.; Ben-Dov, I.Z.; Mauro, M.; Williams, Z. Lowering the quantification limit of the QubitTM RNA HS assay using RNA spike-in. BMC Mol. Biol. 2015, 16, 9. [Google Scholar] [CrossRef] [Green Version]
- Ward, Z.; Schmeier, S.; Saddic, L.; Sigurdsson, M.I.; Cameron, V.A.; Pearson, J.; Miller, A.; Morley-Bunker, A.; Gorham, J.; Seidman, J.G.; et al. Novel and Annotated Long Noncoding RNAs Associated with Ischemia in the Human Heart. Int. J. Mol. Sci. 2021, 22, 11324. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Fastqc Tools. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 December 2017).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rademaker, M.T.; Pilbrow, A.P.; Ellmers, L.J.; Palmer, S.C.; Davidson, T.; Mbikou, P.; Scott, N.J.A.; Permina, E.; Charles, C.J.; Endre, Z.H.; et al. Acute Decompensated Heart Failure and the Kidney: Physiological, Histological and Transcriptomic Responses to Development and Recovery. J. Am. Heart Assoc. 2021, 10, e021312. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Simko, V. R Package “Corrplot”: Visualization of a Correlation Matrix (Version 0.84). 2017. Available online: https://github.com/taiyun/corrplot (accessed on 22 November 2018).
- Zhang, X.O.; Dong, R.; Zhang, Y.; Zhang, J.L.; Luo, Z.; Zhang, J.; Chen, L.L.; Yang, L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef] [Green Version]
- Rodosthenous, R.S.; Hutchins, E.; Reiman, R.; Yeri, A.S.; Srinivasan, S.; Whitsett, T.G.; Ghiran, I.; Silverman, M.G.; Laurent, L.C.; Van Keuren-Jensen, K.; et al. Profiling Extracellular Long RNA Transcriptome in Human Plasma and Extracellular Vesicles for Biomarker Discovery. iScience 2020, 23, 101182. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, R.; Minich, J.J.; Marotz, C.; Cooper, A.; Knight, R.; Weyrich, L.S. Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends Microbiol. 2019, 27, 105–117. [Google Scholar] [CrossRef]
- Glassing, A.; Dowd, S.E.; Galandiuk, S.; Davis, B.; Chiodini, R.J. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog 2016, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Weyrich, L.S.; Farrer, A.G.; Eisenhofer, R.; Arriola, L.A.; Young, J.; Selway, C.A.; Handsley-Davis, M.; Adler, C.J.; Breen, J.; Cooper, A. Laboratory contamination over time during low-biomass sample analysis. Mol. Ecol. Resour. 2019, 19, 982–996. [Google Scholar] [CrossRef]
- Pandey, P.R.; Rout, P.K.; Das, A.; Gorospe, M.; Panda, A.C. RPAD (RNase R treatment, polyadenylation, and poly(A)+ RNA depletion) method to isolate highly pure circular RNA. Methods 2019, 155, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.S.; Wilusz, J.E. An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G-quadruplexes or structured 3′ ends. Nucleic Acids Res. 2019, 47, 8755–8769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef]
