
Neutrophil Activated by the Famous and Potent PMA (Phorbol Myristate Acetate)

{kind=link}
{kind=link}
Abstract
:1. Introduction
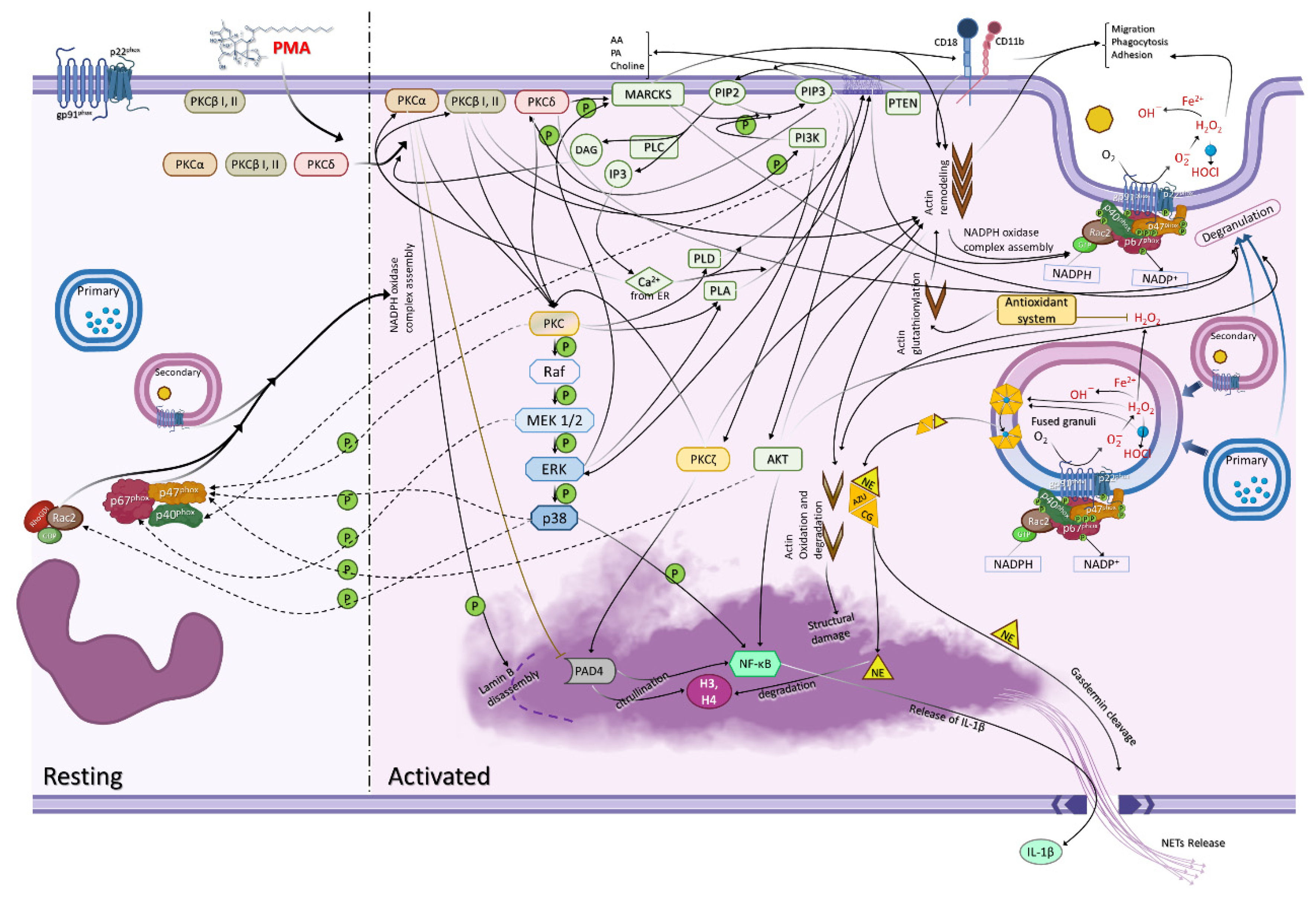
2. Neutrophils Are Activated by PMA
2.1. Protein Kinase C
2.2. Protein Kinase C Isoform Locations and Translocation
2.3. PMA Triggers Degranulation
2.4. Crosstalk between Adhesion/Migration and NADPH Oxidase
2.5. NADPH Oxidase Subunit Phosphorylation and Translocation
3. PI3K Participates in ROS Production
The Relation between Calcium and PMA Activation
4. Neutrophil Extracellular Trap (NET) Formation
4.1. Protein Arginine Deiminase 4 (PAD4)
4.2. Autophagy
4.3. NF-κB
4.4. Gasdermin D
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Requirements for NADPH Oxidase and Myeloperoxidase in Neutrophil Extracellular Trap Formation Differ Depending on the Stimulus. J. Leukoc. Biol. 2012, 92, 841–849. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil Recruitment and Function in Health and Inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Heit, B.; Tavener, S.; Raharjo, E.; Kubes, P. An Intracellular Signaling Hierarchy Determines Direction of Migration in Opposing Chemotactic Gradients. J. Cell Biol. 2002, 159, 91–102. [Google Scholar] [CrossRef]
- Aquino, E.N.; Neves, A.C.D.; Santos, K.C.; Uribe, C.E.; Souza, P.E.N.; Correa, J.R.; Castro, M.S.; Fontes, W. Proteomic Analysis of Neutrophil Priming by PAF. Protein Pept. Lett. 2016, 23, 142–151. [Google Scholar] [CrossRef]
- Gera, N.; Swanson, K.D.; Jin, T. β-Arrestin 1-Dependent Regulation of Rap2 Is Required for FMLP-Stimulated Chemotaxis in Neutrophil-like HL-60 Cells. J. Leukoc. Biol. 2017, 101, 239–251. [Google Scholar] [CrossRef]
- Goel, G.; Makkar, H.P.S.; Francis, G.; Becker, K. Phorbol Esters: Structure, Biological Activity, and Toxicity in Animals. Int. J. Toxicol. 2007, 26, 279–288. [Google Scholar] [CrossRef]
- Pagani, A.; Gaeta, S.; Savchenko, A.I.; Williams, C.M.; Appendino, G. An Improved Preparation of Phorbol from Croton Oil. Beilstein J. Org. Chem. 2017, 13, 1361–1367. [Google Scholar] [CrossRef]
- Steinberg, S.F. Structural Basis of Protein Kinase C Isoform Function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef]
- Fontayne, A.; Dang, P.M.; El Benna, J. Phosphorylation of P47 Phox Sites by PKC R, ΙΙ, δ, and: Effect on Binding to P22 Phox and on NADPH Oxidase Activation. Society 2002, 24, 7743–7750. [Google Scholar]
- Dekker, L.V.; Leitges, M.; Altshuler, G.; Mistry, N.; Mcdermott, A.; Roes, J.; Segal, A.W. Protein Kinase C-β Contributes to NADPH Oxidase Activation in Neutrophils. Biochem. J. 2000, 347, 285–289. [Google Scholar] [CrossRef]
- Brown, G.E.; Stewart, M.Q.; Liu, H.; Ha, V.L.; Yaffe, M.B. A Novel Assay System Implicates PtdIns(3,4)P2, PtdIns(3)P, and PKCδ in Intracellular Production of Reactive Oxygen Species by the NADPH Oxidase. Mol. Cell 2003, 11, 35–47. [Google Scholar] [CrossRef]
- Laudanna, C.; Mochly-Rosen, D.; Liron, T.; Constantin, G.; Butcher, E.C. Evidence of ζ Protein Kinase C Involvement in Polymorphonuclear Neutrophil Integrin-Dependent Adhesion and Chemotaxis. J. Biol. Chem. 1998, 273, 30306–30315. [Google Scholar] [CrossRef] [PubMed]
- Niggli, V.; Djafarzadeh, S.; Keller, H. Stimulus-Induced Selective Association of Actin-Associated Proteins (α-Actinin) and Protein Kinase C Isoforms with the Cytoskeleton of Human Neutrophils. Exp. Cell Res. 1999, 250, 558–568. [Google Scholar] [CrossRef]
- Dang, P.M.C.; Rais, S.; Hakim, J.; Périanin, A. Redistribution of Protein Kinase C Isoforms in Human Neutrophils Stimulated by Formyl Peptides and Phorbol Myristate Acetate. Biochem. Biophys. Res. Commun. 1995, 212, 664–672. [Google Scholar] [CrossRef]
- Hattori, H.; Subramanian, K.K.; Sakai, J.; Jia, Y.; Li, Y.; Porter, T.F.; Loison, F.; Sarraj, B.; Kasorn, A.; Jo, H.; et al. Small-Molecule Screen Identifies Reactive Oxygen Species as Key Regulators of Neutrophil Chemotaxis. Proc. Natl. Acad. Sci. USA 2010, 107, 3546. [Google Scholar] [CrossRef]
- Sakai, J.; Li, J.; Subramanian, K.K.; Mondal, S.; Bajrami, B.; Hattori, H.; Jia, Y.; Dickinson, B.C.; Zhong, J.; Ye, K.; et al. Reactive Oxygen Species-Induced Actin Glutathionylation Controls Actin Dynamics in Neutrophils. Immunity 2012, 37, 1037–1049. [Google Scholar] [CrossRef]
- Fontes, W.; Sousa, M.V.; Aragão, J.B.; Morhy, L. Determination of the Amino Acid Sequence of the Plant Cytolysin Enterolobin. Arch. Biochem. Biophys. 1997, 347, 201–207. [Google Scholar] [CrossRef]
- Belambri, S.A.; Rolas, L.; Raad, H.; Hurtado-Nedelec, M.; Dang, P.M.C.; El-Benna, J. NADPH Oxidase Activation in Neutrophils: Role of the Phosphorylation of Its Subunits. Eur. J. Clin. Investig. 2018, 48, e12951. [Google Scholar] [CrossRef]
- Bertram, A.; Ley, K. Protein Kinase C Isoforms in Neutrophil Adhesion and Activation. Arch. Immunol. Ther. Exp. 2011, 59, 79–87. [Google Scholar] [CrossRef]
