
Network-Based Analysis to Identify Hub Genes Involved in Spatial Root Response to Mechanical Constrains

Abstract
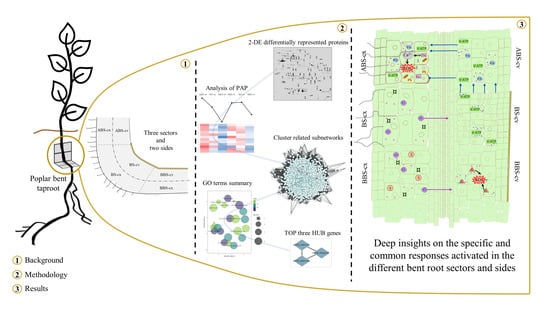
:
1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. K-Means Analysis and Subnetwork Identification
2.3. Gene Set Enrichment Analysis
2.4. Identification of Hub Genes
3. Results
3.1. Cluster Analysis of Protein Abundance Profiles
3.2. Network and Gene Set Enrichment Analysis
3.3. Identification of Subnetwork-Related Hub Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scippa, G.S.; Trupiano, D.; Rocco, M.; Di Iorio, A.; Chiatante, D. Unravelling the response of poplar (Populus nigra) roots to mechanical stress imposed by bending. Plant Biosyst. 2008, 142, 401–413. [Google Scholar] [CrossRef]
- Trupiano, D.; Di Iorio, A.; Montagnoli, A.; Lasserre, B.; Rocco, M.; Grosso, A.; Scaloni, A.; Marra, M.; Chiatante, D.; Scippa, G.S. Involvement of lignin and hormones in the response of woody poplar taproots to mechanical stress. Physiol. Plant. 2012, 146, 39–52. [Google Scholar] [CrossRef] [Green Version]
- Trupiano, D.; Rocco, M.; Renzone, G.; Scaloni, A.; Rossi, M.; Viscosi, V.; Chiatante, D.; Scippa, G.S. Temporal analysis of poplar woody root response to bending stress. Physiol. Plant. 2014, 150, 174–193. [Google Scholar] [CrossRef] [PubMed]
- Trupiano, D.; Rocco, M.; Renzone, G.; Scaloni, A.; Viscosi, V.; Chiatante, D.; Scippa, G.S. The proteome of Populus nigra woody root: Response to bending. Ann. Bot. 2012, 110, 415–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saviano, G.; Paris, D.; Melck, D.; Falasca, A.; Trupiano, D.; Iorizzi, M.; Scippa, G.S.; Motta, A. Monitoring spatial and temporal metabolic dynamics of woody poplar root under mechanical stress conditions by NMR-based metabolomics. Metabolomics 2016, 12, 65. [Google Scholar] [CrossRef]
- Rossi, M.; Trupiano, D.; Tamburro, M.; Ripabelli, G.; Montagnoli, A.; Chiatante, D.; Scippa, G.S. MicroRNAs expression patterns in the response of poplar woody root to bending stress. Planta 2015, 242, 339–351. [Google Scholar] [CrossRef]
- De Zio, E.; Montagnoli, A.; Karady, M.; Terzaghi, M.; Sferra, G.; Antoniadi, I.; Scippa, G.S.; Ljung, K.; Chiatante, D.; Trupiano, D. Reaction Wood Anatomical Traits and Hormonal Profiles in Poplar Bent Stem and Root. Front. Plant Sci. 2020, 11, 590985. [Google Scholar] [CrossRef]
- De Zio, E.; Trupiano, D.; Karady, M.; Antoniadi, I.; Montagnoli, A.; Terzaghi, M.; Chiatante, D.; Ljung, K.; Scippa, G.S. Tissue-specific hormone profiles from woody poplar roots under bending stress. Physiol. Plant. 2019, 165, 101–113. [Google Scholar] [CrossRef] [Green Version]
- De Zio, E.; Trupiano, D.; Montagnoli, A.; Terzaghi, M.; Chiatante, D.; Grosso, A.; Marra, M.; Scaloni, A.; Scippa, G.S. Poplar woody taproot under bending stress: The asymmetric response of the convex and concave sides. Ann. Bot. 2016, 118, 865–883. [Google Scholar] [CrossRef] [Green Version]
- Richter, G.L.; Monshausen, G.B.; Krol, A.; Gilroy, S. Mechanical Stimuli Modulate Lateral Root Organogenesis. Plant Physiol. 2009, 151, 1855–1866. [Google Scholar] [CrossRef]
- Sparke, M.A.; Wünsche, J.N. Mechanosensing of plants. Hortic. Rev. 2020, 47, 43–83. [Google Scholar]
