
Native and Oxidized Low-Density Lipoproteins Increase the Expression of the LDL Receptor and the LOX-1 Receptor, Respectively, in Arterial Endothelial Cells

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
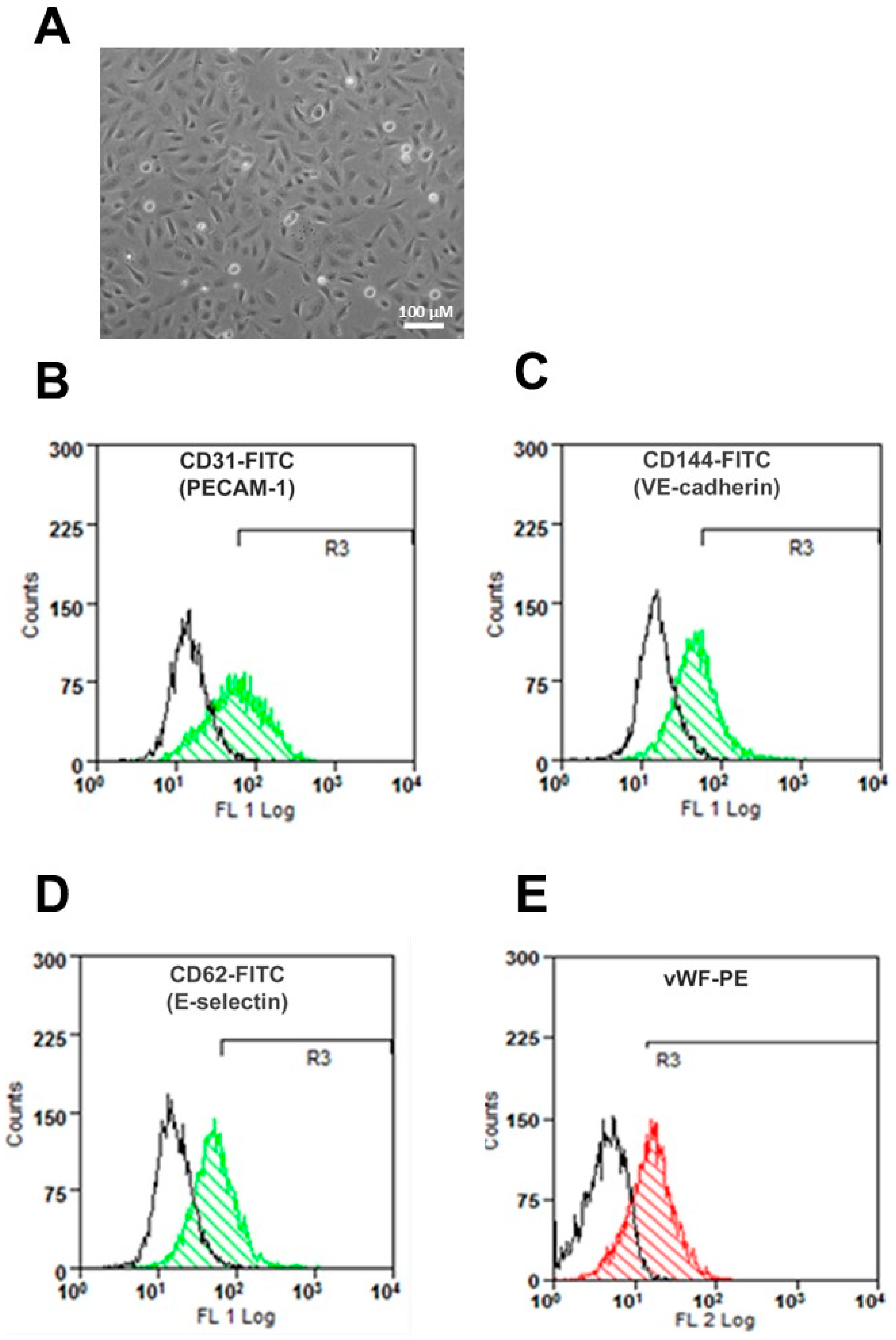
2.1. HUAEC Culture
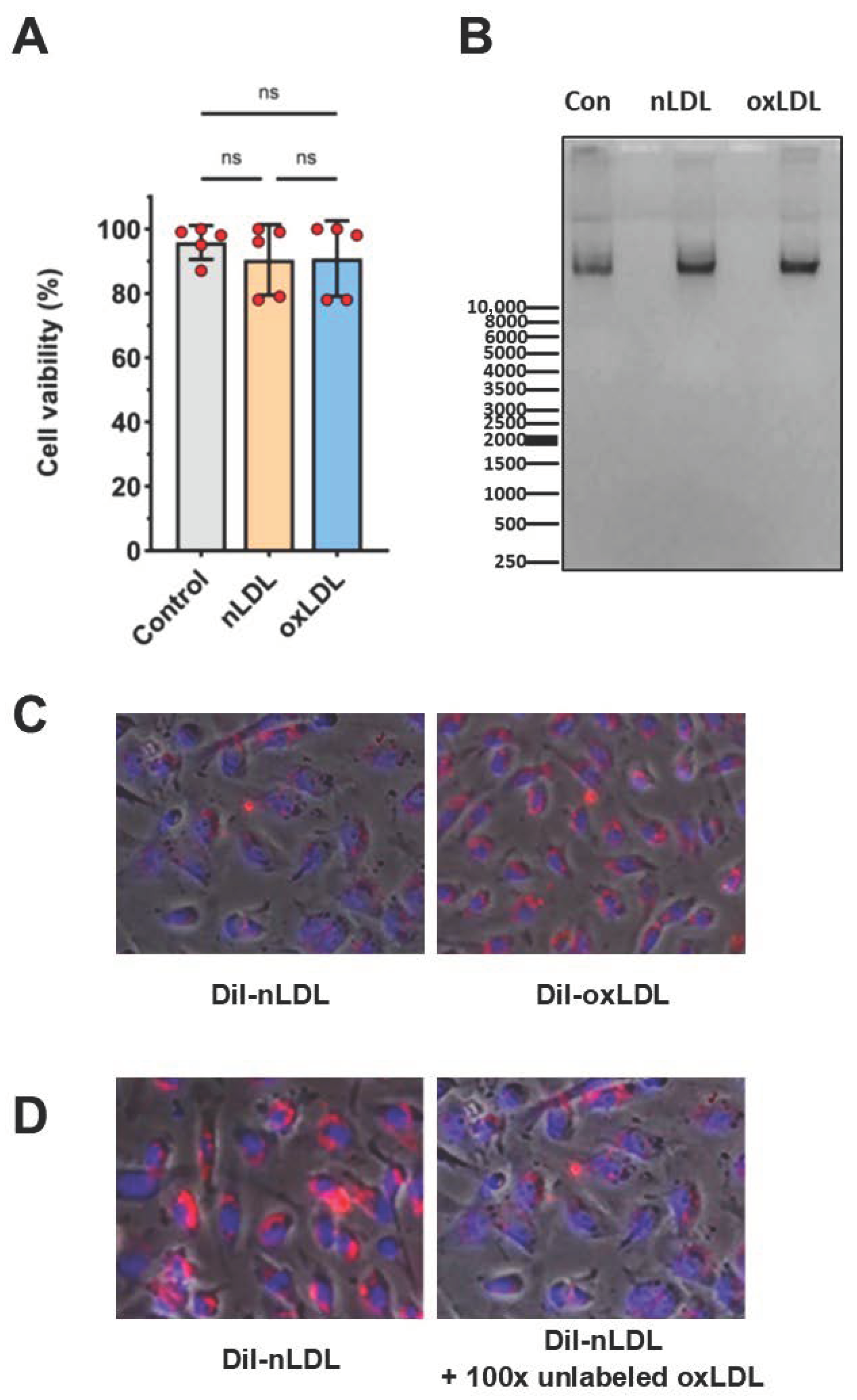
2.2. LDL Oxidation
2.3. Cell Viability and LDL Uptake
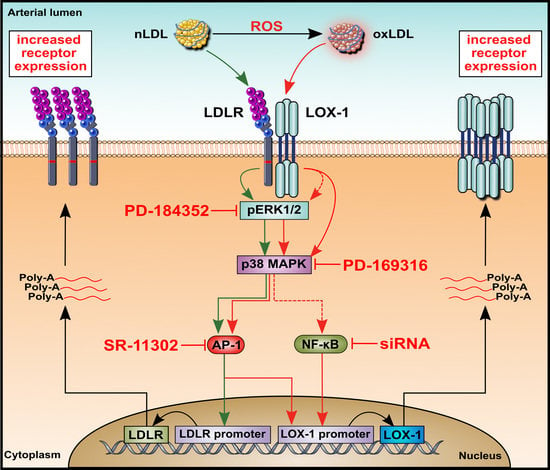
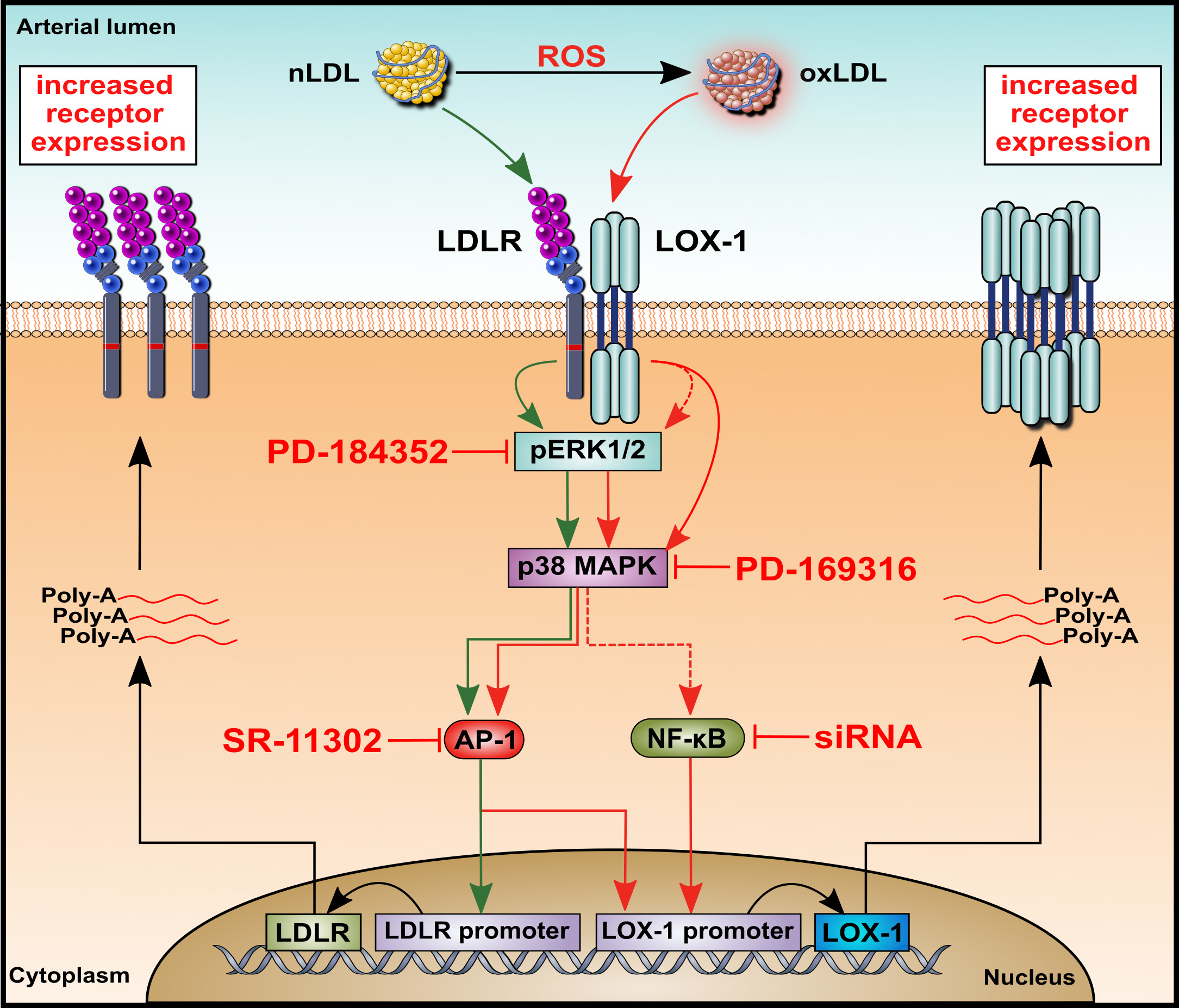
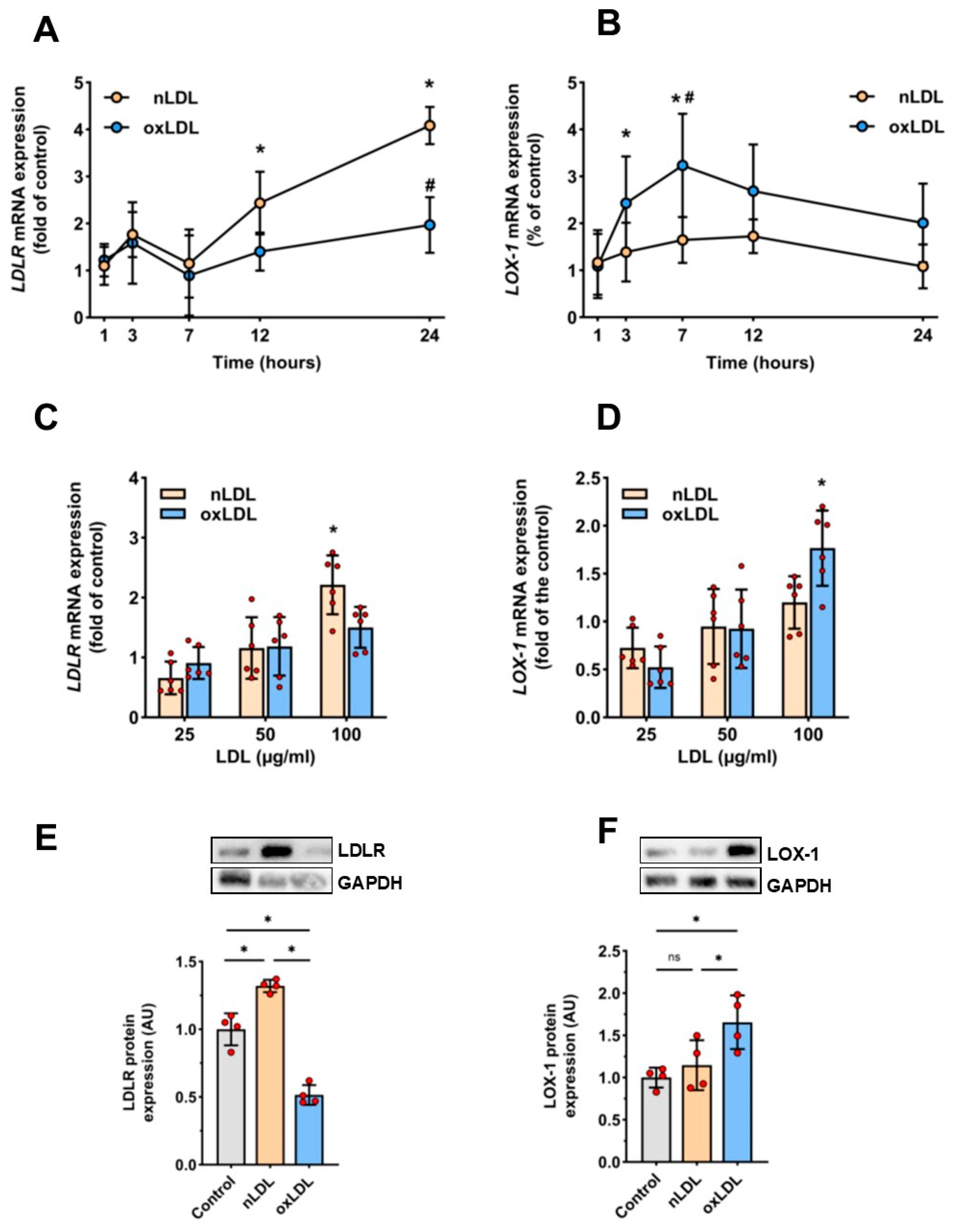
2.4. Effects of nLDL and oxLDL on the Expression of Their Receptors in HUAECs
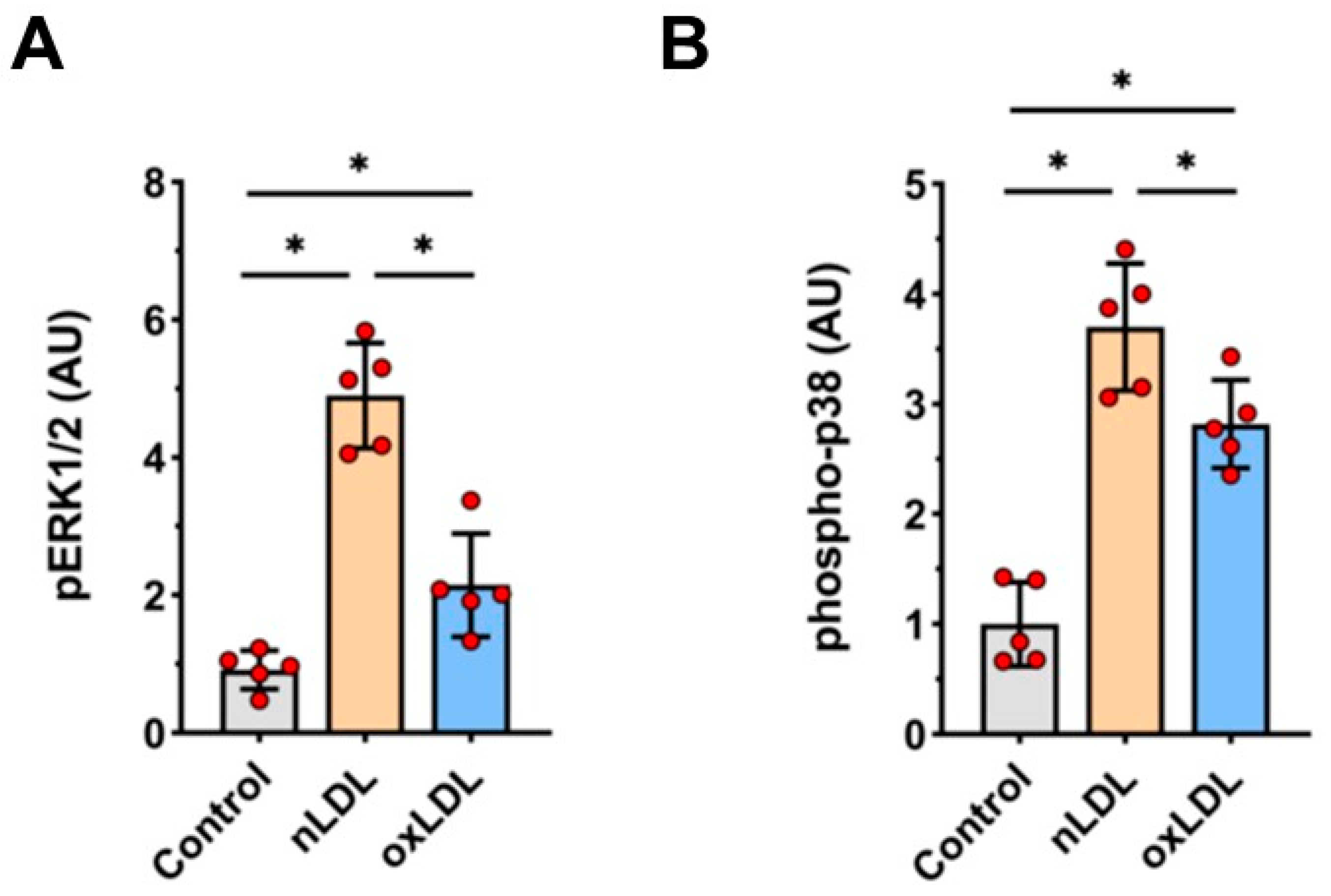
2.5. Effects of nLDL and oxLDL on ERK1/2 and p38 Activation in HUAECs
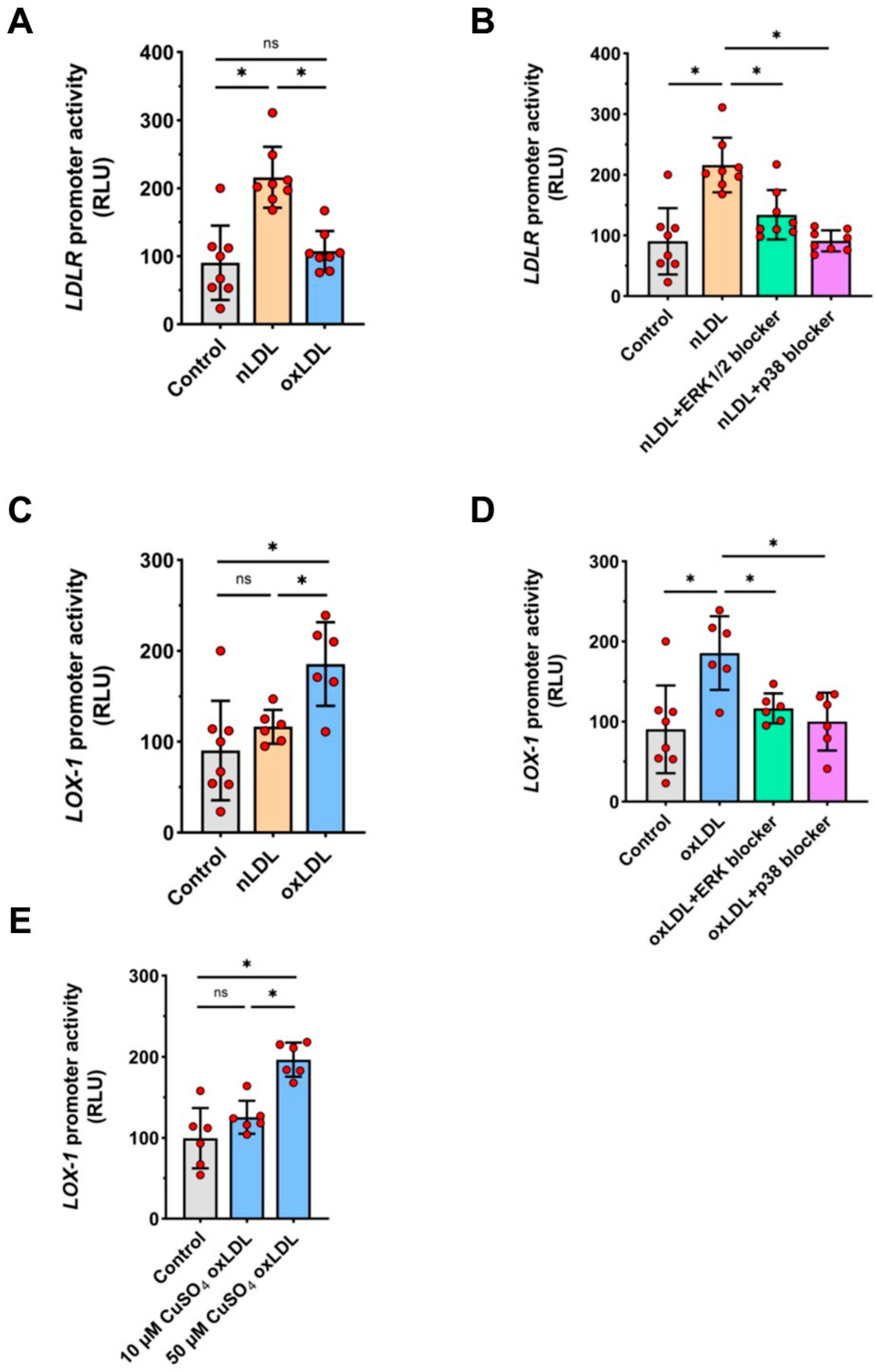
2.6. Effects of nLDL and oxLDL on LDLR and LOX-1 Gene Promoters in HUAECs
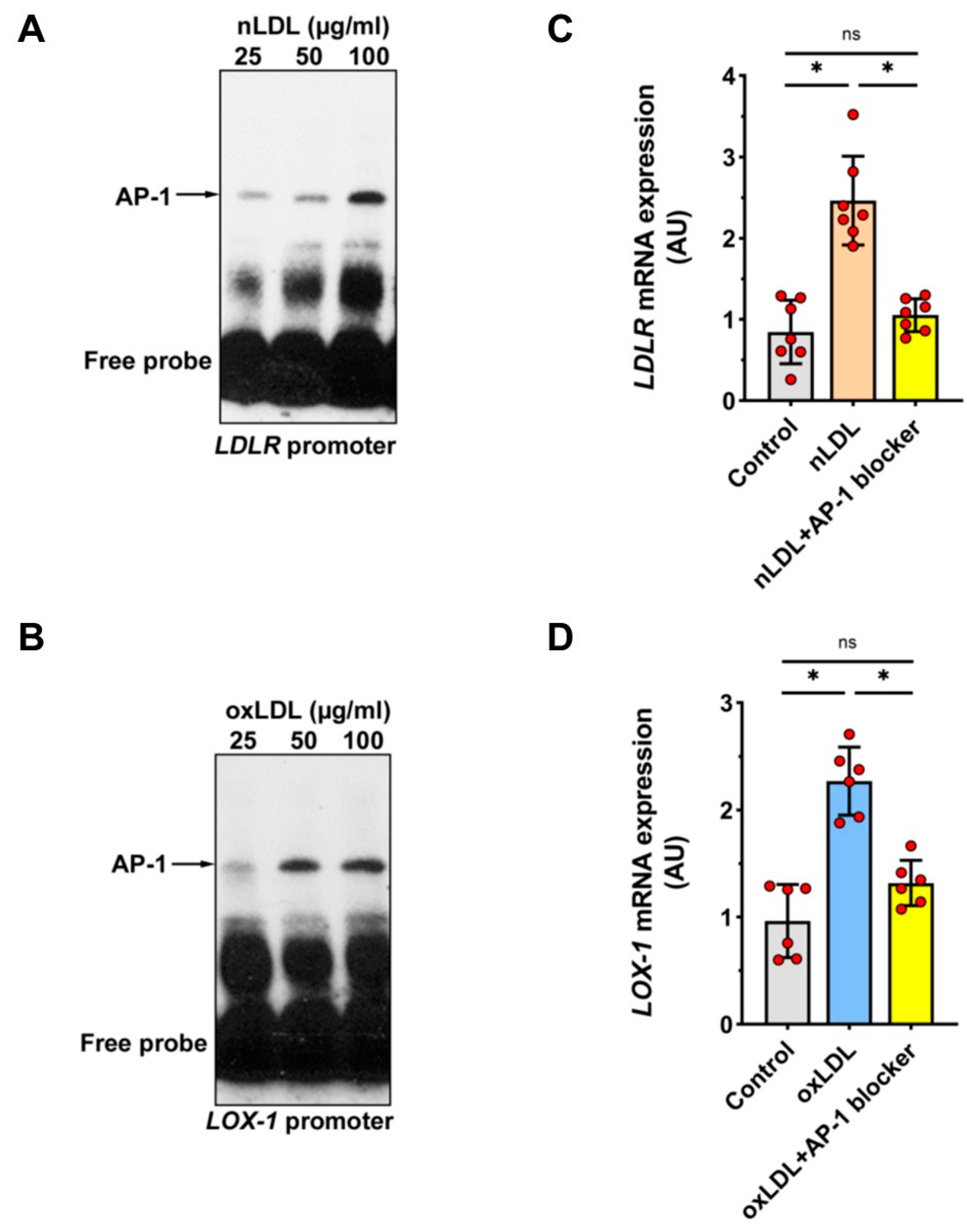
2.7. Involvement of Transcription Factor AP-1 in nLDL-Induced LDLR Expression and oxLDL-Induced LOX-1 Expression
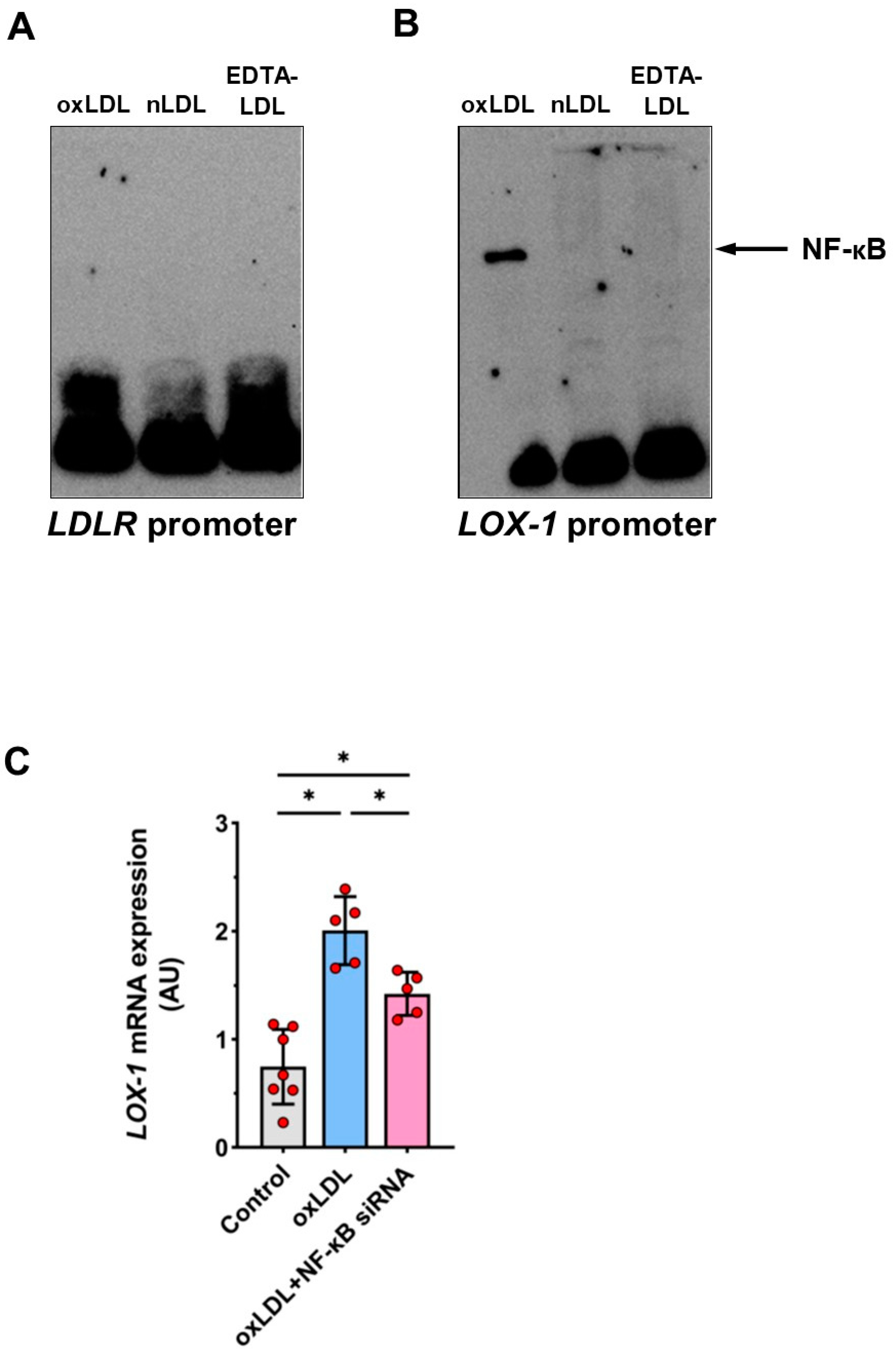
2.8. Involvement of Transcription Factor NF-κB in oxLDL-Induced LOX-1 Expression
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Lipoprotein Isolation and Oxidation
4.4. Assessment of Cell Viability and Apoptosis
4.5. LDL Uptake
4.6. Gene Expression Analysis
4.7. Western Blot Analysis
4.8. Quantification of Protein Phosphorylation
4.9. Promoter Analysis
4.10. Transient Transfections and Luciferase Assays
4.11. Computational Analysis of the LOX-1 and LDLR Promoter
4.12. Nuclear Extracts and Electrophoretic Mobility Shift Assay
4.13. Statistical Analysis
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchetti, F.; Crinelli, R.; Nasoni, M.G.; Benedetti, S.; Palma, F.; Fraternale, A.; Iuliano, L. LDL receptors, caveolae and cholesterol in endothelial dysfunction: oxLDLs accomplices or victims? Br. J. Pharmacol. 2021, 178, 3104–3114. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Witztum, J.L. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2311–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mineo, C. Lipoprotein receptor signalling in atherosclerosis. Cardiovasc. Res. 2020, 116, 1254–1274. [Google Scholar] [CrossRef]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef]
- Chen, M.; Masaki, T.; Sawamura, T. LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: Implications in endothelial dysfunction and atherosclerosis. Pharmacol. Ther. 2002, 95, 89–100. [Google Scholar] [CrossRef]
- Li, D.; Mehta, J.L. Antisense to LOX-1 inhibits oxidized LDL-mediated upregulation of monocyte chemoattractant protein-1 and monocyte adhesion to human coronary artery endothelial cells. Circulation 2000, 101, 2889–2895. [Google Scholar] [CrossRef] [Green Version]
- Akhmedov, A.; Rozenberg, I.; Paneni, F.; Camici, G.G.; Shi, Y.; Doerries, C.; Sledzinska, A.; Mocharla, P.; Breitenstein, A.; Lohmann, C.; et al. Endothelial overexpression of LOX-1 increases plaque formation and promotes atherosclerosis in vivo. Eur. Heart J. 2014, 35, 2839–2848. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.; Bai, Y.P.; Gao, H.C.; Wang, X.; Li, L.F.; Zhang, G.G.; Hu, C.P. Ox-LDL induces endothelial cell apoptosis via the LOX-1-dependent endoplasmic reticulum stress pathway. Atherosclerosis 2014, 235, 310–317. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lee, A.S.; Lu, L.S.; Ke, L.Y.; Chen, W.Y.; Dong, J.W.; Lu, J.; Chen, Z.; Chu, C.S.; Chan, H.C.; et al. Human electronegative LDL induces mitochondrial dysfunction and premature senescence of vascular cells in vivo. Aging Cell 2018, 17, e12792. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Arai, Y.; Kurihara, H.; Kita, T.; Sawamura, T. Overexpression of lectin-like oxidized low-density lipoprotein receptor-1 induces intramyocardial vasculopathy in apolipoprotein E-null mice. Circ. Res. 2005, 97, 176–184. [Google Scholar] [CrossRef] [Green Version]
