

The Neuroprotective Effects of mGlu1 Receptor Antagonists Are Mediated by an Enhancement of GABAergic Synaptic Transmission via a Presynaptic CB1 Receptor Mechanism
 , , , , and
, , , , and 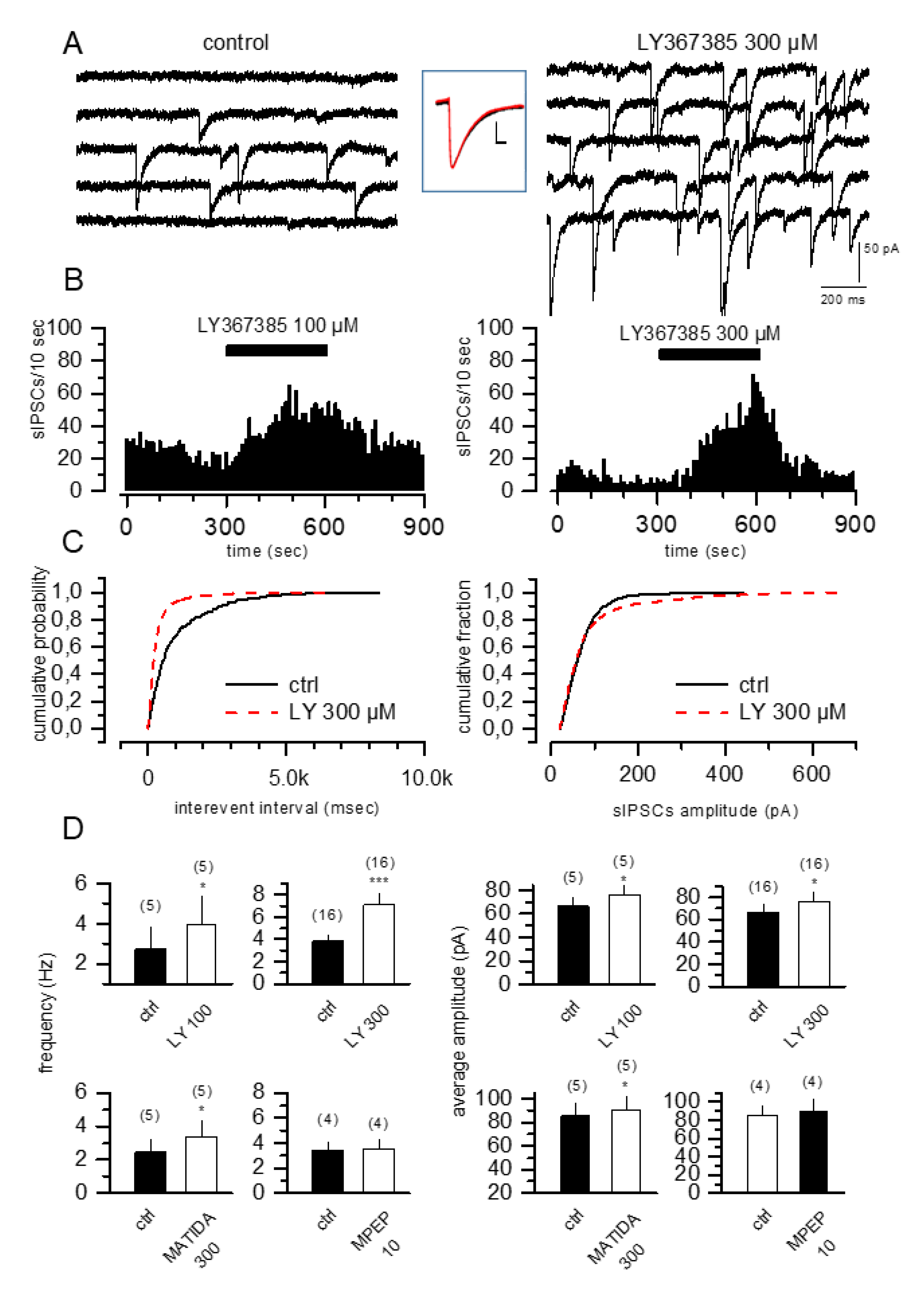{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Electrophysiological sIPSC Recordings from Rat Hippocampal Slices
2.4. Oxygen–Glucose Deprivation (OGD) in Organotypic Hippocampal Cultures
2.5. Immunohistochemistry
2.6. HPLC-Mass Spectrometer Analysis of Anandamide (AEA) and 2-Arachidonoylglicerol (2-AG) in Organotypic Hippocampal Slices
2.7. Microdialysis Studies in Freely Moving Gerbils and HPLC Quantitation of Glutamate and GABA
2.8. Microdialysis Experiments in Gerbils Subjected to Transient Global Ischemia and Assessment of CA1 Pyramidal Cell Injury
2.9. Statistical Analysis
3. Results
3.1. mGlu1 but Not mGlu5 Competitive Antagonists Increase sIPSCs in CA1 Pyramidal Cells
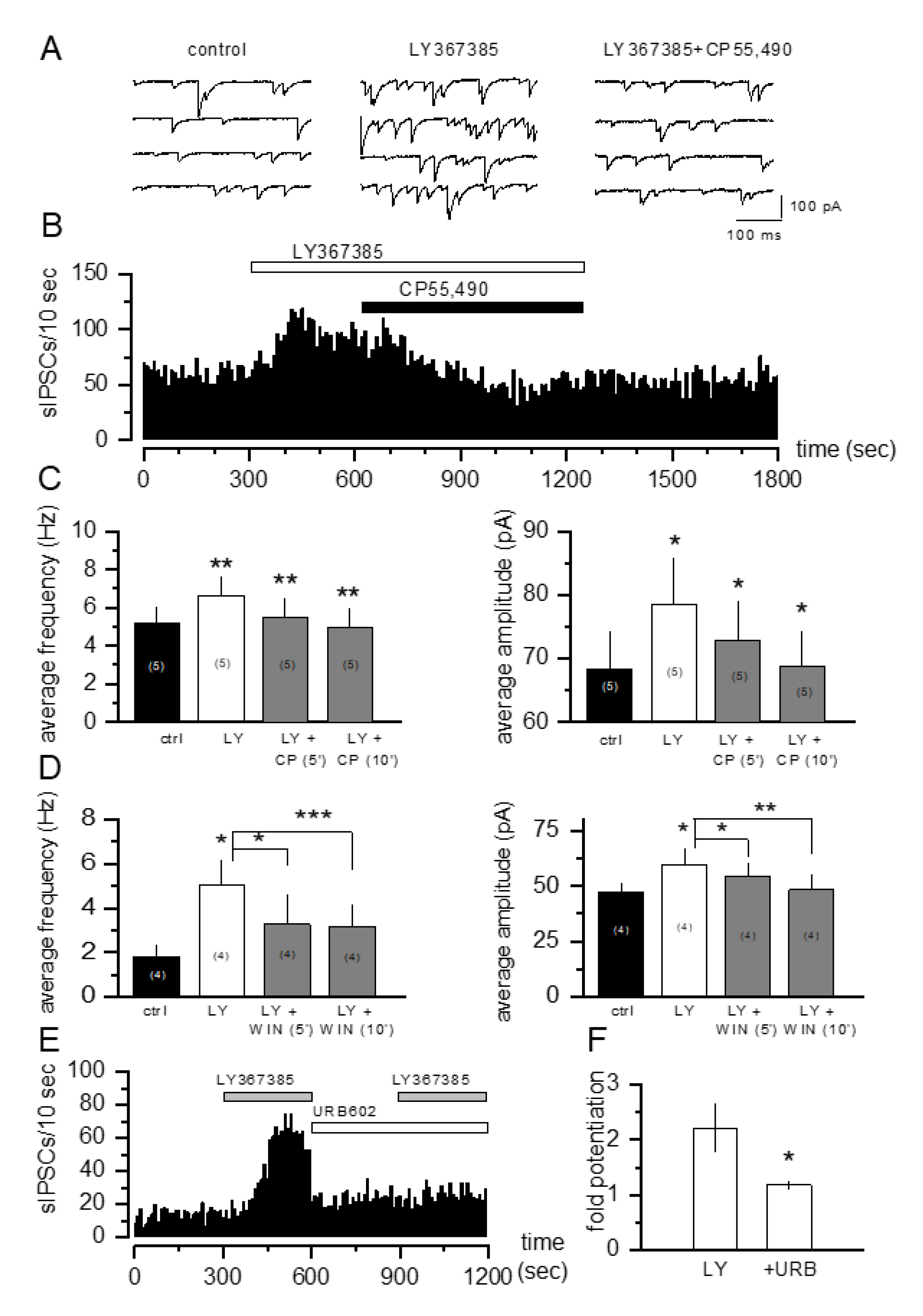
3.2. The Increase in sIPSCs Induced by mGlu1 Receptor Antagonists Is Prevented by CB1 Receptor Activation
3.3. The Neuroprotective Effects of mGlu1 Receptor Antagonists against OGD Toxicity in Organotypic Hippocampal Slices Are Prevented by CB1 Receptor Activation
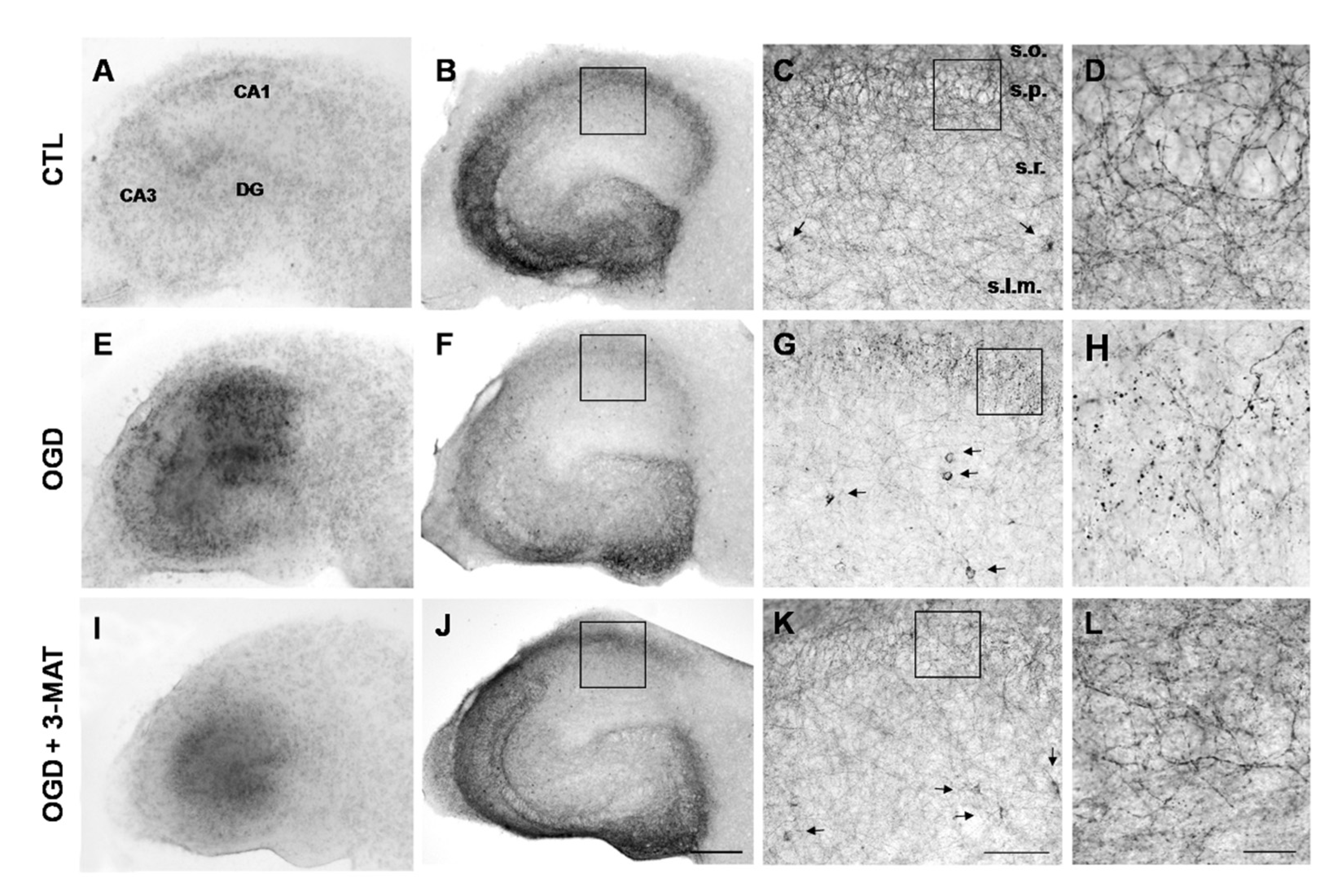
3.4. The Disruption of Cb1-Like Immunoreactivity in Organotypic Hippocampal Slices Exposed to OGD Is Reduced by mGlu1 Receptor Antagonists
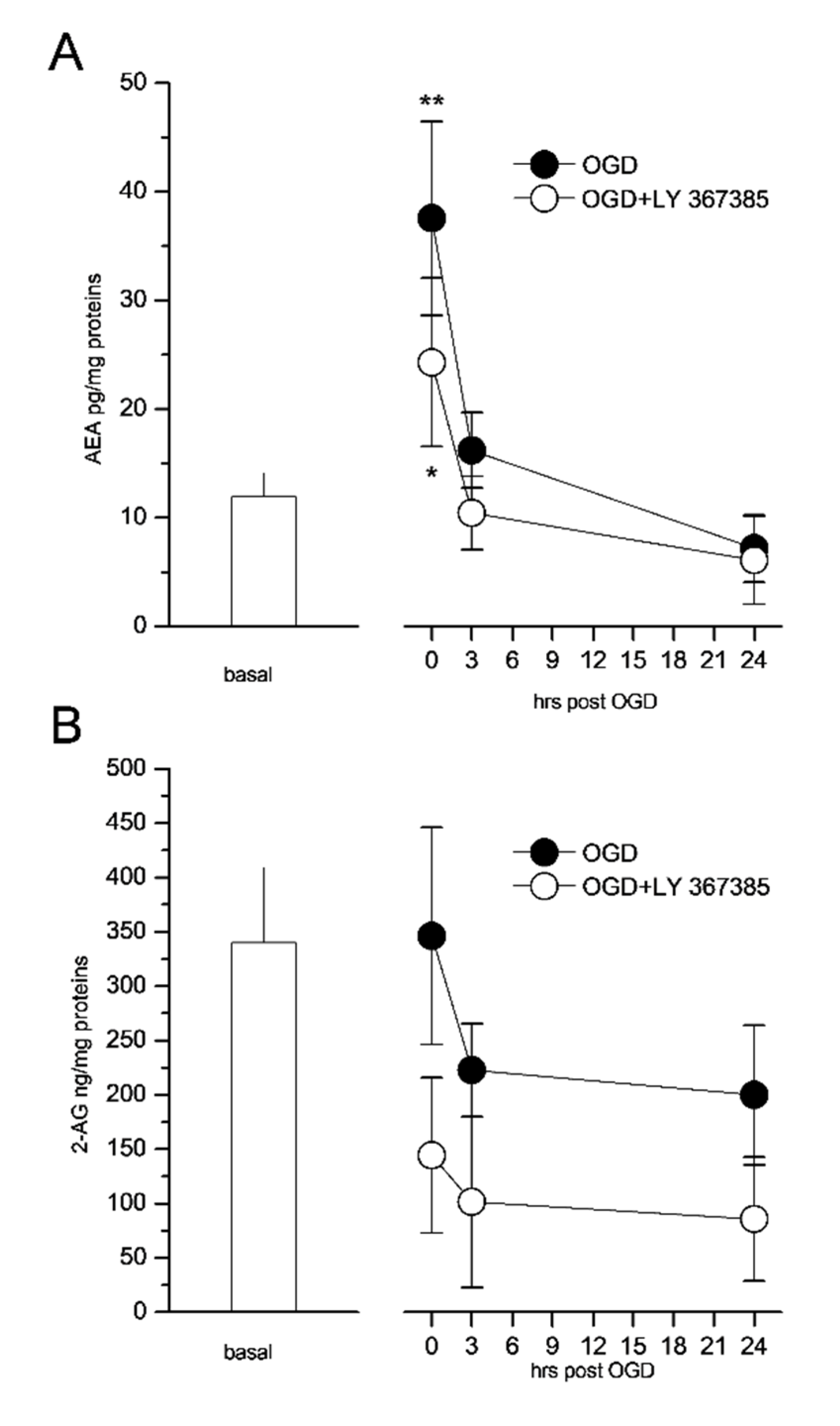
3.5. The mGlu1 Antagonist LY367385 Prevents the Formation of Endocannabinoids in the CA1 Region of Organotypic Hippocampal Slices Exposed to OGD
3.6. The Hippocampal Output of GABA and the Neuroprotective Effects Induced by mGlu1 Receptor Antagonists in Ischemic Gerbils Are Prevented by CB1 Receptor Activation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nakanishi, S. Metabotropic glutamate receptors: Synaptic transmission, modulation, and plasticity. Neuron 1994, 13, 1031–1037. [Google Scholar] [CrossRef]
- Bruno, V.; Copani, A.; Knöpfel, T.; Kuhn, R.; Casabona, G.; Dell’ Albani, P.; Condorelli, D.F.; Nicoletti, F. Activation of metabotropic glutamate receptors coupled to inositol phospholipid hydrolysis amplifies NMDA-induced neuronal degeneration in cultured cortical cells. Neuropharmacology 1995, 34, 1089–1098. [Google Scholar] [CrossRef]