- Monshausen, G.B.; Gilroy, S. Feeling green: Mechanosensing in plants. Trends Cell Biol. 2009, 19, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Senkler, J.; Senkler, M.; Eubel, H.; Hildebrandt, T.; Lengwenus, C.; Schertl, P.; Schwarzländer, M.; Wagner, S.; Wittig, I.; Braun, H.-P. The mitochondrial complexome of Arabidopsis thaliana. Plant J. 2017, 89, 1079–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, N.; Li, W.; Gao, X.; Cha, M.; Qin, L.; Liu, L. Advances in Transcriptomics in the Response to Stress in Plants. Glob. Med. Genet. 2020, 7, 30–34. [Google Scholar] [CrossRef]
- Rives, A.W.; Galitski, T. Modular organization of cellular networks. Proc. Natl. Acad. Sci. USA 2003, 100, 1128–1133. [Google Scholar] [CrossRef] [Green Version]
- Stuart, J.M.; Segal, E.; Koller, D.; Kim, S.K. A gene-coexpression network for global discovery of conserved genetic modules. Science 2003, 302, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Oulas, A.; Minadakis, G.; Zachariou, M.; Sokratous, K.; Bourdakou, M.M.; Spyrou, G.M. Systems Bioinformatics: Increasing precision of computational diagnostics and therapeutics through network-based approaches. Brief. Bioinform. 2019, 20, 806–824. [Google Scholar] [CrossRef]
- Kopecky, D.; Matušíková, I.; Sziderics, A.H.; Trognitz, F.; Spieß, N.; Stierschneider, M.; Fluch, S. In silico search for drought-responsive genes in plants on the basis of scientific data: Case study on poplar roots. Acta Physiol. Plant. 2013, 35, 1955–1966. [Google Scholar] [CrossRef]
- Yoshida, T.; Mogami, J.; Yamaguchi-Shinozaki, K. ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 2014, 21, 133–139. [Google Scholar] [CrossRef]
- Lee, S.; Zhang, C.; Liu, Z.; Klevstig, M.; Mukhopadhyay, B.; Bergentall, M.; Cinar, R.; Ståhlman, M.; Sikanic, N.; Park, J.K.; et al. Network analyses identify liver-specific targets for treating liver diseases. Mol. Syst. Biol. 2017, 13, 938. [Google Scholar] [CrossRef]
- Huang, L.; Injac, S.G.; Cui, K.; Braun, F.; Lin, Q.; Du, Y.; Zhang, H.; Kogiso, M.; Lindsay, H.; Zhao, S.; et al. Systems biology-based drug repositioning identifies digoxin as a potential therapy for groups 3 and 4 medulloblastoma. Sci. Transl. Med. 2018, 10, eaat0150. [Google Scholar] [CrossRef] [PubMed]
- Ashtiani, M.; Salehzadeh-Yazdi, A.; Razaghi-Moghadam, Z.; Hennig, H.; Wolkenhauer, O.; Mirzaie, M.; Jafari, M. A systematic survey of centrality measures for protein-protein interaction networks. BMC Syst. Biol. 2018, 12, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simiele, M.; Sferra, G.; Lebrun, M.; Renzone, G.; Bourgerie, S.; Scippa, G.S.; Morabito, D.; Scaloni, A.; Trupiano, D. In-depth study to decipher mechanisms underlying Arabidopsis thaliana tolerance to metal(loid) soil contamination in association with biochar and/or bacteria. Environ. Exp. Bot. 2021, 182, 104335. [Google Scholar] [CrossRef]
- Qiu, J.; Chen, K.; Zhong, C.; Zhu, S.; Ma, X. Network-based protein-protein interaction prediction method maps perturbations of cancer interactome. PLoS Genet. 2021, 17, e1009869. [Google Scholar] [CrossRef] [PubMed]
- Mihr, C.; Braun, H.-P. Proteomics in Plant Biology. In Handbook of Proteomic Methods; Humana Press: Totowa, NJ, USA, 2003; pp. 409–416. [Google Scholar]
- Bevan, M.; Bancroft, I.; Bent, E.; Love, K.; Goodman, H.; Dean, C.; Bergkamp, R.; Dirkase, W.; Van Staveren, M.; Stiekema, W.; et al. Analysis of 1.9Mb of contiguous sequence from the chromosome 4 of Arabidopsis thaliana. Nature 1998, 391, 485–488. [Google Scholar] [PubMed]