- Sheats, M.K.; Sung, E.J.; Adler, K.B.; Jones, S.L. In Vitro Neutrophil Migration Requires Protein Kinase C-Delta (δ-PKC)-Mediated Myristoylated Alanine-Rich C-Kinase Substrate (MARCKS) Phosphorylation. Inflammation 2015, 38, 1126–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiansen, N.O.; Borregaard, N. Translocation of Protein Kinase C to Subcellular Fractions of Human Neutrophils. Scand. J. Immunol. 1989, 29, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Nixon, J.B.; McPhail, L.C. Protein Kinase C (PKC) Isoforms Translocate to Triton-Insoluble Fractions in Stimulated Human Neutrophils: Correlation of Conventional PKC with Activation of NADPH Oxidase. J. Immunol. 1999, 163, 4574–4582. [Google Scholar]
- Dang, P.M.C.; Hakim, J.; Périanin, A. Immunochemical Identification and Translocation of Protein Kinase C Zeta in Human Neutrophils. FEBS Lett. 1994, 349, 338–342. [Google Scholar] [CrossRef]
- Dang, P.M.-C.; Fontayne, A.; Hakim, J.; El Benna, J.; Périanin, A. Protein Kinase C ζ Phosphorylates a Subset of Selective Sites of the NADPH Oxidase Component P47 Phox and Participates in Formyl Peptide-Mediated Neutrophil Respiratory Burst. J. Immunol. 2001, 166, 1206–1213. [Google Scholar] [CrossRef]
- Kato, M.; Yamaguchi, T.; Tachibana, A.; Kimura, H. Differential Role of an Atypical Protein Kinase C, PKC ζ, in Regulation of Human Eosinophil and Neutrophil Functions. Int. Arch. Allergy Immunol. 2005, 137, 27–34. [Google Scholar] [CrossRef]
- Eckert, R.E.; Neuder, L.E.; Park, J.; Adler, K.B.; Jones, S.L. Myristoylated Alanine-Rich C-Kinase Substrate (MARCKS) Protein Regulation of Human Neutrophil Migration. Am. J. Respir. Cell Mol. Biol. 2010, 42, 586–594. [Google Scholar] [CrossRef]
- Rorvig, S.; Ostergaard, O.; Heegaard, N.H.H.; Borregaard, N. Proteome Profiling of Human Neutrophil Granule Subsets, Secretory Vesicles, and Cell Membrane: Correlation with Transcriptome Profiling of Neutrophil Precursors. J. Leukoc. Biol. 2013, 94, 711–721. [Google Scholar] [CrossRef]
- De Castro, M.S.; Fontes, W.; Morhy, L.; Bloch, C., Jr. Complete Amino Acid Sequences of Two γ-Thionins from Maize (Zea Mays L.) Seeds. Protein Pept. Lett. 1996, 3, 267–274. [Google Scholar] [CrossRef]
- Lominadze, G.; Powell, D.W.; Luerman, G.C.; Link, A.J.; Ward, R.A.; McLeish, K.R. Proteomic Analysis of Human Neutrophil Granules. Mol. Cell. Proteom. 2005, 4, 1503–1521. [Google Scholar] [CrossRef]
- Rorvig, S.; Honore, C.; Larsson, L.-I.; Ohlsson, S.; Pedersen, C.C.; Jacobsen, L.C.; Cowland, J.B.; Garred, P.; Borregaard, N. Ficolin-1 Is Present in a Highly Mobilizable Subset of Human Neutrophil Granules and Associates with the Cell Surface after Stimulation with FMLP. J. Leukoc. Biol. 2009, 86, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N.; Kjeldsen, L.; Rygaard, K.; Bastholm, L.; Nielsen, M.H.; Sengelev, H.; Bjerrum, O.W.; Johnsent, A.H. Stimulus-Dependent Secretion of Plasma Proteins from Human Neutrophils. J. Clin. Investig. 1992, 90, 86–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, A.; Nixon, J.B.; McPhail, L.C. Phorbol Myristate Acetate Induces Neutrophil NADPH-Oxidase Activity by Two Separate Signal Transduction Pathways: Dependent or Independent of Phosphatidylinositol 3-Kinase. J. Leukoc. Biol. 2000, 67, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Degroote, R.L.; Weigand, M.; Hauck, S.M.; Deeg, C.A. IL8 and PMA Trigger the Regulation of Different Biological Processes in Granulocyte Activation. Front. Immunol. 2020, 10, 3064. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.-H.; Choi, D.-S.; Zhang, H.; Mu, D.; Mcmahon, T.; Kharazia, V.N.; Lowell, C.A.; Ferriero, D.M.; Messing, R.O. Neutrophil Protein Kinase Cδ as a Mediator of Stroke-Reperfusion Injury. J. Clin. Investig. 2004, 114, 49–56. [Google Scholar] [CrossRef]
- Takashi, S.; Park, J.; Fang, S.; Koyama, S.; Parikh, I.; Adler, K.B. A Peptide against the N-Terminus of Myristoylated Alanme-Rich C Kinase Substrate Inhibits Degranulation of Human Leukocytes in Vitro. Am. J. Respir. Cell Mol. Biol. 2006, 34, 647–652. [Google Scholar] [CrossRef]
- Mócsai, A.; Ligeti, E.; Lowell, C.A.; Berton, G. Adhesion-Dependent Degranulation of Neutrophils Requires the Src Family Kinases Fgr and Hck. J. Immunol. 1999, 162, 1120–1126. [Google Scholar]
- Chen, J.; Tang, H.; Hay, N.; Xu, J.; Ye, R.D. Akt Isoforms Differentially Regulate Neutrophil Functions. Blood 2010, 115, 4237–4246. [Google Scholar] [CrossRef]
- Abdel-Latif, D.; Steward, M.; Lacy, P. Neutrophil Primary Granule Release and Maximal Superoxide Generation Depend on Rac2 in a Common Signalling Pathway. Can. J. Physiol. Pharmacol. 2005, 83, 69–75. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Rochael, N.C.; Guimarães-Costa, A.B.; De Souza-Vieira, T.S.; Ganilho, J.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. A Metabolic Shift toward Pentose Phosphate Pathway Is Necessary for Amyloid Fibril- and Phorbol 12-Myristate 13-Acetate-Induced Neutrophil Extracellular Trap (NET) Formation. J. Biol. Chem. 2015, 290, 22174–22183. [Google Scholar] [CrossRef]