- White, S.J.; Sala-Newby, G.B.; Newby, A.C. Overexpression of scavenger receptor LOX-1 in endothelial cells promotes atherogenesis in the ApoE(-/-) mouse model. Cardiovasc. Pathol. 2011, 20, 369–373. [Google Scholar] [CrossRef] [Green Version]
- Mehta, J.L.; Sanada, N.; Hu, C.P.; Chen, J.; Dandapat, A.; Sugawara, F.; Satoh, H.; Inoue, K.; Kawase, Y.; Jishage, K.; et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ. Res. 2007, 100, 1634–1642. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Dandapat, A.; Sun, L.; Chen, J.; Marwali, M.R.; Romeo, F.; Sawamura, T.; Mehta, J.L. LOX-1 deletion decreases collagen accumulation in atherosclerotic plaque in low-density lipoprotein receptor knockout mice fed a high-cholesterol diet. Cardiovasc. Res. 2008, 9, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleuren, A.C.A.; van der Ent, M.A.; Jiang, H.; Hunker, K.L.; Yee, A.; Siemieniak, D.R.; Molema, G.; Aird, W.C.; Ganesh, S.K.; Ginsburg, D. The in vivo endothelial cell translatome is highly heterogeneous across vascular beds. Proc. Natl. Acad. Sci. USA 2019, 116, 23618–23624. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.T.; Chang, H.Y.; Haraldsen, G.; Jahnsen, F.L.; Troyanskaya, O.G.; Chang, D.S.; Wang, Z.; Rockson, S.G.; van de Rijn, M.; Botstein, D.; et al. Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. USA 2003, 100, 10623–10628. [Google Scholar] [CrossRef] [Green Version]
- Deng, D.X.; Tsalenko, A.; Vailaya, A.; Ben-Dor, A.; Kundu, R.; Estay, I.; Tabibiazar, R.; Kincaid, R.; Yakhini, Z.; Bruhn, L.; et al. Differences in vascular bed disease susceptibility reflect differences in gene expression response to atherogenic stimuli. Circ. Res. 2006, 98, 200–208. [Google Scholar] [CrossRef]