- Ilic, Z.; Saxena, A.R.; Periasamy, S.; Crawford, D.R. Control (Native) and oxidized (DeMP) mitochondrial RNA are proinflammatory regulators in human. Free Radic. Biol. Med. 2019, 143, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Pacheu-Grau, D.; Bareth, B.; Dudek, J.; Juris, L.; Vogtle, F.N.; Wissel, M.; Leary, S.C.; Dennerlein, S.; Rehling, P.; Deckers, M. Cooperation between COA6 and SCO2 in COX2 maturation during cytochrome c oxidase assembly links two mitochondrial cardiomyopathies. Cell. Metab. 2015, 21, 823–833. [Google Scholar] [CrossRef] [Green Version]
- Ogilvie, I.; Kennaway, N.G.; Shoubridge, E.A. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J. Clin. Invest. 2005, 115, 2784–2792. [Google Scholar] [CrossRef]
- Isakova, T.; Houston, J.; Santacruz, L.; Schiavenato, E.; Somarriba, G.; Harmon, W.G.; Lipshultz, S.E.; Miller, T.L.; Rusconi, P.G. Associations between fibroblast growth factor 23 and cardiac characteristics in pediatric heart failure. Pediatr. Nephrol. 2013, 28, 2035–2042. [Google Scholar] [CrossRef] [Green Version]
- Leifheit-Nestler, M.; Haffner, D. Paracrine Effects of FGF23 on the Heart. Front. Endocrinol. 2018, 9, 278. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.A.; Larsson, A.; Melhus, H.; Lind, L.; Larsson, T.E. Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis 2009, 207, 546–551. [Google Scholar] [CrossRef]
- Parker, B.D.; Schurgers, L.J.; Brandenburg, V.M.; Christenson, R.H.; Vermeer, C.; Ketteler, M.; Shlipak, M.G.; Whooley, M.A.; Ix, J.H. The associations of fibroblast growth factor 23 and uncarboxylated matrix Gla protein with mortality in coronary artery disease: The Heart and Soul Study. Ann. Intern. Med. 2010, 152, 640–648. [Google Scholar] [CrossRef]
- Shibata, K.; Fujita, S.; Morita, H.; Okamoto, Y.; Sohmiya, K.; Hoshiga, M.; Ishizaka, N. Association between circulating fibroblast growth factor 23, alpha-Klotho, and the left ventricular ejection fraction and left ventricular mass in cardiology inpatients. PLoS ONE 2013, 8, e73184. [Google Scholar] [CrossRef]
- De Koninck, M.; Lapi, E.; Badia-Careaga, C.; Cossio, I.; Gimenez-Llorente, D.; Rodriguez-Corsino, M.; Andrada, E.; Hidalgo, A.; Manzanares, M.; Real, F.X.; et al. Essential Roles of Cohesin STAG2 in Mouse Embryonic Development and Adult Tissue Homeostasis. Cell. Rep. 2020, 32, 108014. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.; Zaccagnini, G.; Perfetti, A.; Fuschi, P.; Valaperta, R.; Voellenkle, C.; Castelvecchio, S.; Gaetano, C.; Finato, N.; Beltrami, A.P.; et al. Long noncoding RNA dysregulation in ischemic heart failure. J. Transl. Med. 2016, 14, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, B.; Li, N.; Xu, X.X.; Li, X.X.; Xu, X.J.; Guo, D.; Zhang, D.; Wu, Z.H.; Zhang, S.Y. Long noncoding RNA FTX regulates cardiomyocyte apoptosis by targeting miR-29b-1-5p and Bcl2l2. Biochem. Biophys. Res. Commun. 