- Bruno, V.; Battaglia, G.; Copani, A.; Cespédes, V.M.; Galindo, M.F.; Ceña, V.; Sánchez-Prieto, J.; Gasparini, F.; Kuhn, R.; Flor, P.J.; et al. An activity-dependent switch from facilitation to inhibition in the control of excitotoxicity by group I metabotropic glutamate receptors. Eur. J. Neurosci. 2001, 13, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini-Giampietro, D.E. The distinct role of mGlu1 receptors in post-ischemic neuronal death. Trends Pharmacol. Sci. 2003, 24, 461–470. [Google Scholar] [CrossRef]
- Pellegrini-Giampietro, D.E.; Cozzi, A.; Peruginelli, F.; Leonardi, P.; Meli, E.; Pellicciari, R.; Moroni, F. 1-Aminoindan-1,5-dicarboxylic acid and (S)-(+)-2-(3′-carboxybicyclo[1.1.1] pentyl)-glycine, two mGlu1 receptor-preferring antagonists, reduce neuronal death in in vitro and in vivo models of cerebral ischaemia. Eur. J. Neurosci. 1999, 11, 3637–3647. [Google Scholar] [CrossRef]
- Cozzi, A.; Meli, E.; Carlà, V.; Pellicciari, R.; Moroni, F.; Pellegrini-Giampietro, D.E. Metabotropic glutamate 1 (mGlu1) receptor antagonists enhance GABAergic neurotransmission: A mechanism for the attenuation of post-ischemic injury and epileptiform activity? Neuropharmacology 2002, 43, 119–130. [Google Scholar] [CrossRef]
- Moroni, F.; Attucci, S.; Cozzi, A.; Meli, E.; Picca, R.; Scheideler, M.A.; Pellicciari, R.; Noe, C.; Sarichelou, I.; Pellegrini-Giampietro, D.E. The novel and systemically active metabotropic glutamate 1 (mGlu1) receptor antagonist 3-MATIDA reduces post-ischemic neuronal death. Neuropharmacology 2002, 42, 741–751. [Google Scholar] [CrossRef]
- Werner, C.G.; Scartabelli, T.; Pancani, T.; Landucci, E.; Moroni, F.; Pellegrini-Giampietro, D.E. Differential role of mGlu1 and mGlu5 receptors in rat hippocampal slice models of ischemic tolerance. Eur. J. Neurosci. 2007, 25, 3597–3604. [Google Scholar] [CrossRef]
- Scartabelli, T.; Gerace, E.; Landucci, E.; Moroni, F.; Pellegrini-Giampietro, D.E. Neuroprotection by group I mGlu receptors in a rat hippocampal slice model of cerebral ischemia is associated with the PI3K–Akt signaling pathway: A novel postconditioning strategy? Neuropharmacology 2008, 55, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Gerace, E.; Zianni, E.; Landucci, E.; Scartabelli, T.; Berlinguer Palmini, R.; Iezzi, D.; Moroni, F.; Di Luca, M.; Mannaioni, G.; Gardoni, F.; et al. Differential mechanisms of tolerance induced by NMDA and 3,5-dihydroxyphenylglycine (DHPG) preconditioning. J. Neurochem. 