
TFEB; Beyond Its Role as an Autophagy and Lysosomes Regulator
 , ,
, ,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Post-Translational Control of TFEB Activity
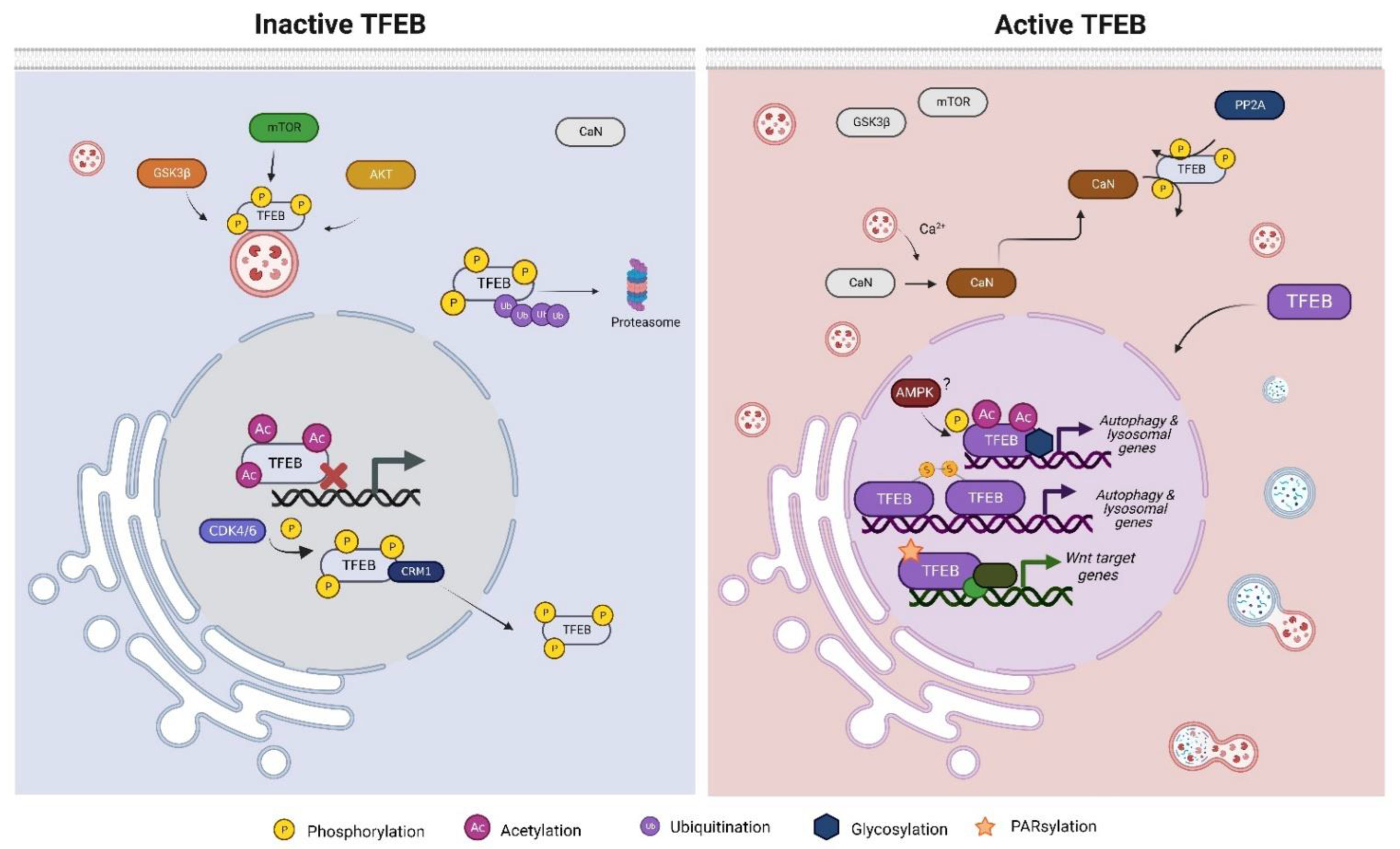
2.1. Phosphorylation
2.2. Acetylation
2.3. Other Post-Translational Modifications of TFEB
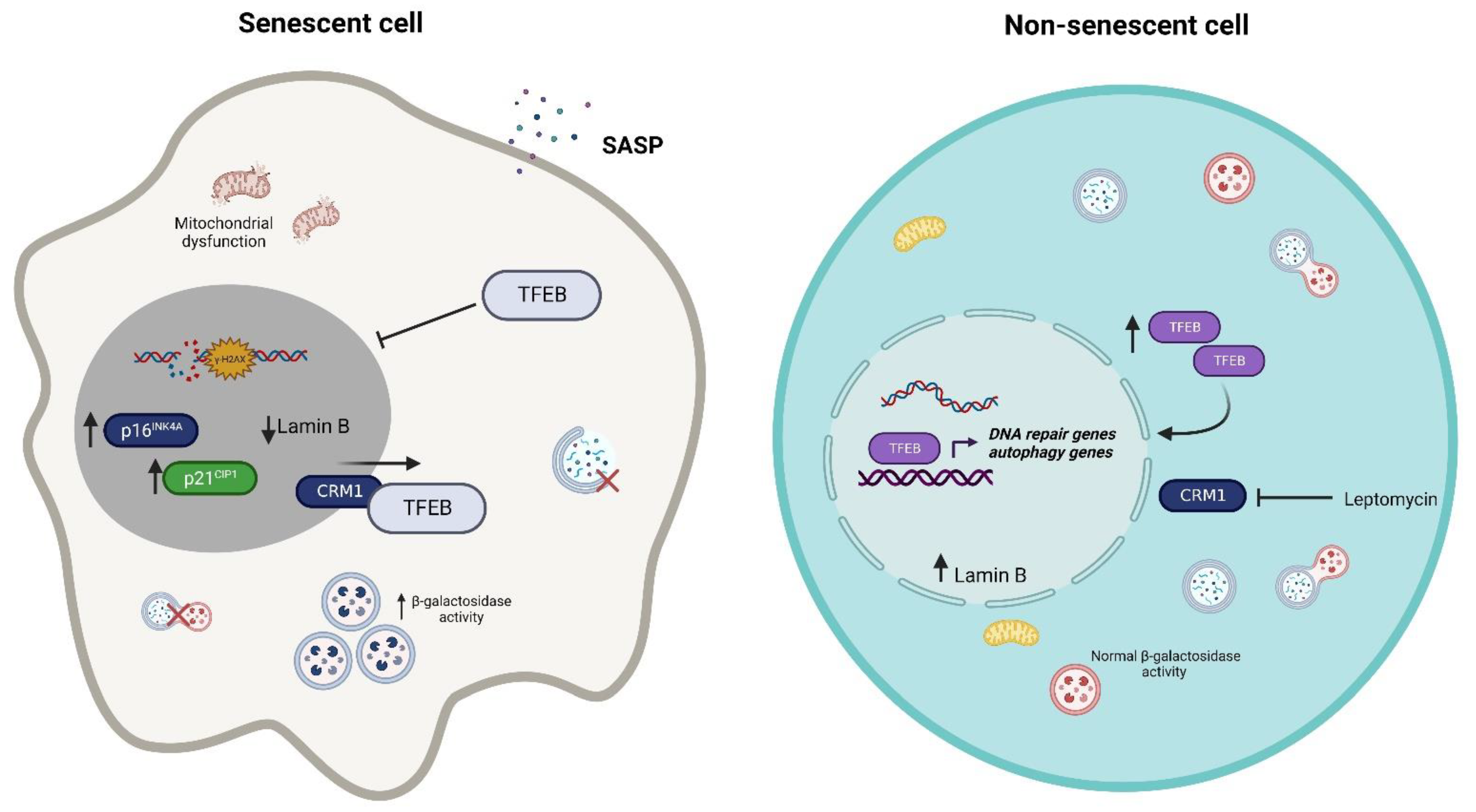
3. TFEB on Senescence
4. TFEB on DNA Damage Repair and Cell Cycle
5. TFEB on WNT Signaling
6. Endoplasmic Reticulum Stress and TFEB
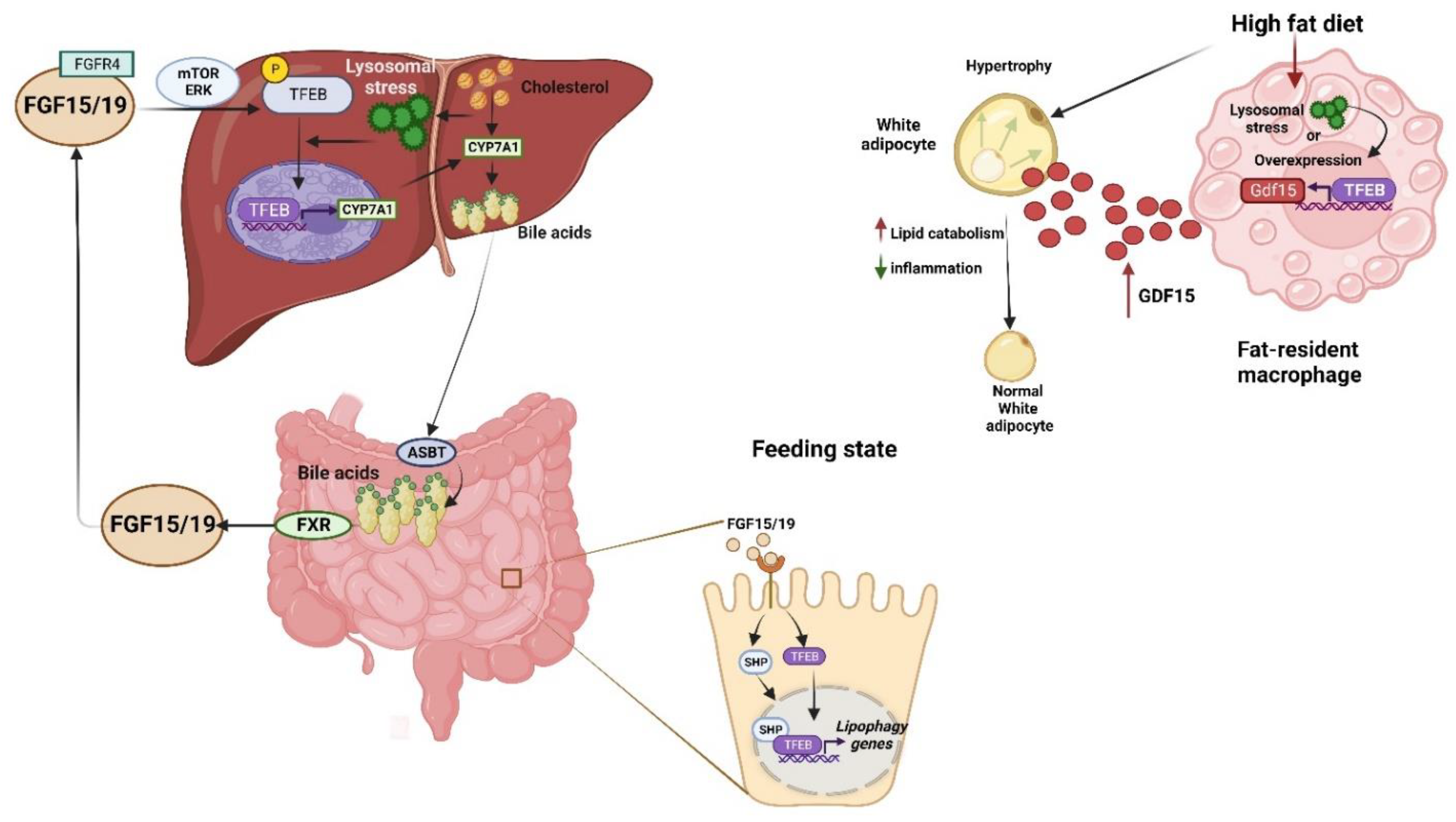
7. TFEB and Its Influence on the Metabolism
7.1. Role in Glucose Metabolism
7.2. Role in Lipid Metabolism
8. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Slade, L.; Pulinilkunnil, T. The MiTF/TFE Family of Transcription Factors: Master Regulators of Organelle Signaling, Metabolism, and Stress Adaptation. Mol. Cancer Res. 2017, 15, 1637–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Spina, M.; Contreras, P.S.; Rissone, A.; Meena, N.K.; Jeong, E.; Martina, J.A. MiT/TFE Family of Transcription Factors: An Evolutionary Perspective. Front. Cell Dev. Biol. 2020, 8, 609683. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.C.; Bartlett, J.J.; Mercer, A.; Slade, L.; Surette, M.; Ballabio, A.; Flibotte, S.; Hussein, B.; Rodrigues, B.; Kienesberger, P.C.; et al. Loss of function of transcription factor EB remodels lipid metabolism and cell death pathways in the cardiomyocyte. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165832. [Google Scholar] [CrossRef]
- Li, L.; Friedrichsen, H.J.; Andrews, S.; Picaud, S.; Volpon, L.; Ngeow, K.; Berridge, G.; Fischer, R.; Borden, K.L.B.; Filippakopoulos, P.; et al. A TFEB nuclear export signal integrates amino acid supply and glucose availability. Nat. Commun. 2018, 9, 2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapierre, L.R.; De Magalhaes Filho, C.D.; McQuary, P.R.; Chu, C.C.; Visvikis, O.; Chang, J.T.; Gelino, S.; Ong, B.; Davis, A.E.; Irazoqui, J.E.; et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 2013, 4, 2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallsson, J.H.; Haflidadottir, B.S.; Stivers, C.; Odenwald, W.; Arnheiter, H.; Pignoni, F.; Steingrimsson, E. The basic helix–loop–helix leucine zipper transcription factor Mitf is conserved in Drosophila and functions in eye development. Genetics 2004, 167, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Lister, J.A.; Lane, B.M.; Nguyen, A.; Lunney, K. Embryonic expression of zebrafish MiT family genes tfe3b, tfeb, and tfec. Dev. Dyn. 2011, 240, 2529–2538. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Wang, F.; Savini, M.; Ake, A.; di Ronza, A.; Sardiello, M.; Segatori, L. TFEB regulates lysosomal proteostasis. Hum. Mol. Genet. 2013, 22, 1994–2009. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Ballabio, A. TFEB regulates autophagy: An integrated coordination of cellular degradation and recycling processes. Autophagy 2011, 7, 1379–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, X.; Wang, S.; Zhao, K.; Li, Y.; Williams, J.A.; Li, T.; Chavan, H.; Krishnamurthy, P.; He, X.C.; Li, L.; et al. Impaired TFEB-Mediated Lysosome Biogenesis and Autophagy Promote Chronic Ethanol-Induced Liver Injury and Steatosis in Mice. Gastroenterology 2018, 155, 865–879.e12. [Google Scholar] [CrossRef]