2018, 495, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Toraih, E.A.; El-Wazir, A.; Alghamdi, S.A.; Alhazmi, A.S.; El-Wazir, M.; Abdel-Daim, M.M.; Fawzy, M.S. Association of long non-coding RNA MIAT and MALAT1 expression profiles in peripheral blood of coronary artery disease patients with previous cardiac events. Genet. Mol. Biol. 2019, 42, 509–518. [Google Scholar] [CrossRef] [Green Version]
- Jiao, L.; Li, M.; Shao, Y.; Zhang, Y.; Gong, M.; Yang, X.; Wang, Y.; Tan, Z.; Sun, L.; Xuan, L.; et al. lncRNA-ZFAS1 induces mitochondria-mediated apoptosis by causing cytosolic Ca(2+) overload in myocardial infarction mice model. Cell. Death Dis. 2019, 10, 942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sun, L.; Xuan, L.; Pan, Z.; Li, K.; Liu, S.; Huang, Y.; Zhao, X.; Huang, L.; Wang, Z.; et al. Reciprocal Changes of Circulating Long Non-Coding RNAs ZFAS1 and CDR1AS Predict Acute Myocardial Infarction. Sci. Rep. 2016, 6, 22384. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Yang, S.T.; Liu, J.; Zhang, K.X.; Leng, J.Y. Silence of LncRNA GAS5 Protects Cardiomyocytes H9c2 against Hypoxic Injury via Sponging miR-142-5p. Mol. Cells 2019, 42, 397–405. [Google Scholar] [CrossRef]
- Hao, S.; Liu, X.; Sui, X.; Pei, Y.; Liang, Z.; Zhou, N. Long non-coding RNA GAS5 reduces cardiomyocyte apoptosis induced by MI through sema3a. Int. J. Biol. Macromol. 2018, 120, 371–377. [Google Scholar] [CrossRef]
- Zhang, Y.; Hou, Y.M.; Gao, F.; Xiao, J.W.; Li, C.C.; Tang, Y. lncRNA GAS5 regulates myocardial infarction by targeting the miR-525-5p/CALM2 axis. J. Cell. Biochem. 2019, 120, 18678–18688. [Google Scholar] [CrossRef]
- Ghirlando, R.; Felsenfeld, G. CTCF: Making the right connections. Genes Dev. 2016, 30, 881–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, P.P.; Leonardi, T.; Han, N.; Vire, E.; Gascoigne, D.K.; Arias-Carrasco, R.; Buscher, M.; Pandolfini, L.; Zhang, A.; Pluchino, S.; et al. Genomic positional conservation identifies topological anchor point RNAs linked to developmental loci. Genome Biol. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Saldana-Meyer, R.; Gonzalez-Buendia, E.; Guerrero, G.; Narendra, V.; Bonasio, R.; Recillas-Targa, F.; Reinberg, D. CTCF regulates the human p53 gene through direct interaction with its natural antisense transcript, Wrap53. Genes Dev. 2014, 28, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Deng, X.; Ma, W.; Berletch, J.B.; Rabaia, N.; Wei, G.; Moore, J.M.; Filippova, G.N.; Xu, J.; Liu, Y.; et