- Ambruso, D.R.; Cusack, N.; Thurman, G. NADPH Oxidase Activity of Neutrophil Specific Granules: Requirements for Cytosolic Components and Evidence of Assembly during Cell Activation. Mol. Genet. Metab. 2004, 81, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, C.; Karlsson, A.; Bylund, J. Intracellular Neutrophil Oxidants: From Laboratory Curiosity to Clinical Reality. J. Immunol. 2019, 202, 3127–3134. [Google Scholar] [CrossRef] [PubMed]
- Björnsdottir, H.; Welin, A.; Dahlgren, C.; Karlsson, A.; Bylund, J. Quantification of Heterotypic Granule Fusion in Human Neutrophils by Imaging Flow Cytometry. Data Br. 2016, 6, 386–393. [Google Scholar] [CrossRef]
- Borregaard, N.; Heiple, J.M.; Simons, E.R.; Clark, R.A. Subcellular Localization of the B-Cytochrome Component of the Human Neutrophil Microbicidal Oxidase: Translocation during Activation. J. Cell Biol. 1983, 97, 52–61. [Google Scholar] [CrossRef]
- Clark, R.A.; Volpp, B.D.; Leidal, K.G.; Nauseef, W.M. Two Cytosolic Components of the Human Neutrophil Respiratory Burst Oxidase Translocate to the Plasma Membrane during Cell Activation. J. Clin. Investig. 1990, 85, 714–721. [Google Scholar] [CrossRef]
- Shaw, S.K.; Ma, S.; Kim, M.B.; Rao, R.M.; Hartman, C.U.; Froio, R.M.; Yang, L.; Jones, T.; Liu, Y.; Nusrat, A.; et al. Coordinated Redistribution of Leukocyte LFA-1 and Endothelial Cell ICAM-1 Accompany Neutrophil Transmigration. J. Exp. Med. 2004, 200, 1571–1580. [Google Scholar] [CrossRef]
- Barnard, J.W.; Biro, M.G.; Lo, S.K.; Ohno, S.; Carozza, M.A.; Moyle, M.; Soule, H.R.; Malik, A.B. Neutrophil Inhibitory Factor Prevents Neutrophil-Dependent Lung Injury. J. Immunol. 1995, 155, 4876–4881. [Google Scholar]
- Campos, V.; Melo, R.C.N.; Silva, L.P.; Aquino, E.N.; Castro, M.S.; Fontes, W. Characterization of Neutrophil Adhesion to Different Titanium Surfaces. Bull. Mater. Sci. 2014, 37, 157–166. [Google Scholar] [CrossRef]
- Videm, V.; Strand, E. Changes in Neutrophil Surface-Receptor Expression after Stimulation with FMLP, Endotoxin, Interleukin-8 and Activated Complement Compared to Degranulation. Scand. J. Immunol. 2004, 59, 25–33. [Google Scholar] [CrossRef]
- Peng, S.; Chen, S.B.; Li, L.D.; Tong, C.F.; Li, N.; Lü, S.Q.; Long, M. Impact of Real-Time Shedding on Binding Kinetics of Membrane-Remaining l-Selectin to PSGL-1. Am. J. Physiol.-Cell Physiol. 2019, 316, C678–C689. [Google Scholar] [CrossRef]
- Alam, C.A.S.; Seed, M.P.; Freemantle, C.; Brown, J.; Perretti, M.; Carrier, M.; Divwedi, A.; West, D.C.; Gustafson, S.; Colville-Nash, P.R.; et al. The Inhibition of Neutrophil-Endothelial Cell Adhesion by Hyaluronan Independent of CD44. Inflammopharmacology 2005, 12, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Davenpeck, K.L.; Brummet, M.E.; Hudson, S.A.; Mayer, R.J.; Bochner, B.S. Activation of Human Leukocytes Reduces Surface P-Selectin Glycoprotein Ligand-1 (PSGL-1, CD162) and Adhesion to P-Selectin In Vitro. J. Immunol. 2000, 165, 2764–2772. [Google Scholar] [CrossRef]
- Lofgren, R.; Ng-Sikorski, J.; Sjolander, A.; Andersson, T. Β2 Integrin Engagement Triggers Actin Polymerization and Phosphatidylinositol Trisphosphate Formation in Non-Adherent Human Neutrophils. J. Cell Biol. 1993, 123, 1597–1605. [Google Scholar] [CrossRef]
- Bengtsson, T.; Orselius, K.; Wetterö, J. Role of the Actin Cytoskeleton during Respiratory Burst in Chemoattractant-Stimulated Neutrophils. Cell Biol. Int. 2006, 30, 154–163. [Google Scholar] [CrossRef]
- Shen, Y.C.; Chen, C.F.; Wang, S.Y.; Sung, Y.J. Impediment to Calcium Influx and Reactive Oxygen Production Accounts for the Inhibition of Neutrophil Mac-1 up-Regulation and Adhesion by Tetrandrine. Mol. Pharmacol. 1999, 55, 186–193. [Google Scholar] [CrossRef]
- Matsumoto, M.; Shigeta, A.; Miyasaka, M.; Hirata, T. CD43 Plays Both Antiadhesive and Proadhesive Roles in Neutrophil Rolling in a Context-Dependent Manner. J. Immunol. 2008, 181, 3628–3635. [Google Scholar] [CrossRef] [PubMed]
- Rieu, P.; Porteu, F.; Bessou, G.; Lesavre, P.; Halbwachs-Mecarelli, L. Human Neutrophils Release Their Major Membrane Sialoprotein, Leukosialin (CD43), during Cell Activation. Eur. J. Immunol. 1992, 22, 3021–3026. [Google Scholar] [CrossRef]
- Remold-O’Donnell, E.; Parent, D. Downregulation of Neutrophil CD43 by Opsonized Zymosan. Blood 1995, 85, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Mambole, A.; Baruch, D.; Nusbaum, P.; Bigot, S.; Suzuki, M.; Lesavre, P.; Fukuda, M.; Halbwachs-Mecarelli, L. The Cleavage of Neutrophil Leukosialin (CD43) by Cathepsin G Releases Its Extracellular Domain and Triggers Its Intramembrane Proteolysis by Presenilin/Gamma-Secretase. J. Biol. Chem. 2008, 283, 23627–23635. [Google Scholar] [CrossRef]