- Stielow, C.; Catar, R.A.; Muller, G.; Wingler, K.; Scheurer, P.; Schmidt, H.H.; Morawietz, H. Novel Nox inhibitor of oxLDL-induced reactive oxygen species formation in human endothelial cells. Biochem. Biophys. Res. Commun. 2006, 344, 200–205. [Google Scholar] [CrossRef]
- Arrio, B.; Bonnefont-Rousselot, D.; Catudioc, J.D.; Packer, L. Electrophoretic mobility changes of oxidized human low density lipoprotein measured by laser Doppler electrophoresis. Biochem. Mol. Biol. Int. 1993, 30, 1101–1114. [Google Scholar] [PubMed]
- Ma, L.; Qian, L.; Ying, Q.; Zhang, Y.; Zhou, C.; Wu, G. I4, a synthetic anti-diabetes agent, attenuates atherosclerosis through its lipid-lowering, anti-inflammatory and anti-apoptosis properties. Mol. Cell. Endocrinol. 2017, 440, 80–92. [Google Scholar] [CrossRef]
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized LDL: Diversity, patterns of recognition, and pathophysiology. Antioxid. Redox. Signal. 2010, 13, 39–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natesampillai, S.; Fernandez-Zapico, M.E.; Urrutia, R.; Veldhuis, J.D. A novel functional interaction between the Sp1-like protein KLF13 and SREBP-Sp1 activation complex underlies regulation of low density lipoprotein receptor promoter function. J. Biol. Chem. 2006, 281, 3040–3047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Huang, Y.; Bandyopadhyay, S.; Virella, G.; Lopes-Virella, M.F. LDL immune complexes stimulate LDL receptor expression in U937 histiocytes via extracellular signal-regulated kinase and AP-1. J. Lipid Res. 2003, 44, 1315–1321. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, T.; Fujiwara, H.; Masaki, T.; Sawamura, T. Induction of lectin-like oxidized LDL receptor by oxidized LDL and lysophosphatidylcholine in cultured endothelial cells. J. Mol. Cell. Cardiol. 1999, 31, 2101–2114. [Google Scholar] [CrossRef]