al. The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol. 2015, 16, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.; Brick, K.; Evrard, Y.; Xiao, T.; Camerini-Otero, R.D.; Felsenfeld, G. Mediation of CTCF transcriptional insulation by DEAD-box RNA-binding protein p68 and steroid receptor RNA activator SRA. Genes Dev. 2010, 24, 2543–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, M.; Zhu, Y.; Qi, L.; Hu, F.; Gao, P. Circular RNA profiling in plasma exosomes from patients with gastric cancer. Oncol. Lett. 2020, 20, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, Q.; Bao, C.; Li, S.; Guo, W.; Zhao, J.; Chen, D.; Gu, J.; He, X.; Huang, S. Circular RNA is enriched and stable in exosomes: A promising biomarker for cancer diagnosis. Cell. Res. 2015, 25, 981–984. [Google Scholar] [CrossRef] [Green Version]
- Vausort, M.; Salgado-Somoza, A.; Zhang, L.; Leszek, P.; Scholz, M.; Teren, A.; Burkhardt, R.; Thiery, J.; Wagner, D.R.; Devaux, Y. Myocardial Infarction-Associated Circular RNA Predicting Left Ventricular Dysfunction. J. Am. Coll. Cardiol. 2016, 68, 1247–1248. [Google Scholar] [CrossRef]
- Research Square. Available online: https://www.researchsquare.com/article/rs-33371/v1 (accessed on 12 June 2022).


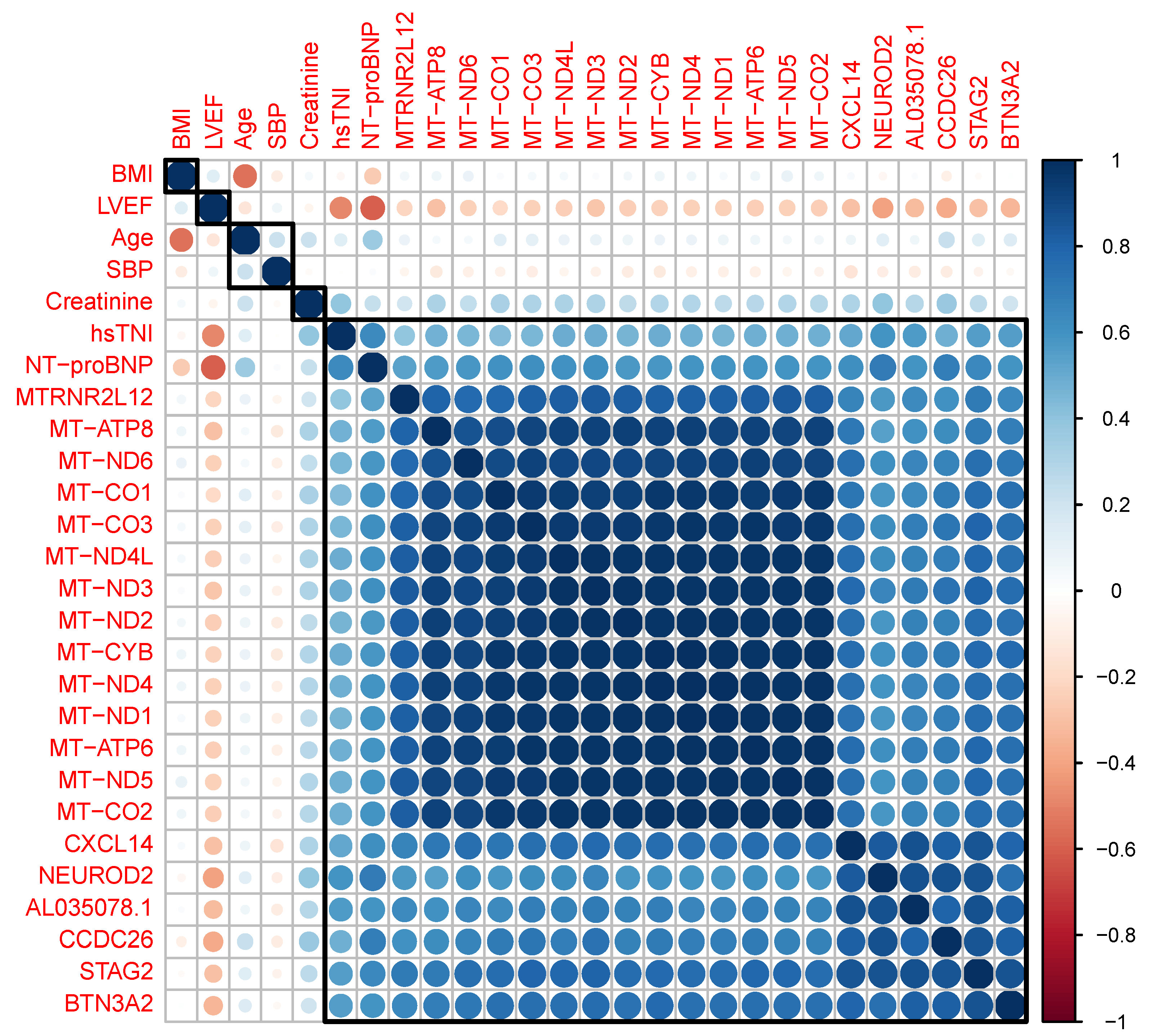{kind=link}
| Controls (n = 30) | HF− (n = 30) | HF+ (n = 29) | |
|---|---|---|---|
| Total reads (M) * | 111.2 (100.1–113.2) | 101.9 (92.3–117.0) | 110.2 (101.4–117.8) |
| Reads uniquely mapped (M) * | 23.9 (9.2–33.1) | 22.4 (7.6–28.7) | 15.0 (9.1–30.3) |
| Reads uniquely mapped (%) * | 18.9 (8.5–29.6) | 19.0 (7.8–27.8) | 13.6 (8.8–29.6) |
| Controls (n = 30) | HF− (n = 30) | HF+ (n = 29) | |
|---|---|---|---|
| Age (years) * | 70 (60–77) | 70 (63–76) | 72 (60–77) |
| Male sex † | 21 (70%) | 22 (73%) | 18 (62%) |
| European ethnicity † | 26 (87%) | 25 (83%) | 21 (72%) |
| Cigarette smoker † | 0 (0%) | 0 (0%) | 1 (3%) |
| SBP (mmHg) * | 140 (130–149) | 135 (121–160) | 130 (111–143) |
| BMI (kg/m2) * | 29 (25–33) | 29 (26–33) | 29 (25–34) |
| Diagnosis † | - | UA: 7 (23%) | UA: 6 (21%) |
| - | NSTEMI: 15 (50%) | NSTEMI: 16 (55%) | |
| - | STEMI: 8 (27%) | STEMI: 7 (24%) | |
| Type 2 Diabetes † | 2 (7%) | 11 (37%) | 11 (38%) |
| Hypertension † | 9 (30%) | 22 (73%) | 22 (76%) |
| Atrial fibrillation † | 0 (0%) | 5 (17%) | 5 (17%) |
| Creatinine (μg/L) * | 92 (78–99) | 97 (85–109) | 102 (99–116) |
| hsTnI (ng/L) * | 2.9 (1.7–5.1) | 8.0 (5.8–14.9) | 11.1 (6.7–24.3) |
| NT-proBNP (pmol/L) * | 8.0 (3.8–25.3) | 92 (47–141) | 146 (67–233) |
| LVEF (%) * | 68 (63–74) | 64 (52–75) | 64 (52–75) |
| Beta blockers † | 3 (10%) | 27 (90%) | 23 (79%) |
| ACE inhibitors or ARBs † | 5 (17%) | 19 (63%) | 18 (62%) |
| Statins † | 8 (27%) | 27 (90%) | 27 (93%) |