2020, 155, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, D.; Landucci, E.; Gerace, E.; Lana, D.; Ugolini, F.; Henley, J.M.; Giovannini, M.G.; Pellegrini-Giampietro, D.E. Neuroprotective effects of mGluR5 activation through the PI3K/Akt pathway and the molecular switch of AMPA receptors. Neuropharmacology 2020, 162, 107810. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, G.; Bruno, V.; Pisani, A.; Centonze, D.; Catania, M.V.; Calabresi, P.; Nicoletti, F. Selective blockade of type-1 metabotropic glutamate receptors induces neuroprotection by enhancing gabaergic transmission. Mol. Cell. Neurosci. 2001, 17, 1071–1083. [Google Scholar] [CrossRef]
- Morishita, W.; Kirov, S.A.; Alger, B.E. Evidence for metabotropic glutamate receptor activation in the induction of depolarization-induced suppression of inhibition in hippocampal CA1. J. Neurosci. 1998, 18, 4870–4882. [Google Scholar] [CrossRef] [PubMed]
- Bushell, T.J.; Lee, C.C.; Shigemoto, R.; Miller, R.J. Modulation of synaptic transmission and differential localisation of mGlus in cultured hippocampal autapses. Neuropharmacology 1999, 38, 1553–1567. [Google Scholar] [CrossRef]
- Mannaioni, G.; Marino, M.J.; Valenti, O.; Traynelis, S.F.; Conn, P.J. Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J. Neurosci. 2001, 21, 5925–5934. [Google Scholar] [CrossRef]
- Schwartz, R.D.; Yu, X.; Katzman, M.R.; Hayden-Hixson, D.M.; Perry, J.M. Diazepam, given postischemia, protects selectively vulnerable neurons in the rat hippocampus and striatum. J. Neurosci. 1995, 15, 529–539. [Google Scholar] [CrossRef]
- Schwartz-Bloom, R.D.; Miller, K.A.; Evenson, D.A.; Crain, B.J.; Nadler, J.V. Benzodiazepines protect hippocampal neurons from degeneration after transient cerebral ischemia: An ultrastructural study. Neuroscience 2000, 98, 471–484. [Google Scholar] [CrossRef]
- Alger, B.E. Retrograde signaling in the regulation of synaptic transmission: Focus on endocannabinoids. Prog. Neurobiol. 2002, 68, 247–286. [Google Scholar] [CrossRef]
- Chevaleyre, V.; Takahashi, K.A.; Castillo, P.E. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu. Rev. Neurosci. 2006, 29, 37–76. [Google Scholar] [CrossRef]