- Morawietz, H.; Rueckschloss, U.; Niemann, B.; Duerrschmidt, N.; Galle, J.; Hakim, K.; Zerkowski, H.R.; Sawamura, T.; Holtz, J. Angiotensin II induces LOX-1, the human endothelial receptor for oxidized low-density lipoprotein. Circulation 1999, 100, 899–902. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Sawamura, T.; Renier, G. Glucose enhances endothelial LOX-1 expression: Role for LOX-1 in glucose-induced human monocyte adhesion to endothelium. Diabetes 2003, 52, 1843–1850. [Google Scholar] [CrossRef] [Green Version]
- Nagase, M.; Abe, J.; Takahashi, K.; Ando, J.; Hirose, S.; Fujita, T. Genomic organization and regulation of expression of the lectin-like oxidized low-density lipoprotein receptor (LOX-1) gene. J. Biol. Chem. 1998, 273, 3702–33707. [Google Scholar] [CrossRef] [Green Version]
- Kume, N.; Murase, T.; Moriwaki, H.; Aoyama, T.; Sawamura, T.; Masaki, T.; Kita, T. Inducible expression of lectin-like oxidized LDL receptor-1 in vascular endothelial cells. Circ. Res. 1998, 83, 322–327. [Google Scholar] [CrossRef] [Green Version]
- Chiba, Y.; Ogita, T.; Ando, K.; Fujita, T. PPARgamma ligands inhibit TNF-alpha-induced LOX-1 expression in cultured endothelial cells. Biochem. Biophys. Res. Commun. 2001, 286, 541–546. [Google Scholar] [CrossRef]
- Hofnagel, O.; Luechtenborg, B.; Stolle, K.; Lorkowski, S.; Eschert, H.; Plenz, G.; Robenek, H. Proinflammatory cytokines regulate LOX-1 expression in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1789–1795. [Google Scholar] [CrossRef] [Green Version]
- Thum, T.; Borlak, J. LOX-1 receptor blockade abrogates oxLDL-induced oxidative DNA damage and prevents activation of the transcriptional repressor Oct-1 in human coronary arterial endothelium. J. Biol. Chem. 2008, 283, 19456–19464. [Google Scholar] [CrossRef] [Green Version]