- Tsunemi, T.; Ashe, T.D.; Morrison, B.E.; Soriano, K.R.; Au, J.; Roque, R.A.; Lazarowski, E.R.; Damian, V.A.; Masliah, E.; La Spada, A.R. PGC-1alpha rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 2012, 4, 142ra97. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.K.; Kotia, V.; Ghosh, D.; Mohite, G.M.; Kumar, A.; Maji, S.K. Curcumin modulates alpha-synuclein aggregation and toxicity. ACS Chem. Neurosci. 2013, 4, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.D.; Zhao, J.J. TFEB Participates in the Aβ-Induced Pathogenesis of Alzheimer’s Disease by Regulating the Autophagy-Lysosome Pathway. DNA Cell Biol. 2015, 34, 661–668. [Google Scholar] [CrossRef]
- Visvikis, O.; Ihuegbu, N.; Labed, S.A.; Luhachack, L.G.; Alves, A.F.; Wollenberg, A.C.; Stuart, L.M.; Stormo, G.D.; Irazoqui, J.E. Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity 2014, 40, 896–909. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Cai, Z.; Tao, K.; Zeng, W.; Lu, F.; Yang, R.; Feng, D.; Gao, G.; Yang, Q. Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy 2016, 12, 1215–1228. [Google Scholar] [CrossRef]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef]
- Sha, Y.; Rao, L.; Settembre, C.; Ballabio, A.; Eissa, N.T. STUB1 regulates TFEB-induced autophagy-lysosome pathway. EMBO J. 2017, 36, 2544–2552. [Google Scholar] [CrossRef] [PubMed]
- Vega-Rubin-de-Celis, S.; Pena-Llopis, S.; Konda, M.; Brugarolas, J. Multistep regulation of TFEB by MTORC1. Autophagy 2017, 13, 464–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, M.; Ding, X.; Yan, C.; Song, Z.; Chen, L.; Huang, X.; Wang, X.; Jian, Y.; Tang, G.; et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat. Cell Biol. 2016, 18, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barski, M.S.; Minnell, J.J.; Maertens, G.N. PP2A Phosphatase as an Emerging Viral Host Factor. Front. Cell Infect. Microbiol. 2021, 11, 725615. [Google Scholar] [CrossRef] [PubMed]
- Martina, J.A.; Puertollano, R. Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress. J. Biol. Chem 2018, 293, 12525–12534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestrini, M.J.; Johnson, J.R.; Kumar, A.V.; Thakurta, T.G.; Blais, K.; Neill, Z.A.; Marion, S.W.; St Amand, V.; Reenan, R.A.; Lapierre, L.R. Nuclear Export Inhibition Enhances HLH-30/TFEB Activity, Autophagy, and Lifespan. Cell Rep. 2018, 23, 1915–1921. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Jian, Y.; Xu, M.; Huang, X.; Wang, N.; Liu, Z.; Li, Q.; Li, J.; Zhou, H.; Xu, L.; et al. CDK4/6 regulate lysosome biogenesis through TFEB/TFE3. J. Cell Biol. 2020, 219, e201911036. [Google Scholar] [CrossRef]
- Paquette, M.; El-Houjeiri, L.; CZirden, L.; Puustinen, P.; Blanchette, P.; Jeong, H.; Dejgaard, K.; Siegel, P.M.; Pause, A. AMPK-dependent phosphorylation is required for transcriptional activation of TFEB and TFE3. Autophagy 2021, 17, 3957–3975. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yu, L.; Xue, H.; Yang, Z.; Yin, Y.; Zhang, B.; Chen, M.; Ma, H. Nuclear AMPK regulated CARM1 stabilization impacts autophagy in aged heart. Biochem. Biophys. Res. Commun. 