- Fratini, F.; Raggi, C.; Sferra, G.; Birago, C.; Sansone, A.; Grasso, F.; Curra, C.; Olivieri, A.; Pace, T.; Mochi, S.; et al. An integrated approach to explore composition and dynamics of cholesterol-rich membrane microdomains in sexual stages of malaria parasite. Mol. Cell. Proteom. 2017, 16, 1801–1814. [Google Scholar] [CrossRef] [Green Version]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. 2020. Available online: https://cran.microsoft.com/snapshot/2016-11-30/web/packages/factoextra/factoextra.pdf (accessed on 4 December 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Bozhilova, L.V.; Whitmore, A.V.; Wray, J.; Reinert, G.; Deane, C.M. Measuring rank robustness in scored protein interaction networks. BMC Bioinform. 2019, 20, 446. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models. Genome Res. 2003, 13, 2499–2504. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists. Nucleic Acids Res. 2019, 47, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Carbon, S.; Douglass, E.; Dunn, N.; Good, B.; Harris, N.L.; Lewis, S.E.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; et al. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, 330–338. [Google Scholar]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, V.G.; Da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, 480–489. [Google Scholar]
- Boratyn, G.M.; Camacho, C.; Cooper, P.S.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.L.; Matten, W.T.; McGinnis, S.D.; Merezhuk, Y.; et al. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 1, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Sundell, D.; Mannapperuma, C.; Netotea, S.; Delhomme, N.; Lin, Y.-C.; Sjödin, A.; Peer, Y.; Van de Jansson, S.; Hvidsten, T.R.; Street, N.R. The Plant Genome Integrative Explorer Resource: PlantGenIE.org. New Phytol. 2015, 208, 1149–1156. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, M.; Grieneisen, V.A.; Hofhuis, H.; Ten Hove, C.A.; Hogeweg, P.; Marée, A.F.M.; Scheres, B. Root System Architecture from Coupling Cell Shape to Auxin Transport. PLoS Biol. 2008, 6, 307. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Rodríguez, C.; Persson, S. Regulation of cell wall formation by membrane traffic. In Plant Cell Wall Patterning and Cell Shape, 1st ed.; Fukuda, H., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 35–64. [Google Scholar]
- Bassani, M.; Neumann, P.M.; Gepstein, S. Differential expression profiles of growth-related genes in the elongation zone of maize primary roots. Plant Mol. Biol. 2004, 56, 367–380. [Google Scholar] [CrossRef]
- Clark, G.; Cantero-Garcia, A.; Butterfield, T.; Dauwalder, M.; Roux, S.J. Secretion as a key component of gravitropic growth: Implications for annexin involvement in differential growth. Gravit. Space Biol. Bull. 2005, 18, 113–114. [Google Scholar]
- Clark, G.B.; Lee, D.; Dauwalder, M.; Roux, S.J. Immunolocalization and histochemical evidence for the association of two different Arabidopsis annexins with secretion during early seedling growth and development. Planta 2005, 220, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, J.C.; Laohavisit, A.; Macpherson, N.; Webb, A.; Brownlee, C.; Battey, N.H.; Davies, J.M. Annexins: Multifunctional components of growth and adaptation. J. Exp. Bot. 2008, 59, 533–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueoka-Nakanishi, H.; Sazuka, T.; Nakanishi, Y.; Maeshima, M.; Mori, H.; Hisabori, T. Thioredoxin h regulates calcium dependent protein kinases in plasma membranes. FEBS J. 2003, 280, 3220–3231. [Google Scholar] [CrossRef] [PubMed]