| Diuretics † | 3 (10%) | 8 (27%) | 10 (35%) |
| Gene Name | Transcript Type | Log 2 Fold Change (Standard Error) | p-Value * |
|---|---|---|---|
| STAG2 | Protein coding | 1.61 (0.12) | 1.62 × 10−36 |
| NEUROD2 | Protein coding | 1.89 (0.15) | 1.48 × 10−31 |
| MT-ND3 | Protein coding | 2.09 (0.18) | 5.75 × 10−29 |
| MT-ND5 | Protein coding | 2.13 (0.19) | 5.80 × 10−28 |
| MT-CO2 | Protein coding | 2.08 (0.18) | 1.00 × 10−27 |
| MT-ND6 | Protein coding | 2.22 (0.20) | 1.64 × 10−27 |
| MT-CYB | Protein coding | 2.10 (0.19) | 3.29 × 10−27 |
| MT-ND1 | Protein coding | 2.16 (0.19) | 1.65 × 10−26 |
| MT-ATP6 | Protein coding | 2.00 (0.18) | 3.33 × 10−26 |
| MT-ND4L | Protein coding | 1.98 (0.18) | 5.77 × 10−26 |
| AL035078.1 | lncRNA | 1.77 (0.16) | 6.48 × 10−26 |
| CXCL14 | Protein coding | 1.98 (0.18) | 1.40 × 10−25 |
| MT-CO1 | Protein coding | 2.04 (0.19) | 1.84 × 10−25 |
| CCDC26 | lncRNA | 1.72 (0.16) | 4.51 × 10−25 |
| MT-ND2 | Protein coding | 1.98 (0.18) | 6.49 × 10−25 |
| MT-ND4 | Protein coding | 2.01 (0.19) | 9.42 × 10−25 |
| MT-CO3 | Protein coding | 2.01 (0.19) | 1.82 × 10−24 |
| BTN3A2 | Protein coding | 1.39 (0.13) | 6.75 × 10−24 |
| MT-ATP8 | Protein coding | 2.05 (0.20) | 3.25 × 10−22 |
| MTRNR2L12 | Protein coding | 2.13 (0.22) | 8.15 × 10−20 |
| Gene Name | Chromosomal Position (Strand) | Log 2 Fold Change (Standard Error) | p-Value * |
|---|---|---|---|
| MSTRG.752033.2 MSTRG.76602.1 | 7: 113240035−113240441 10: 3408557−3409012 | 1.69 (0.40) 2.44 (0.60) | 1.66 × 10−3 3.36 × 10−3 |
| Gene Name | Chromosomal Position (Strand) | Log 2 Fold Change (Standard Error) | p-Value * |
|---|---|---|---|
| UBCA2 | 13:99238426−99244624 (+) | 0.43 (0.16) | 4.70 × 10−2 |
| CLNS1A | 11:77619605−77625818 (-) | 0.74 (0.28) | 6.50 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ward, Z.; Schmeier, S.; Pearson, J.; Cameron, V.A.; Frampton, C.M.; Troughton, R.W.; Doughty, R.N.; Richards, A.M.; Pilbrow, A.P. Identifying Candidate Circulating RNA Markers for Coronary Artery Disease by Deep RNA-Sequencing in Human Plasma. Cells 2022, 11, 3191. https://doi.org/10.3390/cells11203191
Ward Z, Schmeier S, Pearson J, Cameron VA, Frampton CM, Troughton RW, Doughty RN, Richards AM, Pilbrow AP. Identifying Candidate Circulating RNA Markers for Coronary Artery Disease by Deep RNA-Sequencing in Human Plasma. Cells. 2022; 11(20):3191. https://doi.org/10.3390/cells11203191
Chicago/Turabian StyleWard, Zoe, Sebastian Schmeier, John Pearson, Vicky A Cameron, Chris M Frampton, Richard W Troughton, Rob N Doughty, A. Mark Richards, and Anna P Pilbrow. 2022. "Identifying Candidate Circulating RNA Markers for Coronary Artery Disease by Deep RNA-Sequencing in Human Plasma" Cells 11, no. 20: 3191. https://doi.org/10.3390/cells11203191