2017, 486, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Zheng, L.; Zhang, Q.; Li, X.; Zhang, X.; Li, Z.; Bai, X.; Zhang, Z.; Huo, W.; Zhao, X.; et al. Deacetylation of TFEB promotes fibrillar Aβ degradation by upregulating lysosomal biogenesis in microglia. Protein Cell 2016, 7, 417–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, J.; Zhou, Z.; Park, J.E.; Wang, L.; Wu, S.; Sun, X.; Lu, L.; Wang, T.; Lin, Q.; et al. Importance of TFEB acetylation in control of its transcriptional activity and lysosomal function in response to histone deacetylase inhibitors. Autophagy 2018, 14, 1043–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Huang, Y.; Liu, J.; Zhang, J.; Xu, M.; You, Z.; Peng, C.; Gong, Z.; Liu, W. Acetyltransferase GCN5 regulates autophagy and lysosome biogenesis by targeting TFEB. EMBO Rep. 2020, 21, e48335. [Google Scholar] [CrossRef]
- Brijmohan, A.S.; Batchu, S.N.; Majumder, S.; Alghamdi, T.A.; Thieme, K.; McGaugh, S.; Liu, Y.; Advani, S.L.; Bowskill, B.B.; Kabir, M.G.; et al. HDAC6 Inhibition Promotes Transcription Factor EB Activation and Is Protective in Experimental Kidney Disease. Front. Pharmacol 2018, 9, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.J.; Levy, C.; Davis, I.J.; Razin, E.; Fisher, D.E. Sumoylation of MITF and its related family members TFE3 and TFEB. J. Biol. Chem. 2005, 280, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Pahi, Z.G.; Borsos, B.N.; Pantazi, V.; Ujfaludi, Z.; Pankotai, T. PARylation During Transcription: Insights into the Fine-Tuning Mechanism and Regulation. Cancers 2020, 12, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.K. Novel insight into the function of tankyrase. Oncol. Lett. 2018, 16, 6895–6902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Song, G.; Lee, T.; Kim, M.; Kim, J.; Kwon, H.; Kim, J.; Jeong, W.; Lee, U.; Na, C.; et al. PARsylated transcription factor EB (TFEB) regulates the expression of a subset of Wnt target genes by forming a complex with beta-catenin-TCF/LEF1. Cell Death Differ. 2021, 28, 2555–2570. [Google Scholar] [CrossRef] [PubMed]
- Beck, W.H.J.; Kim, D.; Das, J.; Yu, H.; Smolka, M.B.; Mao, Y. Glucosylation by the Legionella Effector SetA Promotes the Nuclear Localization of the Transcription Factor TFEB. iScience 2020, 23, 101300. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Santamarina, S.; Boronat, S.; Hidalgo, E. Reversible cysteine oxidation in hydrogen peroxide sensing and signal transduction. Biochemistry 2014, 53, 2560–2580. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Guerrero-Gomez, D.; Gomez-Orte, E.; Antonio Barcena, J.; Cabello, J.; Miranda-Vizuete, A.; Puertollano, R. A conserved cysteine-based redox mechanism sustains TFEB/HLH-30 activity under persistent stress. EMBO J. 2021, 40, e105793. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Roy, A.L.; Sierra, F.; Howcroft, K.; Singer, D.S.; Sharpless, N.; Hodes, R.J.; Wilder, E.L.; Anderson, J.M. A Blueprint for Characterizing Senescence. Cell 2020, 183, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Yoon, M.H.; Kang, S.M.; Lee, S.J.; Woo, T.G.; Oh, A.Y.; Park, S.; Ha, N.C.; Park, B.J. p53 induces senescence through Lamin A/C stabilization-mediated nuclear deformation. Cell Death Dis. 2019, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Burton, D.G.; Krizhanovsky, V. Physiological and pathological consequences of cellular senescence. Cell Mol. Life Sci. 2014, 71, 4373–4386. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Inagaki, N.; Kondoh, H. Cellular Senescence in Diabetes Mellitus: Distinct Senotherapeutic Strategies for Adipose Tissue and Pancreatic β Cells. Front. Endocrinol. 2022, 13, 869414. [Google Scholar] [CrossRef]
- McCulloch, K.; Litherland, G.J.; Rai, T.S. Cellular senescence in osteoarthritis pathology. Aging Cell 2017, 16, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, M.; Hong, D.; Zeng, M.; Zhang, X. The Paradoxical Role of Cellular Senescence in Cancer. Front. Cell Dev. Biol. 2021, 9, 722205. [Google Scholar] [CrossRef]
- Martinez-Cue, C.; Rueda, N. Cellular Senescence in Neurodegenerative Diseases. Front. Cell Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef]
- Kang, H.T.; Lee, K.B.; Kim, S.Y.; Choi, H.R.; Park, S.C. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS ONE 2011, 6, e23367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Alzahrani, A.M.; Hanieh, H.N.; Kumar, S.A.; Ben Ammar, R.; Rengarajan, T.; Alhoot, M.A. Autophagy and senescence: A new insight in selected human diseases. J. Cell Physiol. 2019, 234, 21485–21492. [Google Scholar] [CrossRef]
- Zheng, G.; Pan, Z.; Zhan, Y.; Tang, Q.; Zheng, F.; Zhou, Y.; Wu, Y.; Zhou, Y.; Chen, D.; Chen, J.; et al. TFEB protects nucleus pulposus cells against apoptosis and senescence via restoring autophagic flux. Osteoarthr. Cartil. 2019, 27, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Wang, H.; Muthu Karuppan, M.K.; Devadoss, D.; Nair, M.; Chand, H.S.; Lakshmana, M.K. TFEB protein expression is reduced in aged brains and its overexpression mitigates senescence-associated biomarkers and memory deficits in mice. Neurobiol. Aging 2021, 106, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Gorostieta-Salas, E.; Moreno-Blas, D.; Geronimo-Olvera, C.; Cisneros, B.; Court, F.A.; Castro-Obregon, S. Enhanced Activity of Exportin-1/CRM1 in Neurons Contributes to Autophagy Dysfunction and Senescent Features in Old Mouse Brain. Oxid. Med. Cell Longev. 2021, 2021, 6682336. [Google Scholar] [CrossRef] [PubMed]
- Bodas, M.; Patel, N.; Silverberg, D.; Walworth, K.; Vij, N. Master Autophagy Regulator Transcription Factor EB Regulates Cigarette Smoke-Induced Autophagy Impairment and Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis. Antioxid. Redox Signal. 2017, 27, 150–167. [Google Scholar] [CrossRef] [PubMed]
- Carusillo, A.; Mussolino, C. DNA Damage: From Threat to Treatment. Cells 2020, 9, 1665. [Google Scholar] [CrossRef] [PubMed]
- Feroz, W.; Sheikh, A.M. Exploring the multiple roles of guardian of the genome: P53. Egypt. J. Med. Hum. Genet. 2020, 21, 1–23. [Google Scholar] [CrossRef]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [Green Version]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Brady, O.A.; Jeong, E.; Martina, J.A.; Pirooznia, M.; Tunc, I.; Puertollano, R. The transcription factors TFE3 and TFEB amplify p53 dependent transcriptional programs in response to DNA damage. Elife 2018, 7, e40856. [Google Scholar]