- Doherty, J.; Dingledine, R. Functional interactions between cannabinoid and metabotropic glutamate receptors in the central nervous system. Curr. Opin. Pharmacol. 2003, 3, 46–53. [Google Scholar] [CrossRef]
- Freund, T.F.; Katona, I.; Piomelli, D. Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 2003, 83, 1017–1066. [Google Scholar] [CrossRef]
- Kano, M.; Ohno-Shosaku, T.; Maejima, T. Retrograde signaling at central synapses via endogenous cannabinoids. Mol. Psychiatry 2002, 7, 234–235. [Google Scholar] [CrossRef]
- Ohno-Shosaku, T.; Maejima, T.; Kano, M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 2001, 29, 729–738. [Google Scholar] [CrossRef]
- Wilson, R.I.; Nicoll, R.A. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 2001, 410, 588–592. [Google Scholar] [CrossRef]
- Gereau IV, R.W.; Conn, P.J. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J. Neurosci. 1995, 15, 6879–6889. [Google Scholar] [CrossRef]
- Neu, A.; Földy, C.; Soltesz, I. Postsynaptic origin of CB1-dependent tonic inhibition of GABA release at cholecystokinin-positive basket cell to pyramidal cell synapses in the CA1 region of the rat hippocampus. J. Physiol. 2007, 578, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Shosaku, T.; Tsubokawa, H.; Mizushima, I.; Yoneda, N.; Zimmer, A.; Kano, M. Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J. Neurosci. 2002, 22, 3864–3872. [Google Scholar] [CrossRef] [PubMed]
- Varma, N.; Brager, D.H.; Morishita, W.; Lenz, R.A.; London, B.; Alger, B. Presynaptic factors in the regulation of DSI expression in hippocampus. Neuropharmacology 2002, 43, 550–562. [Google Scholar] [CrossRef]
- Chevaleyre, V.; Castillo, P.E. Heterosynaptic LTD of hippocampal GABAergic synapses: A novel role of endocannabinoids in regulating excitability. Neuron 2003, 38, 461–472. [Google Scholar] [CrossRef]
- Chen, K.; Neu, A.; Howard, A.L.; Földy, C.; Echegoyen, J.; Hilgenberg, L.; Smith, M.; Mackie, K.; Soltesz, I. Prevention of plasticity of endocannabinoid signaling inhibits persistent limbic hyperexcitability caused by developmental seizures. J. Neurosci. 2007, 27, 46–58. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.; Lv, X.; Maksymetz, J.; Stansley, B.J.; Ghoshal, A.; Gogliotti, R.G.; Niswender, C.M.; Lindsley, C.W.; Conn, P.J. mGlu 5 positive allosteric modulators facilitate long-term potentiation via disinhibition mediated by mGlu 5-endocannabinoid signaling. ACS Pharmacol. Transl. Sci. 2019, 2, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Landucci, E.; Boscia, F.; Gerace, E.; Scartabelli, T.; Cozzi, A.; Moroni, F.; Mannaioni, G.; Pellegrini-Giampietro, D.E. Chapter 23 Involvement of endocannabinoid signaling in the neuroprotective effects of subtype 1 metabotropic glutamate receptor antagonists in models of cerebral ischemia. Int. Rev. Neurobiol. 2009, 85, 337–350. [Google Scholar] [PubMed]
- Berlinguer-Palmini, R.; Masi, A.; Narducci, R.; Cavone, L.; Maratea, D.; Cozzi, A.; Sili, M.; Moroni, F.; Mannaioni, G. GPR35 activation reduces Ca2+ transients and contributes to the kynurenic acid-dependent reduction of synaptic activity at CA3-CA1 synapses. PLoS ONE 2013, 8, e82180. [Google Scholar] [CrossRef] [PubMed]
- Gerace, E.; Landucci, E.; Scartabelli, T.; Moroni, F.; Pellegrini-Giampietro, D.E. Rat hippocampal slice culture models for the evaluation of neuroprotective agents. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 846, pp. 343–354. ISBN 9781617795350. [Google Scholar]
- Landucci, E.; Mazzantini, C.; Lana, D.; Davolio, P.L.; Giovannini, M.G.; Pellegrini-Giampietro, D.E. Neuroprotective effects of cannabidiol but not Δ9-tetrahydrocannabinol in rat hippocampal slices exposed to oxygen-glucose deprivation: Studies with cannabis extracts and selected cannabinoids. Int. J. Mol. Sci. 2021, 22, 9773. [Google Scholar] [CrossRef]
- Boscia, F.; Ferraguti, F.; Moroni, F.; Annunziato, L.; Pellegrini-Giampietro, D.E. mGlu1alpha receptors are co-expressed with CB1 receptors in a subset of interneurons in the CA1 region of organotypic hippocampal slice cultures and adult rat brain. Neuropharmacology 2008, 55, 428–439. [Google Scholar] [CrossRef]
- Lombardi, G.; Moroni, F. GM1 ganglioside reduces ischemia-induced excitatory amino acid output: A microdialysis study in the gerbil hippocampus. Neurosci. Lett. 1992, 134, 171–174. [Google Scholar] [CrossRef]
- Cozzi, N.V.; Nichols, D.E. 5-HT2A receptor antagonists inhibit potassium-stimulated gamma-aminobutyric acid release in rat frontal cortex. Eur. J. Pharmacol. 1996, 309, 25–31. [Google Scholar] [CrossRef]