- Kerr, E.M.; Fry, S.C. Extracellular cross-linking of xylan and xyloglucan in maize cell-suspension cultures: The role of oxidative phenolic coupling. Planta 2004, 219, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Ditengou, F.A.; Teale, W.D.; Kochersperger, P.; Flittner, K.A.; Kneuper, I.; Van der Graaff, E.; Nziengui, H.; Pinosa, F.; Li, X.; Nitschke, R.; et al. Mechanical induction of lateral root initiation in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2008, 105, 18818–18823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler-Lee, J.; Geisler, M.; Coutinho, P.M.; Segerman, B.; Nishikubo, N.; Takahashi, J.; Aspeborg, H.; Djerbi, S.; Master, E.; Andersoon-Gunneras, S.; et al. Poplar carbohydrate-active enzymes. Gene identification and expression analyses. Plant Physiol. 2006, 140, 946–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Van Nocker, S. Analysis of promoter activity of members of the PECTATE LYASE-LIKE (PLL) gene family in cell separation in Arabidopsis. BMC Plant Biol. 2010, 10, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, F.J.; Herranz, R. Microgravity environment uncouples cell growth and cell proliferation in root meristematic cells: The mediator role of auxin. Plant Signal. Behav. 2010, 5, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.-W.; Miller, N.D.; Dai, C.; Spalding, E.P.; Monshausen, G.B. The receptor-like kinase FERONIA is required for mechanical signal transduction in Arabidopsis seedlings. Curr. Biol. 2014, 24, 1887–1892. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, J.A.; Shirasu, K.; Deng, X.W. The diverse roles of ubiquitin and the 26S proteasome in the life of plants. Nat. Rev. Genet. 2003, 4, 948–958. [Google Scholar] [CrossRef]
- Liu, J.-X.; Howell, S.H. Endoplasmic Reticulum Protein Quality Control and Its Relationship to Environmental Stress Responses in Plants. Plant Cell 2010, 22, 2930–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Shi, Y.; Zhang, X.; Du, H.; Xu, B.; Huang, B. Melatonin suppression of heat-induced leaf senescence involves changes in abscisic acid and cytokinin biosynthesis and signaling pathways in perennial ryegrass (Lolium perenne L.). Environ. Exp. Bot. 2017, 138, 36–45. [Google Scholar] [CrossRef]
- Zhu, J.K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haak, D.C.; Fukao, T.; Grene, R.; Hua, Z.; Ivanov, R.; Perrella, G.; Li, S. Multilevel regulation of abiotic stress responses in plants. Front. Plant Sci. 2017, 8, 1564. [Google Scholar] [CrossRef]
- Waidmann, S.; Kleine-Vehn, J. Asymmetric cytokinin signaling opposes gravitropism in roots. J. Integr. Plant Biol. 2020, 62, 882–886. [Google Scholar] [CrossRef]
- Monshausen, G.B.; Haswell, E.S. A force of nature: Molecular mechanisms of mechanoperception in plants. J. Exp. Bot. 2013, 64, 4663–4680. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, F.P.; Tinturier, E.; Julien, J.-L.; Leblanc-Fournier, N. Between Stress and Response: Function and Localization of Mechanosensitive Ca 2+ Channels in Herbaceous and Perennial Plants. Int. J. Mol. Sci. 2021, 22, 11043. [Google Scholar] [CrossRef]
- Choi, W.-G.; Hilleary, R.; Swanson, S.J.; Kim, S.-H.; Gilroy, S. Rapid, long-distance electrical and calcium signaling in plants. Ann. Rev. Plant Biol. 2016, 67, 287–307. [Google Scholar] [CrossRef]
- Tintuier, E.; Badel, E.; Leblanc-Fournier, N.; Julien, J.L. Stem bending generates electrical response in poplar. Physiol. Plant. 2021, 173, 954–960. [Google Scholar] [CrossRef]