- Cominacini, L.; Pasini, A.F.; Garbin, U.; Davoli, A.; Tosetti, M.L.; Campagnola, M.; Rigoni, A.; Pastorino, A.M.; Lo Cascio, V.; Sawamura, T. Oxidized low density lipoprotein (ox-LDL) binding to ox-LDL receptor-1 in endothelial cells induces the activation of NF-kappaB through an increased production of intracellular reactive oxygen species. J. Biol. Chem. 2000, 275, 12633–12638. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, T.; Hokari, S.; Koyama, I.; Harada, T.; Komoda, T. NF-kappa B activation in endothelial cells treated with oxidized high-density lipoprotein. Biochem. Biophys. Res. Commun. 2003, 303, 313–319. [Google Scholar] [CrossRef]
- Mentrup, T.; Cabrera-Cabrera, F.; Schröder, B. Proteolytic Regulation of the Lectin-Like Oxidized Lipoprotein Receptor LOX-1. Front. Cardiovasc. Med. 2020, 7, 594441. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Mehta, J.L.; Haider, N.; Zhang, X.; Narula, J.; Li, D. Role of caspases in Ox-LDL-induced apoptotic cascade in human coronary artery endothelial cells. Circ. Res. 2004, 94, 370–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryoo, S.; Bhunia, A.; Chang, F.; Shoukas, A.; Berkowitz, D.E.; Romer, L.H. OxLDL-dependent activation of arginase II is dependent on the LOX-1 receptor and downstream RhoA signaling. Atherosclerosis 2011, 214, 279–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taye, A.; Saad, A.H.; Kumar, A.H.; Morawietz, H. Effect of apocynin on NADPH oxidase-mediated oxidative stress-LOX-1-eNOS pathway in human endothelial cells exposed to high glucose. Eur. J. Pharmacol. 2010, 627, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Moll, G.; Alm, J.J.; Davies, L.C.; von Bahr, L.; Heldring, N.; Stenbeck-Funke, L.; Hamad, O.A.; Hinsch, R.; Ignatowicz, L.; Locke, M.; et al. Do cryopreserved mesenchymal stromal cells display impaired immunomodulatory and therapeutic properties? Stem Cells 2014, 32, 2430–2442. [Google Scholar] [CrossRef] [Green Version]