- Pellegrini-Giampietro, D.E.; Cherici, G.; Alesiani, M.; Carla, V.; Moroni, F. Excitatory amino acid release and free radical formation may cooperate in the genesis of ischemia-induced neuronal damage. J. Neurosci. 1990, 10, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini-Giampietro, D.E.; Cozzi, A.; Moroni, F. The glycine antagonist and free radical scavenger 7-Cl-thio-kynurenate reduces CA1 ischemic damage in the gerbil. Neuroscience 1994, 63, 701–709. [Google Scholar] [CrossRef]
- Maccarrone, M.; Valverde, O.; Barbaccia, M.L.; Castañé, A.; Maldonado, R.; Ledent, C.; Parmentier, M.; Finazzi-Agrò, A. Age-related changes of anandamide metabolism in CB1 cannabinoid receptor knockout mice: Correlation with behaviour. Eur. J. Neurosci. 2002, 15, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.F.; Lupica, C.R. Mechanisms of cannabinoid inhibition of GABA(A) synaptic transmission in the hippocampus. J. Neurosci. 2000, 20, 2470–2479. [Google Scholar] [CrossRef]
- Hájos, N.; Freund, T.F. Pharmacological separation of cannabinoid sensitive receptors on hippocampal excitatory and inhibitory fibers. Neuropharmacology 2002, 43, 503–510. [Google Scholar] [CrossRef]
- Landucci, E.; Scartabelli, T.; Gerace, E.; Moroni, F.; Pellegrini-Giampietro, D.E. CB1 receptors and post-ischemic brain damage: Studies on the toxic and neuroprotective effects of cannabinoids in rat organotypic hippocampal slices. Neuropharmacology 2011, 60, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Manahan-Vaughan, D.; Herrero, I.; Reymann, K.G.; Sánchez-Prieto, J. Presynaptic group 1 metabotropic glutamate receptors may contribute to the expression of long-term potentiation in the hippocampal CA1 region. Neuroscience 1999, 94, 71–82. [Google Scholar] [CrossRef]
- Moroni, F.; Cozzi, A.; Lombardi, G.; Sourtcheva, S.; Leonardi, P.; Carfì, M.; Pellicciari, R. Presynaptic mGlu1 type receptors potentiate transmitter output in the rat cortex. Eur. J. Pharmacol. 1998, 347, 189–195. [Google Scholar] [CrossRef]
- Gereau IV, R.W.; Conn, P.J. Roles of specific metabotropic glutamate receptor subtypes in regulation of hippocampal CA1 pyramidal cell excitability. J. Neurophysiol. 1995, 74, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.J.; Wittmann, M.; Bradley, S.R.; Hubert, G.W.; Smith, Y.; Conn, P.J. Activation of group I metabotropic glutamate receptors produces a direct excitation and disinhibition of GABAergic projection neurons in the substantia nigra pars reticulata. J. Neurosci. 2001, 21, 7001–7012. [Google Scholar] [CrossRef]