- Slade, L.; Biswas, D.; Ihionu, F.; El Hiani, Y.; Kienesberger, P.C.; Pulinilkunnil, T. A lysosome independent role for TFEB in activating DNA repair and inhibiting apoptosis in breast cancer cells. Biochem. J. 2020, 477, 137–160. [Google Scholar] [CrossRef]
- Pisonero-Vaquero, S.; Soldati, C.; Cesana, M.; Ballabio, A.; Medina, D.L. TFEB Modulates p21/WAF1/CIP1 during the DNA Damage Response. Cells 2020, 9, 1186. [Google Scholar] [CrossRef]
- Doronzo, G.; Astanina, E.; Cora, D.; Chiabotto, G.; Comunanza, V.; Noghero, A.; Neri, F.; Puliafito, A.; Primo, L.; Spampanato, C.; et al. TFEB controls vascular development by regulating the proliferation of endothelial cells. EMBO J. 2019, 38, e98250. [Google Scholar] [CrossRef] [PubMed]
- Pastore, N.; Huynh, T.; Herz, N.J.; Calcagni, A.; Klisch, T.J.; Brunetti, L.; Kim, K.H.; De Giorgi, M.; Hurley, A.; Carissimo, A.; et al. TFEB regulates murine liver cell fate during development and regeneration. Nat. Commun. 2020, 11, 2461. [Google Scholar] [CrossRef] [PubMed]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, J.; Dannappel, M.; Wan, C.; Firestein, R. Transcriptional Regulation of Wnt/β-Catenin Pathway in Colorectal Cancer. Cells 2020, 9, 2125. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.F.; Kaur, P.; Bunnag, N.; Suresh, J.; Sung, I.C.H.; Tan, Q.H.; Gruber, J.; Tolwinski, N.S. WNT Signaling in Disease. Cells 2019, 8, 826. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Liu, F.; Xu, L.; Li, C.; Yang, X.; Guo, B.; Gu, J.; Wang, L. Wnt/β-Catenin Signaling Axis Is Required for TFEB-Mediated Gastric Cancer Metastasis and Epithelial-Mesenchymal Transition. Mol. Cancer Res. 2020, 18, 1650–1659. [Google Scholar] [CrossRef]
- Liang, J.; Jia, X.; Wang, K.; Zhao, N. High expression of TFEB is associated with aggressive clinical features in colorectal cancer. Onco Targets Ther. 2018, 11, 8089–8098. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Lei, H.; Yi, T.; Su, P.; Tang, S.; Tong, Y.; Dong, B.; Ruan, G.; Mustea, A.; Sehouli, J.; et al. Lipid reprogramming induced by the TFEB-ERRα axis enhanced membrane fluidity to promote EC progression. J. Exp. Clin. Cancer Res. 2022, 41, 28. [Google Scholar] [CrossRef]
- Zhu, X.; Zhuo, Y.; Wu, S.; Chen, Y.; Ye, J.; Deng, Y.; Feng, Y.; Liu, R.; Cai, S.; Zou, Z.; et al. TFEB Promotes Prostate Cancer Progression via Regulating ABCA2-Dependent Lysosomal Biogenesis. Front. Oncol. 2021, 11, 632524. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Kalamida, D.; Sivridis, E.; Karagounis, I.V.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Increased expression of transcription factor EB (TFEB) is associated with autophagy, migratory phenotype and poor prognosis in non-small cell lung cancer. Lung Cancer 2015, 90, 98–105. [Google Scholar] [CrossRef]
- He, R.; Wang, M.; Zhao, C.; Shen, M.; Yu, Y.; He, L.; Zhao, Y.; Chen, H.; Shi, X.; Zhou, M.; et al. TFEB-driven autophagy potentiates TGF-β induced migration in pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 340. [Google Scholar] [CrossRef] [PubMed]