- Sheppard, F.R.; Kelher, M.R.; Moore, E.E.; McLaughlin, N.J.D.; Banerjee, A.; Silliman, C.C. Structural Organization of the Neutrophil NADPH Oxidase: Phosphorylation and Translocation during Priming and Activation. J. Leukoc. Biol. 2005, 78, 1025–1042. [Google Scholar] [CrossRef]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef] [PubMed]
- Grogan, A.; Reeves, E.; Keep, N.; Wientjes, F.; Totty, N.F.; Burlingame, A.L.; Justin Hsuan, J.; Segal, A.W. Cytosolic Phox Proteins Interact with and Regulate the Assembly of Coronin in Neutrophils. J. Cell Sci. 1997, 110, 3071–3081. [Google Scholar] [CrossRef] [PubMed]
- Morimatsu, T.; Kawagoshi, A.; Yoshida, K.; Tamura, M. Actin Enhances the Activation of Human Neutrophil NADPH Oxidase in a Cell-Free System. Biochem. Biophys. Res. Commun. 1997, 230, 206–210. [Google Scholar] [CrossRef] [PubMed]
- El Benna, J.; Dang, P.M.C.; Andrieu, V.; Vergnaud, S.; Dewas, C.; Cachia, O.; Fay, M.; Morel, F.; Chollet-Martin, S.; Hakim, J.; et al. P40(Phox) Associates with the Neutrophil Triton X-100-Insoluble Cytoskeletal Fraction and PMA-Activated Membrane Skeleton: A Comparative Study with P67(Phox) and P47(Phox). J. Leukoc. Biol. 1999, 66, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Hattori, H.; Subramanian, K.K.; Sakai, J.; Luo, H.R. Reactive Oxygen Species as Signaling Molecules in Neutrophil Chemotaxis. Commun. Integr. Biol. 2010, 3, 278. [Google Scholar] [CrossRef]
- Keller, H.U.; Niggli, V. The PKC-Inhibitor RO 31-8220 Selectively Suppresses PMA- and Diacylglycerol-Induced Fluid Pinocytosis and Actin Polymerization in PMNS. Biochem. Biophys. Res. Commun. 1993, 194, 1111–1116. [Google Scholar] [CrossRef]
- Downey, G.P.; Chan, C.K.; Lea, P.; Takai, A.; Grinstein, S. Phorbol Ester-Induced Actin Assembly in Neutrophils: Role of Protein Kinase C. J. Cell Biol. 1992, 116, 695–706. [Google Scholar] [CrossRef]
- Hendey, B.; Zhu, C.L.; Greenstein, S. Fas Activation Opposes PMA-Stimulated Changes in the Localization of PKCδ: A Mechanism for Reducing Neutrophil Adhesion to Endothelial Cells. J. Leukoc. Biol. 2002, 71, 863–870. [Google Scholar] [CrossRef]
- Belambri, S.A.; Hurtado-Nedelec, M.; Senator, A.; Makni-Maalej, K.; Fay, M.; Gougerot-Pocidalo, M.-A.; Marie, J.-C.; Dang, P.M.-C.; El-Benna, J. Phosphorylation of P47phox Is Required for Receptor-Mediated NADPH Oxidase/NOX2 Activation in Epstein-Barr Virus-Transformed Human B Lymphocytes. Am. J. Blood Res. 2012, 2, 187–193. [Google Scholar]
- Bouin, A.P.; Grandvaux, N.; Vignais, P.V.; Fuchs, A. P40(Phox) Is Phosphorylated on Threonine 154 and Serine 315 during Activation of the Phagocyte NADPH Oxidase: Implication of a Protein Kinase C- Type Kinase in the Phosphorylation Process. J. Biol. Chem. 1998, 273, 30097–30103. [Google Scholar] [CrossRef]
- Dang, P.M.C.; Morel, F.; Gougerot-Pocidalo, M.A.; El Benna, J. Phosphorylation of the NADPH Oxidase Component P67PHOX by ERK2 and P38MAPK: Selectivity of Phosphorylated Sites and Existence of an Intramolecular Regulatory Domain in the Tetratricopeptide-Rich Region. Biochemistry 2003, 42, 4520–4526. [Google Scholar] [CrossRef] [PubMed]
- Raad, H.; Paclet, M.H.; Boussetta, T.; Kroviarski, Y.; Morel, F.; Quinn, M.T.; Gougerot-Pocidalo, M.A.; Dang, P.M.C.; El-Benna, J. Regulation of the Phagocyte NADPH Oxidase Activity: Phosphorylation of Gp91phox/NOX2 by Protein Kinase C Enhances Its Diaphorase Activity and Binding to Rac2, P67phox, and P47phox. FASEB J. 2009, 23, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Regier, D.S.; Greene, D.G.; Sergeant, S.; Jesaitis, A.J.; McPhail, L.C. Phosphorylation of P22(Phox) Is Mediated by Phospholipase D-Dependent and -Independent Mechanisms. Correlation of NADPH Oxidase Activity and P22(Phox) Phosphorylation. J. Biol. Chem. 2000, 275, 28406–28412. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Rossi, M.W.; Fujiki, T.; Phillips, W.A.; Disa, S.; Queen, C.F.; Johnston, R.B., Jr.; Rosen, O.M.; Corkey, B.E.; Korchak, H. Protein Kinase C Isotypes and Signaling in Neutrophils. J. Biol. Chem. 1991, 266, 9285–9294. [Google Scholar] [CrossRef]
- Kelher, M.R.; McLaughlin, N.J.D.; Banerjee, A.; Elzi, D.J.; Gamboni, F.; Khan, S.Y.; Meng, X.; Mitra, S.; Silliman, C.C. LysoPCs Induce Hck- and PKCδ-Mediated Activation of PKCγ Causing P47 Phox Phosphorylation and Membrane Translocation in Neutrophils. J. Leukoc. Biol. 2016, 101, 261–273. [Google Scholar] [CrossRef]
- DeCoursey, T.E.; Ligeti, E. Regulation and Termination of NADPH Oxidase Activity. Cell. Mol. Life Sci. 2005, 62, 2173–2193. [Google Scholar] [CrossRef]
- Dusi, S.; Della Bianca, V.; Grzeskowiak, M.; Rossi, F. Relationship between Phosphorylation and Translocation to the Plasma Membrane of P47phox and P67phox and Activation of the NADPH Oxidase in Normal and Ca(2+)-Depleted Human Neutrophils. Biochem. J. 1993, 290, 173–178. [Google Scholar] [CrossRef]