- Choi, S.; Lovinger, D.M. Metabotropic glutamate receptor modulation of voltage-gated Ca2+ channels involves multiple receptor subtypes in cortical neurons. J. Neurosci. 1996, 16, 36–45. [Google Scholar] [CrossRef]
- Fiorillo, C.D.; Williams, J.T. Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Nature 1998, 394, 78–82. [Google Scholar] [CrossRef]
- Romano, C.; Sesma, M.A.; McDonald, C.T.; O’malley, K.; van den Pol, A.N.; Olney, J.W. Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J. Comp. Neurol. 1995, 355, 455–469. [Google Scholar] [CrossRef]
- Hubert, G.W.; Paquet, M.; Smith, Y. Differential subcellular localization of mGluR1a and mGluR5 in the rat and monkey Substantia nigra. J. Neurosci. 2001, 21, 1838–1847. [Google Scholar] [CrossRef]
- Rae, M.G.; Irving, A.J. Both mGluR1 and mGluR5 mediate Ca2+ release and inward currents in hippocampal CA1 pyramidal neurons. Neuropharmacology 2004, 46, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Ferraguti, F.; Conquet, F.; Corti, C.; Grandes, P.; Kuhn, R.; Knopfel, T. Immunohistochemical localization of the mGluR1beta metabotropic glutamate receptor in the adult rodent forebrain: Evidence for a differential distribution of mGluR1 splice variants. J. Comp. Neurol. 1998, 400, 391–407. [Google Scholar] [CrossRef]
- Berthele, A.; Laurie, D.J.; Platzer, S.; Zieglgänsberger, W.; Tölle, T.R.; Sommer, B. Differential expression of rat and human type I metabotropic glutamate receptor splice variant messenger RNAs. Neuroscience 1998, 85, 733–749. [Google Scholar] [CrossRef]
- Leão, R.N.; Mikulovic, S.; Leão, K.E.; Munguba, H.; Gezelius, H.; Enjin, A.; Patra, K.; Eriksson, A.; Loew, L.M.; Tort, A.B.L.; et al. OLM interneurons differentially modulate CA3 and entorhinal inputs to hippocampal CA1 neurons. Nat. Neurosci. 2012, 15, 1524–1530. [Google Scholar] [CrossRef] [PubMed]
- Ferraguti, F.; Cobden, P.; Pollard, M.; Cope, D.; Shigemoto, R.; Watanabe, M.; Somogyi, P. Immunolocalization of metabotropic glutamate receptor 1alpha (mGluR1alpha) in distinct classes of interneuron in the CA1 region of the rat hippocampus. Hippocampus 2004, 14, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Katona, I.; Sperlágh, B.; Sík, A.; Käfalvi, A.; Vizi, E.S.; Mackie, K.; Freund, T.F. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J. Neurosci. 1999, 19, 4544–4558. [Google Scholar] [CrossRef]