- Ariano, C.; Riganti, C.; Cora, D.; Valdembri, D.; Mana, G.; Astanina, E.; Serini, G.; Bussolino, F.; Doronzo, G. TFEB controls integrin-mediated endothelial cell adhesion by the regulation of cholesterol metabolism. Angiogenesis 2022, 25, 471–492. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markovinovic, A.; Greig, J.; Martin-Guerrero, S.M.; Salam, S.; Paillusson, S. Endoplasmic reticulum-mitochondria signaling in neurons and neurodegenerative diseases. J. Cell Sci. 2022, 135, jcs248534. [Google Scholar] [CrossRef]
- Bell, M.C.; Meier, S.E.; Ingram, A.L.; Abisambra, J.F. PERK-opathies: An Endoplasmic Reticulum Stress Mechanism Underlying Neurodegeneration. Curr. Alzheimer Res. 2016, 13, 150–163. [Google Scholar] [CrossRef]
- Rivas, A.; Vidal, R.L.; Hetz, C. Targeting the unfolded protein response for disease intervention. Expert Opin. Ther. Targets 2015, 19, 1203–1218. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallee, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Srinivasan, S.; Ohsugi, M.; Liu, Z.; Fatrai, S.; Bernal-Mizrachi, E.; Permutt, M.A. Endoplasmic reticulum stress-induced apoptosis is partly mediated by reduced insulin signaling through phosphatidylinositol 3-kinase/Akt and increased glycogen synthase kinase-3β in mouse insulinoma cells. Diabetes 2005, 54, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Sykes, E.K.; Mactier, S.; Christopherson, R.I. Melanoma and the Unfolded Protein Response. Cancers 2016, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R., 3rd; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Diab, H.I.; Brady, O.A.; Puertollano, R. TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 2016, 35, 479–495. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.M.; Pae, H.O.; Zheng, M.; Park, R.; Kim, Y.M.; Chung, H.T. Carbon monoxide induces heme oxygenase-1 via activation of protein kinase R-like endoplasmic reticulum kinase and inhibits endothelial cell apoptosis triggered by endoplasmic reticulum stress. Circ. Res. 2007, 101, 919–927. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Joe, Y.; Rah, S.Y.; Kim, S.K.; Park, S.U.; Park, J.; Kim, J.; Ryu, J.; Cho, G.J.; Surh, Y.J.; et al. Carbon monoxide-induced TFEB nuclear translocation enhances mitophagy/mitochondrial biogenesis in hepatocytes and ameliorates inflammatory liver injury. Cell Death Dis. 2018, 9, 1060. [Google Scholar] [CrossRef] [Green Version]
- Do, M.; Park, J.; Chen, Y.; Rah, S.Y.; Nghiem, T.T.; Gong, J.H.; Ju, S.A.; Kim, B.S.; Yu, R.; Park, J.W.; et al. PERK activation by SB202190 ameliorates amyloidogenesis via the TFEB-induced autophagy-lysosomal pathway. Aging 2022, 14, 1233–1252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qian, Q.; Li, M.; Shao, F.; Ding, W.X.; Lira, V.A.; Chen, S.X.; Sebag, S.C.; Hotamisligil, G.S.; Cao, H.; et al. The unfolded protein response regulates hepatic autophagy by sXBP1-mediated activation of TFEB. Autophagy 2021, 17, 1841–1855. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Lee, Y.; Jeong, J.H.; Ryu, J.H.; Kim, K.I. Inhibition of autophagy sensitizes lignan-induced endoplasmic reticulum stress-mediated cell death. Biochem. Biophys. Res. Commun. 2020, 526, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kwon, J.; Jeong, J.H.; Ryu, J.H.; Kim, K.I. Kazinol C from Broussonetia kazinoki stimulates autophagy via endoplasmic reticulum stress-mediated signaling. Anim. Cells Syst. 2022, 26, 28–36. [Google Scholar] [CrossRef]