- Lopez, R.; Badel, E.; Peraudeau, S.; Leblanc-Fournier, N.; Beaujard, F.; Julien, J.L.; Cochard, H.; Moulia, B. Tree shoot bending generates hydraulic pressure pulses: A new long-distance signal? J. Exp. Bot. 2014, 65, 1997–2008. [Google Scholar] [CrossRef] [Green Version]
- Chiatante, D.; Montagnoli, A.; Trupiano, D.; Sferra, G.; Bryant, J.; Rost, T.L.; Scippa, G.S. Meristematic connectome: A cellular coordinator of plant responses to environmental signals? Cells 2021, 10, 2544. [Google Scholar] [CrossRef]
- Eljebbawi, A.; Guerrero, Y.d.C.R.; Dunand, C.; Estevez, J.M. Highlighting reactive oxygen species as multitaskers in root development. iScience 2021, 24, 101978. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, F.K.; Rivero, R.M.; Blumwald, E.; Mittler, R. Reactive oxygen species, abiotic stress and stress combination. Plant J. 2017, 90, 856–867. [Google Scholar] [CrossRef]
- Seagull, R.W.; Gunning, B. The plant cytoskeleton. CRC Crit. Rev. Plant Sci. 1989, 8, 131–167. [Google Scholar] [CrossRef]
- Singh, R.; Singh, S.; Parihar, P.; Mishra, R.K.; Tripathi, D.K.; Singh, V.P.; Chauhan, D.K.; Prasad, S.M. Reactive oxygen species (ROS): Beneficial companions of plants’ developmental processes. Front. Plant Sci. 2016, 7, 1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittler, R. ROS Are Good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- Hasanuzzaman, M.; Nahar, K.; Anee, T.I.; Fujita, M. Glutathione in plants: Biosynthesis and physiological role in environmental stress tolerance. Physiol. Mol. Biol. Plants 2017, 23, 249–268. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Hai, G.; Wang, C.; Cao, S.; Xu, W.; Jia, Z.; Yang, C.; Wang, J.P.; Dai, S.; Cheng, Y. Comparative proteomic analysis of Populus trichocarpa early stem from primary to secondary growth. J. Proteom. 2015, 126, 94–108. [Google Scholar] [CrossRef]
- Koprivova, A.; Mugford, S.T.; Kopriva, S. Arabidopsis root growth dependence on glutathione is linked to auxin transport. Plant Cell Rep. 2010, 29, 1157–1167. [Google Scholar] [CrossRef]
- Pasternak, T.; Palme, K.; Paponov, I.A. Glutathione enhances auxin sensitivity in Arabidopsis roots. Biomolecules 2020, 10, 1550. [Google Scholar] [CrossRef] [PubMed]
- Tomaz, T.; Bagard, M.; Pracharoenwattana, I.; Lindén, P.; Lee, C.P.; Carroll, A.J.; Ströher, E.; Smith, S.M.; Gardeström, P.; Millar, A.H. Mitochondrial Malate Dehydrogenase Lowers Leaf Respiration and Alters Photorespiration and Plant Growth in Arabidopsis. Plant Physiol. 2010, 154, 1143–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, M.; Miyake, H. Redox-shuttling between chloroplast and cytosol: Integration of intra-chloroplast and extra-chloroplast metabolism. Curr. Opin. Plant Biol. 2012, 15, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Grauvogel, C.; Brinkmann, H.; Petersen, J. Evolution of the Glucose-6-Phosphate Isomerase: The Plasticity of Primary Metabolism in Photosynthetic Eukaryotes. Mol. Biol. Evol. 2007, 24, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S. Nitrogen assimilation, abiotic stress and glucose 6-phosphate dehydrogenase: The full circle of reductants. Plants 2016, 5, 24. [Google Scholar] [CrossRef]
- Obudulu, O.; Mähler, N.; Skotare, T.; Bygdell, J.; Abreu, I.N.; Ahnlund, M.; Gandla, M.L.; Petterle, A.; Moritz, T.; Hvidsten, T.R.; et al. A multi-omics approach reveals function of Secretory Carrier-Associated Membrane Proteins in wood formation of Populus trees. BMC Genom. 2018, 19, 11. [Google Scholar] [CrossRef]