- Stolk, J.; Hiltermann, T.J.; Dijkman, J.H.; Verhoeven, A.J. Characteristics of the Inhibition of NADPH Oxidase Activation in Neutrophils by Apocynin, a Methoxy-Substituted Catechol. Am. J. Respir. Cell Mol. Biol. 1994, 11, 95–102. [Google Scholar] [CrossRef]
- Soroush, F.; Tang, Y.; Guglielmo, K.; Engelmann, A.; Liverani, E.; Patel, A.; Langston, J.; Sun, S.; Kunapuli, S.; Kiani, M.F.; et al. Protein Kinase C-Delta (PKCδ) Tyrosine Phosphorylation Is a Critical Regulator of Neutrophil-Endothelial Cell Interaction in Inflammation. Shock 2019, 51, 538–547. [Google Scholar] [CrossRef]
- Rabani, R.; Cossette, C.; Graham, F.; Powell, W.S. Protein Kinase C Activates NAD Kinase in Human Neutrophils. Free Radic. Biol. Med. 2020, 161, 50–59. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus Aureus Florian. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.B.H.; Kuiper, J.W.P.; Katchky, A.; Goldberg, H.; Glogauer, M. Rac2 Is Required for the Formation of Neutrophil Extracellular Traps. J. Leukoc. Biol. 2011, 90, 771–776. [Google Scholar] [CrossRef]
- de Bont, C.M.; Koopman, W.J.H.; Boelens, W.C.; Pruijn, G.J.M. Stimulus-Dependent Chromatin Dynamics, Citrullination, Calcium Signalling and ROS Production during NET Formation. Biochim. Biophys. Acta-Mol. Cell Res. 2018, 1865, 1621–1629. [Google Scholar] [CrossRef]
- Tatsiy, O.; McDonald, P.P. Physiological Stimuli Induce PAD4-Dependent, ROS-Independent NETosis, with Early and Late Events Controlled by Discrete Signaling Pathways. Front. Immunol. 2018, 9, 2036. [Google Scholar] [CrossRef] [PubMed]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-Activated Protein Kinase: Conservation of a Three-Kinase Module From Yeast to Human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Liu, H.T. MAPK Signal Pathways in the Regulation of Cell Proliferation in Mammalian Cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Chiang, C.H.; Chuang, C.H.; Liu, S.L. Apocynin Attenuates Ischemia-Reperfusion Lung Injury in an Isolated and Perfused Rat Lung Model. Transl. Res. 2011, 158, 17–29. [Google Scholar] [CrossRef]
- Mouzaoui, S.; Djerdjouri, B.; Makhezer, N.; Kroviarski, Y.; El-Benna, J.; Dang, P.M.C. Tumor Necrosis Factor-α-Induced Colitis Increases NADPH Oxidase 1 Expression, Oxidative Stress, and Neutrophil Recruitment in the Colon: Preventive Effect of Apocynin. Mediat. Inflamm. 2014, 2014, 312484. [Google Scholar] [CrossRef]
- Arumugam, S.; Girish Subbiah, K.; Kemparaju, K.; Thirunavukkarasu, C. Neutrophil Extracellular Traps in Acrolein Promoted Hepatic Ischemia Reperfusion Injury: Therapeutic Potential of NOX2 and P38MAPK Inhibitors. J. Cell. Physiol. 2017, 233, 3244–3261. [Google Scholar] [CrossRef]
- Dang, P.M.C.; Raad, H.; Derkawi, R.A.; Boussetta, T.; Paclet, M.H.; Belambri, S.A.; Makni-Maalej, K.; Kroviarski, Y.; Morel, F.; Gougerot-Pocidalo, M.A.; et al. The NADPH Oxidase Cytosolic Component P67phox Is Constitutively Phosphorylated in Human Neutrophils: Regulation by a Protein Tyrosine Kinase, MEK1/2 and Phosphatases 1/2A. Biochem. Pharmacol. 2011, 82, 1145–1152. [Google Scholar] [CrossRef]
- Dang, P.M.C.; Stensballe, A.; Boussetta, T.; Raad, H.; Dewas, C.; Kroviarski, Y.; Hayem, G.; Jensen, O.N.; Gougerot-Pocidalo, M.A.; El-Benna, J. A Specific P47phox-Serine Phosphorylated by Convergent MAPKs Mediates Neutrophil NADPH Oxidase Priming at Inflammatory Sites. J. Clin. Investig. 2006, 116, 2033–2043. [Google Scholar] [CrossRef] [PubMed]
- Schönwaßer, D.C.; Palmer, R.H.; Herget, T.; Parker, P.J. P42 MAPK Phosphorylates 80 KDa MARCKS at Ser-113. FEBS Lett. 1996, 395, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Arbuzova, A.; Schmitz, A.A.P.; Vergères, G. Cross-Talk Unfolded: MARCKS Proteins. Biochem. J. 2002, 362, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ziemba, B.P.; Burke, J.E.; Masson, G.; Williams, R.L.; Falke, J.J. Regulation of PI3K by PKC and MARCKS: Single-Molecule Analysis of a Reconstituted Signaling Pathway. Biophys. J. 2016, 110, 1811–1825. [Google Scholar] [CrossRef]
- Takami, M.; Terry, V.; Petruzzelli, L. Signaling Pathways Involved in IL-8-Dependent Activation of Adhesion Through Mac-1. J. Immunol. 2002, 168, 4559–4566. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, P.T.; Stephens, L.R.; Suire, S.; Wilson, M. PI3K Signaling in Neutrophils. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2010; pp. 183–202. ISBN 978-3-642-13663-4. [Google Scholar]
- Puri, K.D.; Doggett, T.A.; Huang, C.Y.; Douangpanya, J.; Hayflick, J.S.; Turner, M.; Penninger, J.; Diacovo, T.G. The Role of Endothelial PI3Kγ Activity in Neutrophil Trafficking. Blood 2005, 106, 150–157. [Google Scholar] [CrossRef]
- Walser, R.; Burke, J.E.; Gogvadze, E.; Bohnacker, T.; Zhang, X.; Hess, D.; Küenzi, P.; Leitges, M.; Hirsch, E.; Williams, R.L.; et al. PKCβ Phosphorylates PI3Kγ to Activate It and Release It from GPCR Control. PLoS Biol. 2013, 11, e1001587. [Google Scholar] [CrossRef]