- Moll, G.; Jitschin, R.; von Bahr, L.; Rasmusson-Duprez, I.; Sundberg, B.; Lonnies, L.; Elgue, G.; Nilsson-Ekdahl, K.; Mougiakakos, D.; Lambris, J.D.; et al. Mesenchymal stromal cells engage complement and complement receptor bearing innate effector cells to modulate immune responses. PLoS ONE 2011, 6, e21703. [Google Scholar] [CrossRef]
- Moll, G.; Hult, A.; von Bahr, L.; Alm, J.J.; Heldring, N.; Hamad, O.A.; Stenbeck-Funke, L.; Larsson, S.; Teramura, Y.; Roelofs, H.; et al. Do ABO blood group antigens hamper the therapeutic efficacy of mesenchymal stromal cells? PLoS ONE 2014, 9, e85040. [Google Scholar]
- Moll, G.; Ignatowicz, L.; Catar, R.; Luecht, C.; Sadeghi, B.; Hamad, O.; Jungebluth, P.; Dragun, D.; Schmidtchen, A.; Ringden, O. Different Procoagulant Activity of Therapeutic Mesenchymal Stromal Cells Derived from Bone Marrow and Placental Decidua. Stem Cells Dev. 2015, 24, 2269–2279. [Google Scholar] [CrossRef]
- Korff, T.; Dandekar, G.; Pfaff, D.; Fuller, T.; Goettsch, W.; Morawietz, H.; Schaffner, F.; Augustin, H.G. Endothelial ephrinB2 is controlled by microenvironmental determinants and associates context-dependently with CD31. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 468–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locher, R.; Brandes, R.P.; Vetter, W.; Barton, M. Native LDL induces proliferation of human vascular smooth muscle cells via redox-mediated activation of ERK 1/2 mitogen-activated protein kinases. Hypertension 2002, 39, 645–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albina, J.E.; Cui, S.; Mateo, R.B.; Reichner, J.S. Nitric oxide-mediated apoptosis in murine peritoneal macrophages. J. Immunol. 1993, 150, 5080–5085. [Google Scholar] [PubMed]
- Schubert, A.; Cattaruzza, M.; Hecker, M.; Darmer, D.; Holtz, J.; Morawietz, H. Shear stress-dependent regulation of the human beta-tubulin folding cofactor D gene. Circ. Res. 2000, 87, 1188–1194. [Google Scholar] [CrossRef] [Green Version]