- Jin, F.; Li, J.; Ding, Q.; Wang, Q.S.; He, X.Q. Proteomic analysis provides insights into changes in the central metabolism of the cambium during dormancy release in poplar. J. Plant Physiol. 2017, 208, 26–39. [Google Scholar] [CrossRef]
- Salleo, S.; Trifil, P.; Esposito, S.; Nardini, A.; Lo Gullo, M.A. Starch-to-sugar conversion in wood parenchyma of field-growing Laurus nobilis plants: A component of the signal pathway for embolism repair? Funct. Plant Biol. 2009, 36, 815–825. [Google Scholar] [CrossRef]
- Secchi, F.; Gilbert, M.E.; Zwieniecki, M.A. Transcriptome Response to Embolism Formation in Stems of Populus trichocarpa Provides Insight into Signaling and the Biology of Refilling. Plant Physiol. 2011, 157, 1419–1429. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bent Root Sectors/Sides | Cluster Size (N° Proteins) | Withinss (Within Cluster Sum of Square) | ||||||
|---|---|---|---|---|---|---|---|---|
| ABS-cx | BS-cx | BBS-cx | ABS-cv | BS-cv | BBS-cv | |||
| Cluster I | 0.7581557 | −0.29911 | −2.06396 | 0.191851 | 0.266578 | −1.21004 | 12 | 36.9506 |
| Cluster II | −0.1136721 | −6.2065 | −4.59749 | 0.250547 | −0.32258 | 0.729957 | 4 | 42.15465 |
| Cluster III | −0.7552579 | 0.793721 | 0.46037 | −0.56888 | 0.069973 | −6.89288 | 11 | 38.7308 |
| Cluster IV | 1.0195613 | −2.84078 | −3.94615 | 0.464451 | 0.864264 | −7.14603 | 3 | 42.71795 |
| Cluster V | 0.4441395 | −1.41837 | −0.12500 | 0.422867 | 0.09185 | −1.34692 | 16 | 65.03039 |
| Cluster VI | −0.3790747 | −0.02729 | 0.300643 | −0.92325 | −0.24844 | −0.05900 | 20 | 137.90456 |
| Cluster | Specific GO-BP | Specific GO-MF | Specific GO-CC |
|---|---|---|---|
| I | GO:0046940, GO:0009141, GO:0009123, GO:0009144, GO:0009199, GO:0009142, GO:0009201, GO:0009058, GO:0006979, GO:1901576, GO:0000461. | GO:0030570, GO:0016837, GO:0008379. | |
| II | GO:0050896, GO:0042221. | GO:0004364, GO:0016765, GO:1900750, GO:0043295, GO:0072341, GO:1901681, GO:0042277, GO:0033218, GO:0015036, GO:0016667, GO:0016740, GO:0005515, GO:0015035. | |
| III | GO:0006086, GO:0006085, GO:0035384, GO:0034033, GO:0006084, GO:0033866, GO:0071616, GO:0034030, GO:0044088, GO:0032889, GO:1901652, GO:0071375, GO:0032869, GO:1901653, GO:0032868, GO:0043434, GO:0044282, GO:0006793, GO:0006796, GO:0016310, GO:1901616, GO:0046174, GO:0046164. | GO:0016624, GO:0051287, GO:0004634, GO:0042132, GO:0016860. | GO:1990204, GO:0070469, GO:0031975, GO:0031967, GO:0005746, GO:0005747, GO:0019866, GO:0098803, GO:0005743, GO:0005740, GO:0031966, GO:0045271, GO:0030964. |
| IV | GO:0051179, GO:0098655, GO:1902600, GO:0006812, GO:0098660, GO:0034220, GO:0098662, GO:0006811, GO:0055085, GO:0006810, GO:0051234. | GO:0046961, GO:0044769, GO:0042625, GO:0009678, GO:0019829, GO:0015078, GO:0042626, GO:0015399, GO:0016887, GO:0022853, GO:0022890, GO:0008324, GO:0022804, GO:0015318, GO:0015075, GO:0022857, GO:0005215, GO:0008553. | GO:0016469, GO:0033176, GO:0033179, GO:0033177, GO:0033180, GO:0005773, GO:0005774, GO:0098588, GO:0031090. |
| V | GO:0010499, GO:0043632, GO:0030163, GO:0043161, GO:0010498, GO:0044265, GO:0006511, GO:0044257, GO:0051603, GO:0019941, GO:2000144, GO:0045899, GO:0060260, GO:0045898, GO:0060261, GO:0043933, GO:0006807, GO:0044238, GO:0071704, GO:0006508, GO:0044267, GO:0019538, GO:1901565, GO:0009057, GO:0044248. | GO:0004298, GO:0070003, GO:0004175, GO:0008233, GO:0036402. | GO:0000502, GO:1905368, GO:0031597, GO:0005838, GO:0019773, GO:0022624, GO:0008541, GO:0005839, GO:0008540, GO:1905369, GO:0140535. |