- Luo, H.R.; Mondal, S. Molecular Control of PtdIns(3,4,5)P3 Signaling in Neutrophils. EMBO Rep. 2015, 16, 149–163. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt Pathway. Cold Spring Harb. Lab. 2016, 4, a011189. [Google Scholar]
- DeSouza-Vieira, T.; Guimarães-Costa, A.; Rochael, N.C.; Lira, M.N.; Nascimento, M.T.; de Lima-Gomez, P.S.; Mariante, R.M.; Persechini, P.M.; Saraiva, E.M. Neutrophil Extracellular Traps Release Induced by Leishmania: Role of PI3Kγ, ERK, PI3Kσ, PKC, and [Ca 2+ ]. J. Leukoc. Biol. 2016, 100, 801–810. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, H.; Xie, W.; Zuchuan, Z.; Smrcka, A.V.; Wu, D. Roles of PLC-β 2 and -Β3 and PI3Kγ in Chemoattractant- Mediated Signal Transduction. Science 2000, 287, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Wrann, C.D.; Tabriz, N.A.; Barkhausen, T.; Klos, A.; van Griensven, M.; Pape, H.C.; Kendoff, D.O.; Guo, R.; Ward, P.A.; Krettek, C.; et al. The Phosphatidylinositol 3-Kinase Signaling Pathway Exerts Protective Effects during Sepsis by Controlling C5a-Mediated Activation of Innate Immune Functions. J. Immunol. 2007, 178, 5940–5948. [Google Scholar] [CrossRef] [PubMed]
- Ellson, C.; Davidson, K.; Anderson, K.; Stephens, L.R.; Hawkins, P.T. PtdIns3P Binding to the PX Domain of P40phox Is a Physiological Signal in NADPH Oxidase Activation. EMBO J. 2006, 25, 4468–4478. [Google Scholar] [CrossRef] [PubMed]
- Ellson, C.D.; Gobert-Gosse, S.; Anderson, K.E.; Davidson, K.; Erdjument-Bromage, H.; Tempst, P.; Thuring, J.W.; Cooper, M.A.; Lim, Z.Y.; Holmes, A.B.; et al. Ptdlns(3)P Regulates the Neutrophil Oxidase Complex by Binding to the PX Domain of P40phox. Nat. Cell Biol. 2001, 3, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Araki, M. Tumor Suppressor PTEN: Modulator of Cell Signaling, Growth, Migration and Apoptosis. J. Cell Sci. 2001, 114, 2375–2382. [Google Scholar] [CrossRef]
- Subramanian, K.K.; Jia, Y.; Zhu, D.; Simms, B.T.; Jo, H.; Hattori, H.; You, J.; Mizgerd, J.P.; Luo, H.R. Tumor Suppressor PTEN Is a Physiologic Suppressor of Chemoattractant- Mediated Neutrophil Functions. Blood 2007, 109, 4028–4037. [Google Scholar] [CrossRef] [Green Version]
- Teimourian, S.; Moghanloo, E. Role of PTEN in Neutrophil Extracellular Trap Formation. Mol. Immunol. 2015, 66, 319–324. [Google Scholar] [CrossRef]
- Antal, C.E.; Newton, A.C. Spatiotemporal Dynamics of Phosphorylation in Lipid Second Messenger Signaling. Mol. Cell. Proteomics 2013, 12, 3498–3508. [Google Scholar] [CrossRef]
- Immler, R.; Simon, S.I.; Sperandio, M. Calcium Signalling and Related Ion Channels in Neutrophil Recruitment and Function. Eur. J. Clin. Investig. 2018, 48, e12964. [Google Scholar] [CrossRef]
- Essin, K.; Salanova, B.; Kettritz, R.; Sausbier, M.; Luft, F.C.; Kraus, D.; Bohn, E.; Autenrieth, I.B.; Peschel, A.; Ruth, P.; et al. Large-Conductance Calcium-Activated Potassium Channel Activity Is Absent in Human and Mouse Neutrophils and Is Not Required for Innate Immunity. Am. J. Physiol.-Cell Physiol. 2007, 293, 45–54. [Google Scholar] [CrossRef]
- Berg, K.; Ericsson, M.; Lindgren, M.; Gustafsson, H. A High Precision Method for Quantitative Measurements of Reactive Oxygen Species in Frozen Biopsies. PLoS ONE 2014, 9, e90964. [Google Scholar] [CrossRef]
- Mahomed, A.G.; Anderson, R. Activation of Human Neutrophils with Chemotactic Peptide, Opsonized Zymosan and the Calcium Ionophore A23187, but Not with a Phorbol Ester, Is Accompanied by Efflux and Store-Operated Influx of Calcium. Inflammation 2000, 24, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Caimi, G.; Ferrara, F.; Montana, M.; Meli, F.; Canino, B.; Carollo, C.; Lo Presti, R. Acute Ischemic Stroke: Polymorphonuclear Leukocyte Membrane Fluidity and Cytosolic Ca2+ Concentration at Baseline and after Chemotactic Activation. Stroke 2000, 31, 1578–1582. [Google Scholar] [CrossRef]
- Bei, L.; Hu, T.; Qian, Z.M.; Shen, X. Extracellular Ca2+ Regulates the Respiratory Burst of Human Neutrophils. Biochim. Biophys. Acta-Mol. Cell Res. 1998, 1404, 475–483. [Google Scholar] [CrossRef]
- Schenten, V.; Melchior, C.; Steinckwich, N.; Tschirhart, E.J.; Brechard, S. Sphingosine Kinases Regulate NOX2 Activity via P38 MAPK-Dependent Translocation of S100A8/A9. J. Leukoc. Biol. 2011, 89, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Dusi, S.; Donini, M.; Rossi, F. Tyrosine Phosphorylation and Activation of NADPH Oxidase in Human Neutrophils: A Possible Role for MAP Kinases and for a 75 KDa Protein. Biochem. J. 1994, 304, 243–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Liu, Z.; Shen, X. Roles of Phospholipase D in Phorbol Myristate Acetate-Stimulated Neutrophil Respiratory Burst. J. Cell. Mol. Med. 2011, 15, 647–653. [Google Scholar] [CrossRef]