- Tsou, K.; Mackie, K.; Sañudo-Peña, M.C.; Walker, J.M. Cannabinoid CB1 receptors are localized primarily on cholecystokinin-containing GABAergic interneurons in the rat hippocampal formation. Neuroscience 1999, 93, 969–975. [Google Scholar] [CrossRef]
- Losonczy, A.; Biró, Á.A.; Nusser, Z. Persistently active cannabinoid receptors mute a subpopulation of hippocampal interneurons. Proc. Natl. Acad. Sci. USA 2004, 101, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini-Giampietro, D.E.; Mannaioni, G.; Bagetta, G. Post-ischemic brain damage: The endocannabinoid system in the mechanisms of neuronal death. FEBS J. 2009, 276, 2–12. [Google Scholar] [CrossRef]
- Navarrete, M.; Araque, A. Endocannabinoids mediate neuron-astrocyte communication. Neuron 2008, 57, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Hájos, N.; Ledent, C.; Freund, T.F. Novel cannabinoid-sensitive receptor mediates inhibition of glutamatergic synaptic transmission in the hippocampus. Neuroscience 2001, 106, 1–4. [Google Scholar] [CrossRef]
- Honoré, E.; Khlaifia, A.; Bosson, A.; Lacaille, J.-C. Hippocampal somatostatin interneurons, long-term synaptic plasticity and memory. Front. Neural Circuits 2021, 15, 687558. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landucci, E.; Berlinguer-Palmini, R.; Baccini, G.; Boscia, F.; Gerace, E.; Mannaioni, G.; Pellegrini-Giampietro, D.E. The Neuroprotective Effects of mGlu1 Receptor Antagonists Are Mediated by an Enhancement of GABAergic Synaptic Transmission via a Presynaptic CB1 Receptor Mechanism. Cells 2022, 11, 3015. https://doi.org/10.3390/cells11193015
Landucci E, Berlinguer-Palmini R, Baccini G, Boscia F, Gerace E, Mannaioni G, Pellegrini-Giampietro DE. The Neuroprotective Effects of mGlu1 Receptor Antagonists Are Mediated by an Enhancement of GABAergic Synaptic Transmission via a Presynaptic CB1 Receptor Mechanism. Cells. 2022; 11(19):3015. https://doi.org/10.3390/cells11193015
Chicago/Turabian StyleLanducci, Elisa, Rolando Berlinguer-Palmini, Gilda Baccini, Francesca Boscia, Elisabetta Gerace, Guido Mannaioni, and Domenico E. Pellegrini-Giampietro. 2022. "The Neuroprotective Effects of mGlu1 Receptor Antagonists Are Mediated by an Enhancement of GABAergic Synaptic Transmission via a Presynaptic CB1 Receptor Mechanism" Cells 11, no. 19: 3015. https://doi.org/10.3390/cells11193015