| VI | GO:0070887, GO:0009060. | GO:0008135, GO:0090079, GO:0045182, GO:0030060, GO:0016615. |
| Cluster | MCC | cytoHubba | UniProt | Blast | PopGenie | |
|---|---|---|---|---|---|---|
| Protein | Gene | |||||
| I | 9.22 × 1013 | POPTR_0013s13220 | N/A | N/A | 60S ribosomal protein L5 | Potri.013G128600 |
| 9.22 × 1013 | POPTR_0014s17230 | Ribosomal_L18_c domain-containing protein | POPTR_014G174000 | 60S ribosomal protein L5 | Potri.014G174000 | |
| 9.22 × 1013 | POPTR_0019s13040 | Ribosomal_L18_c domain-containing protein | POPTR_019G099000 | 60S ribosomal protein L5 | Potri.019G099000 | |
| II | 222,240 | POPTR_0001s14480 | N/A | N/A | Glutathione reductase, chloroplastic isoform X1 | Potri.001G050000 |
| 226,235 | POPTR_0003s12620 | Glutathione peroxidase | N/A | Probable phospholipid hydroperoxide glutathione peroxidase | Potri.003G126100 | |
| 212,160 | POPTR_0003s17670 | Glutathione reductase | POPTR_003G178200 | Glutathione reductase, chloroplastic isoform X1 | Potri.003G178200 | |
| III | 9.22 × 1013 | POPTR_0008s10700 | N/A | N/A | Dihydrolipoyl dehydrogenase 2, chloroplastic isoform X2 | Potri.008G107600 |
| 9.22 × 1013 | POPTR_0010s15200 | Uncharacterized protein | POPTR_010G142100 | Dihydrolipoyl dehydrogenase 2, chloroplastic | Potri.010G142100 | |
| 9.22 × 1013 | POPTR_0010s16120 | N/A | N/A | Dihydrolipoyl dehydrogenase 2, mitochondrial OR lipoamide dehydrogenase | Potri.010G151400 | |
| IV | 9.22 × 1013 | POPTR_0008s00560 | V-ATPase 69 kDa subunit | POPTR_008G005000 | V-type proton ATPase catalytic subunit A | Potri.008G005000 |
| 9.22 × 1013 | POPTR_0017s11530 | V-type proton ATPase subunit | POPTR_017G079200 | V-type proton ATPase subunit d2 | Potri.017G079200 | |
| 9.22 × 1013 | POPTR_0017s11540 | N/A | N/A | V-type proton ATPase subunit d2 | Potri.017G079200 | |
| V | 9.22 × 1013 | POPTR_0006s14260 | Proteasome subunit alpha type | POPTR_006G140400 | Proteasome subunit alpha type-6 | Potri.006G140400 |
| 9.22 × 1013 | POPTR_0008s15530 | Proteasome subunit beta | POPTR_008G155500 | Proteasome subunit beta type-2-A | Potri.008G155500 | |
| 9.22 × 1013 | POPTR_0016s14640 | Proteasome subunit alpha type | POPTR_016G139600 | Proteasome subunit alpha type-6 | Potri.016G139600 | |
| VI | 9.22 × 1013 | POPTR_0002s10420 | Glucose-6-phosphate isomerase | POPTR_002G104000 | Glucose-6-phosphate isomerase 1, chloroplastic | Potri.002G104000 |
| 9.22 × 1013 | POPTR_0005s07990 | N/A | N/A | Uncharacterized protein LOC7477096 | Potri.005G078100 | |
| 9.22 × 1013 | POPTR_0007s11330 | Uncharacterized protein | POPTR_007G040700 | Phosphoglycerate mutase-like protein 4 | Potri.007G040700 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimitrova, A.; Sferra, G.; Scippa, G.S.; Trupiano, D. Network-Based Analysis to Identify Hub Genes Involved in Spatial Root Response to Mechanical Constrains. Cells 2022, 11, 3121. https://doi.org/10.3390/cells11193121
Dimitrova A, Sferra G, Scippa GS, Trupiano D. Network-Based Analysis to Identify Hub Genes Involved in Spatial Root Response to Mechanical Constrains. Cells. 2022; 11(19):3121. https://doi.org/10.3390/cells11193121
Chicago/Turabian StyleDimitrova, Anastazija, Gabriella Sferra, Gabriella Stefania Scippa, and Dalila Trupiano. 2022. "Network-Based Analysis to Identify Hub Genes Involved in Spatial Root Response to Mechanical Constrains" Cells 11, no. 19: 3121. https://doi.org/10.3390/cells11193121