- Björnsdottir, H.; Granfeldt, D.; Welin, A.; Bylund, J.; Karlsson, A. Inhibition of Phospholipase A2 Abrogates Intracellular Processing of NADPH-Oxidase Derived Reactive Oxygen Species in Human Neutrophils. Exp. Cell Res. 2013, 319, 761–774. [Google Scholar] [CrossRef]
- Hazan, I.; Dana, R.; Granot, Y.; Levy, R. Cytosolic Phospholipase A2 and Its Mode of Activation in Human Neutrophils by Opsonized Zymosan: Correlation between 42/44 KDa Mitogen-Activated Protein Kinase, Cytosolic Phospholipase A2 and NADPH Oxidase. Biochem. J. 1997, 326, 867–876. [Google Scholar] [CrossRef]
- Syrbu, S.I.; Waterman, W.H.; Molski, T.F.P.; Nagarkatti, D.; Hajjar, J.J.; Sha’afi, R.I. Phosphorylation of Cytosolic Phospholipase A2 and the Release of Arachidonic Acid in Human Neutrophils. J. Immunol. 1999, 162, 2334–2340. [Google Scholar]
- Bougoin, S.; Poubelle, P.E.; Liao, N.W.; Emezawa, K.; Borgeat, P.; Naccache, P.H. Granulocyte-Macrophage Colony-Stimulating Factor Primes Phospholipase D Activity in Human Nerutrophils in Vitro: Role of Calcium, G-Proteins and Tyrosine Kinases. Cell. Signal. 1992, 4, 487–500. [Google Scholar] [CrossRef]
- Lopez, I.; Burns, D.J.; Lambeth, J.D. Regulation of Phospholipase D by Protein Kinase C in Human Neutrophils. Conventional Isoforms of Protein Kinase C Phosphorylate a Phospholipase D- Related Component in the Plasma Membrane. J. Biol. Chem. 1995, 270, 19465–19472. [Google Scholar] [CrossRef]
- Whatmore, J.; Cronin, P.; Cockcroft, S. ARF1-Regulated Phospholipase D in Human Neutrophils Is Enhanced by PMA and MgATP. FEBS Lett. 1994, 352, 113–117. [Google Scholar] [CrossRef]
- D’Souza-Schorey, C.; Chavrier, P. ARF Proteins: Roles in Membrane Traffic and Beyond. Nat. Rev. Mol. Cell Biol. 2006, 7, 347–358. [Google Scholar] [CrossRef]
- Zhang, H.; Garlichs, C.D.; Mügge, A.; Daniel, W.G. Role of Ca2+-ATPase Inhibitors in Activation of Cytosolic Phospholipase A2 in Human Polymorphonuclear Neutrophils. Eur. J. Pharmacol. 1999, 364, 229–237. [Google Scholar] [CrossRef]
- Gupta, A.K.; Giaglis, S.; Hasler, P.; Hahn, S. Efficient Neutrophil Extracellular Trap Induction Requires Mobilization of Both Intracellular and Extracellular Calcium Pools and Is Modulated by Cyclosporine A. PLoS ONE 2014, 9, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Suh, B.-C.; Kim, J.-S.; Namgung, U.; Ha, H.; Kim, K.-T. P2X7 Nucleotide Receptor Mediation of Membrane Pore Formation and Superoxide Generation in Human Promyelocytes and Neutrophils. J. Immunol. 2001, 166, 6754–6763. [Google Scholar] [CrossRef] [Green Version]
- Kenny, E.F.; Herzig, A.; Kruger, R.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; von Bernuth, H.; Zychlinsky, A. Diverse Stimuli Engage Different Neutrophil Extracellular Trap Pathways Elaine. Elife 2017, 6, e24437. [Google Scholar] [CrossRef]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 Channel and Mitochondrial ROS Mediate NADPH Oxidase-Independent NETosis Induced by Calcium Influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-Triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef]
- Ackermann, M.; Anders, H.J.; Bilyy, R.; Bowlin, G.L.; Daniel, C.; De Lorenzo, R.; Egeblad, M.; Henneck, T.; Hidalgo, A.; Hoffmann, M.; et al. Patients with COVID-19: In the Dark-NETs of Neutrophils. Cell Death Differ. 2021, 28, 3125–3139. [Google Scholar] [CrossRef]
- Brinkmann, V. Neutrophil Extracellular Traps in the Second Decade. J. Innate Immun. 2018, 10, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil Extracellular Traps Contain Calprotectin, a Cytosolic Protein Complex Involved in Host Defense against Candida Albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [PubMed]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Maximiliano, G.; Brown, G.D.; Papayannopoulos, V. Neutrophils Sense Microbial Size and Selectively Release Neutrophil Extracellular Traps in Response to Large Pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable Neutrophils Release Mitochondrial DNA to Form Neutrophil Extracellular Traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Chatfield, S.M.; Grebe, K.; Whitehead, L.W.; Rogers, K.L.; Nebl, T.; Murphy, J.M.; Wicks, I.P. Monosodium Urate Crystals Generate Nuclease-Resistant Neutrophil Extracellular Traps via a Distinct Molecular Pathway. J. Immunol. 2018, 200, 1801–1816. [Google Scholar] [CrossRef]
- Della Coletta, A.M.; Bachiega, T.F.; de Quaglia e Silva, J.C.; Victoriano de Campos Soares, Â.M.; De Faveri, J.; Marques, S.A.; Marques, M.E.A.; Ximenes, V.F.; Dias-Melicio, L.A. Neutrophil Extracellular Traps Identification in Tegumentary Lesions of Patients with Paracoccidioidomycosis and Different Patterns of NETs Generation In Vitro. PLoS Negl. Trop. Dis. 2015, 9, e0004037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; Garcia-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef]
- Konig, M.F.; Andrade, F. A Critical Reappraisal of Neutrophil Extracellular Traps and NETosis Mimics Based on Differential Requirements for Protein Citrullination. Front. Immunol. 2016, 7, 461. [Google Scholar] [CrossRef]