- Wu, Q.; Tian, A.L.; Durand, S.; Aprahamian, F.; Nirmalathasan, N.; Xie, W.; Liu, P.; Zhao, L.; Zhang, S.; Pan, H.; et al. Isobacachalcone induces autophagy and improves the outcome of immunogenic chemotherapy. Cell Death Dis. 2020, 11, 1015. [Google Scholar] [CrossRef] [PubMed]
- Bisicchia, E.; Mastrantonio, R.; Nobili, A.; Palazzo, C.; La Barbera, L.; Latini, L.; Millozzi, F.; Sasso, V.; Palacios, D.; D’Amelio, M.; et al. Restoration of ER proteostasis attenuates remote apoptotic cell death after spinal cord injury by reducing autophagosome overload. Cell Death Dis. 2022, 13, 381. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, Z.; Chen, S.; Qiu, J.; Li, Q.; Wang, S.; Zhou, W.; Chen, D.; Yang, G.; Guo, L. Transcription factor EB-mediated autophagy promotes dermal fibroblast differentiation and collagen production by regulating endoplasmic reticulum stress and autophagy-dependent secretion. Int. J. Mol. Med. 2021, 47, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Yoneshima, E.; Okamoto, K.; Sakai, E.; Nishishita, K.; Yoshida, N.; Tsukuba, T. The Transcription Factor EB (TFEB) Regulates Osteoblast Differentiation Through ATF4/CHOP-Dependent Pathway. J. Cell Physiol. 2016, 231, 1321–1333. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M.; Loo, D.D.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansueto, G.; Armani, A.; Viscomi, C.; D’Orsi, L.; De Cegli, R.; Polishchuk, E.V.; Lamperti, C.; Di Meo, I.; Romanello, V.; Marchet, S.; et al. Transcription Factor EB Controls Metabolic Flexibility during Exercise. Cell Metab. 2017, 25, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Xie, J.; Fiskesund, R.; Dong, W.; Liang, X.; Lv, J.; Jin, X.; Liu, J.; Mo, S.; Zhang, T.; et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun. 2018, 9, 873. [Google Scholar] [CrossRef]
- Fernandez, M.R.; Schaub, F.X.; Yang, C.; Li, W.; Yun, S.; Schaub, S.K.; Dorsey, F.C.; Liu, M.; Steeves, M.A.; Ballabio, A.; et al. Disrupting the MYC-TFEB Circuit Impairs Amino Acid Homeostasis and Provokes Metabolic Anergy. Cancer Res. 2022, 82, 1234–1250. [Google Scholar] [CrossRef]
- Kubota, T.; Kubota, N.; Kadowaki, T. The role of endothelial insulin signaling in the regulation of glucose metabolism. Rev. Endocr. Metab. Disord. 2013, 14, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Quon, M.J. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J. Clin. Investig. 1996, 98, 894–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Lu, H.; Liang, W.; Zhao, G.; Ren, L.; Hu, D.; Chang, Z.; Liu, Y.; Garcia-Barrio, M.T.; Zhang, J.; et al. Endothelial TFEB (Transcription Factor EB) Improves Glucose Tolerance via Upregulation of IRS (Insulin Receptor Substrate) 1 and IRS2. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, C.G.; Zhou, X.; Ding, S. Transcription factor EB enhances autophagy and ameliorates palmitate-induced insulin resistance at least partly via upregulating AMPK activity in skeletal muscle cells. Clin. Exp. Pharmacol. Physiol. 2022, 49, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Nielson, J.R.; Rutter, J.P. Lipid-mediated signals that regulate mitochondrial biology. J. Biol. Chem. 2018, 293, 7517–7521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, T.D.; Zhang, X.; Jeong, S.J.; He, A.; Song, E.; Bhattacharya, S.; Holloway, K.B.; Lodhi, I.J.; Razani, B. TFEB drives PGC-1α expression in adipocytes to protect against diet-induced metabolic dysfunction. Sci. Signal. 2019, 12, eaau2281. [Google Scholar] [CrossRef] [PubMed]