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R., III; Le Beau, M.M.; Earp, H.S.; Liu, E.T. Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar] [PubMed] [Green Version]
- Versteeg, H.H.; Nijhuis, E.; van den Brink, G.R.; Evertzen, M.; Pynaert, G.N.; van Deventer, S.J.; Coffer, P.J.; Peppelenbosch, M.P. A new phosphospecific cell-based ELISA for p42/p44 mitogen-activated protein kinase (MAPK), p38 MAPK, protein kinase B and cAMP-response-element-binding protein. Biochem. J. 2000, 350, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Catar, R.; Witowski, J.; Zhu, N.; Lücht, C.; Derrac Soria, A.; Uceda Fernandez, J.; Chen, L.; Jones, S.A.; Fielding, C.A.; Rudolf, A.; et al. IL-6 Trans-Signaling Links Inflammation with Angiogenesis in the Peritoneal Membrane. J. Am. Soc. Nephrol. 2017, 28, 1188–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catar, R.; Chen, L.; Zhao, H.; Wu, D.; Kamhieh-Milz, J.; Lücht, C.; Zickler, D.; Krug, A.W.; Ziegler, C.G.; Morawietz, H.; et al. Native and Oxidized Low-Density Lipoproteins Increase the Expression of the LDL Receptor and the LOX-1 Receptor, Respectively, in Arterial Endothelial Cells. Cells 2022, 11, 204. https://doi.org/10.3390/cells11020204
Catar R, Chen L, Zhao H, Wu D, Kamhieh-Milz J, Lücht C, Zickler D, Krug AW, Ziegler CG, Morawietz H, et al. Native and Oxidized Low-Density Lipoproteins Increase the Expression of the LDL Receptor and the LOX-1 Receptor, Respectively, in Arterial Endothelial Cells. Cells. 2022; 11(2):204. https://doi.org/10.3390/cells11020204
Chicago/Turabian StyleCatar, Rusan, Lei Chen, Hongfan Zhao, Dashan Wu, Julian Kamhieh-Milz, Christian Lücht, Daniel Zickler, Alexander W. Krug, Christian G. Ziegler, Henning Morawietz, and et al. 2022. "Native and Oxidized Low-Density Lipoproteins Increase the Expression of the LDL Receptor and the LOX-1 Receptor, Respectively, in Arterial Endothelial Cells" Cells 11, no. 2: 204. https://doi.org/10.3390/cells11020204