- Pastore, N.; Vainshtein, A.; Klisch, T.J.; Armani, A.; Huynh, T.; Herz, N.J.; Polishchuk, E.V.; Sandri, M.; Ballabio, A. TFE3 regulates whole-body energy metabolism in cooperation with TFEB. EMBO Mol. Med. 2017, 9, 605–621. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, E.J.; Ruvkun, G. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat. Cell Biol. 2013, 15, 668–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco-Juarez, B.; Gomez-Manzo, S.; Hernandez-Ochoa, B.; Cardenas-Rodriguez, N.; Arreguin-Espinosa, R.; Perez de la Cruz, V.; Ortega-Cuellar, D. Effects of High Dietary Carbohydrate and Lipid Intake on the Lifespan of C. elegans. Cells 2021, 10, 2359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yan, S.; Khambu, B.; Ma, F.; Li, Y.; Chen, X.; Martina, J.A.; Puertollano, R.; Li, Y.; Chalasani, N.; et al. Dynamic MTORC1-TFEB feedback signaling regulates hepatic autophagy, steatosis and liver injury in long-term nutrient oversupply. Autophagy 2018, 14, 1779–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gunewardena, S.; Li, F.; Matye, D.J.; Chen, C.; Chao, X.; Jung, T.; Zhang, Y.; Czerwinski, M.; Ni, H.M.; et al. An FGF15/19-TFEB regulatory loop controls hepatic cholesterol and bile acid homeostasis. Nat. Commun 2020, 11, 3612. [Google Scholar] [CrossRef]
- Seok, S.; Kim, Y.C.; Zhang, Y.; Kong, B.; Guo, G.; Ma, J.; Kemper, B.; Kemper, J.K. Feeding activates FGF15-SHP-TFEB-mediated lipophagy in the gut. EMBO J. 2022, 41, e109997. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, S.H.; Kang, H.; Lee, S.; Park, S.Y.; Cho, Y.; Lim, Y.M.; Ahn, J.W.; Kim, Y.H.; Chung, S.; et al. TFEB-GDF15 axis protects against obesity and insulin resistance as a lysosomal stress response. Nat. Metab. 2021, 3, 410–427. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco-Juárez, B.; Coronel-Cruz, C.; Hernández-Ochoa, B.; Gómez-Manzo, S.; Cárdenas-Rodríguez, N.; Arreguin-Espinosa, R.; Bandala, C.; Canseco-Ávila, L.M.; Ortega-Cuellar, D. TFEB; Beyond Its Role as an Autophagy and Lysosomes Regulator. Cells 2022, 11, 3153. https://doi.org/10.3390/cells11193153
Franco-Juárez B, Coronel-Cruz C, Hernández-Ochoa B, Gómez-Manzo S, Cárdenas-Rodríguez N, Arreguin-Espinosa R, Bandala C, Canseco-Ávila LM, Ortega-Cuellar D. TFEB; Beyond Its Role as an Autophagy and Lysosomes Regulator. Cells. 2022; 11(19):3153. https://doi.org/10.3390/cells11193153
Chicago/Turabian StyleFranco-Juárez, Berenice, Cristina Coronel-Cruz, Beatriz Hernández-Ochoa, Saúl Gómez-Manzo, Noemi Cárdenas-Rodríguez, Roberto Arreguin-Espinosa, Cindy Bandala, Luis Miguel Canseco-Ávila, and Daniel Ortega-Cuellar. 2022. "TFEB; Beyond Its Role as an Autophagy and Lysosomes Regulator" Cells 11, no. 19: 3153. https://doi.org/10.3390/cells11193153