- Wang, Y.; Wysocka, J.; Sayegh, J.; Lee, Y.; Perlin, J.R.; Leonelli, L.; Sonbuchner, L.S.; Mcdonald, C.H.; Cook, R.G.; Roeder, R.C.; et al. Human PAD4 Regulates Histone Arginine Methylation Levels via Demethylimination. Science 2004, 306, 279–283. [Google Scholar] [CrossRef]
- Farley, K.; Stolley, J.M.; Zhao, P.; Cooley, J.; Remold-O’Donnell, E. A SerpinB1 Regulatory Mechanism Is Essential for Restricting Neutrophil Extracellular Trap Generation. J. Immunol. 2012, 189, 4574–4581. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil Elastase and Myeloperoxidase Regulate the Formation of Neutrophil Extracellular Traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Stojkov, D.; Amini, P.; Oberson, K.; Sokollik, C.; Duppenthaler, A.; Simon, H.U.; Yousefi, S. ROS and Glutathionylation Balance Cytoskeletal Dynamics in Neutrophil Extracellular Trap Formation. J. Cell Biol. 2017, 216, 4073–4090. [Google Scholar] [CrossRef] [PubMed]
- Neubert, E.; Meyer, D.; Rocca, F.; Günay, G.; Kwaczala-Tessmann, A.; Grandke, J.; Senger-Sander, S.; Geisler, C.; Egner, A.; Schön, M.P.; et al. Chromatin Swelling Drives Neutrophil Extracellular Trap Release. Nat. Commun. 2018, 9, 3767. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, M.; Weigel, B.; Mall, M.; Werth, V.P.; Liu, M.-L. Nuclear Envelope Rupture and NET Formation Is Driven by PKCα-mediated Lamin B Disassembly. EMBO Rep. 2020, 21, e48779. [Google Scholar] [CrossRef]
- Xie, K.; Varatnitskaya, M.; Maghnouj, A.; Bader, V.; Winklhofer, K.F.; Hahn, S.; Leichert, L.I. Activation Leads to a Significant Shift in the Intracellular Redox Homeostasis of Neutrophil-like Cells. Redox Biol. 2020, 28, 101344. [Google Scholar] [CrossRef]
- Douda, D.N.; Yip, L.; Khan, M.A.; Grasemann, H.; Palaniyar, N. To the Editor: Akt Is Essential to Induce NADPH-Dependent NETosis and to Switch the Neutrophil Death to Apoptosis. Blood 2014, 123, 597–600. [Google Scholar] [CrossRef]
- Neeli, I.; Radic, M. Opposition between PKC Isoforms Regulates Histone Deimination and Neutrophil Extracellular Chromatin Release. Front. Immunol. 2013, 4, 38. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y. Peptidylarginine Deiminases in Citrullination, Gene Regulation, Health and Pathogenesis. Biochim. Biophys. Acta 2013, 1829, 1126–1135. [Google Scholar] [CrossRef]
- Nakashima, K.; Hagiwara, T.; Yamada, M. Nuclear Localization of Peptidylarginine Deiminase V and Histone Deimination in Granulocytes. J. Biol. Chem. 2002, 277, 49562–49568. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone Hypercitrullination Mediates Chromatin Decondensation and Neutrophil Extracellular Trap Formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 Is Essential for Antibacterial Innate Immunity Mediated by Neutrophil Extracellular Traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.L.; Shim, D.; Kernien, J.; Johnson, C.J.; Nett, J.E.; Shelef, M.A. Insight into Neutrophil Extracellular Traps through Systematic Evaluation of Citrullination and Peptidylarginine Deiminases. J. Immunol. Res. 2019, 2019, 2160192. [Google Scholar] [CrossRef] [PubMed]
- Hawez, A.; Al-Haidari, A.; Madhi, R.; Rahman, M.; Thorlacius, H. MiR-155 Regulates PAD4-Dependent Formation of Neutrophil Extracellular Traps. Front. Immunol. 2019, 10, 2462. [Google Scholar] [CrossRef]
- Chargui, A.; Cesaro, A.; Mimouna, S.; Fareh, M.; Brest, P. Subversion of Autophagy in Adherent Invasive Escherichia Coli-Infected Neutrophils Induces Inflammation and Cell Death. PLoS ONE 2012, 7, e51727. [Google Scholar] [CrossRef]
- Mitroulis, I.; Kourtzelis, I.; Kambas, K.; Rafail, S.; Chrysanthopoulou, A.; Speletas, M.; Ritis, K. Regulation of the Autophagic Machinery in Human Neutrophils. Eur. J. Immunol. 2010, 40, 1461–1472. [Google Scholar] [CrossRef]
- Narni-Mancinelli, E.; Soudja, S.M.; Crozat, K.; Dalod, M.; Gounon, P. Inflammatory Monocytes and Neutrophils Are Licensed to Kill during Memory Responses In Vivo. PLoS Pathog. 2011, 7, 1002457. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Martinez, J.; Malireddi, R.K.S.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.L.; Tan, H.; Peng, J.; et al. Molecular Characterization of LC3-Associated Phagocytosis Reveals Distinct Roles for Rubicon, NOX2 and Autophagy Proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Jin, Y.; Kim, J.; Kim, J.H.; Jung, J.; Kang, S.; Kim, I.Y.; Kim, J.; Cheong, H.; Song, H.K. The C-Terminal Region of ATG101 Bridges ULK1 and PtdIns3K Complex in Autophagy Initiation. Autophagy 2018, 14, 2104–2116. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitroulis, I.; Ritis, K. Autophagy in Neutrophils: From Granulopoiesis to Neutrophil Extracellular Traps. Front. Cell Dev. Biol. 2018, 6, 109. [Google Scholar] [CrossRef]
- Liang, C.; Lee, J.-S.; Inn, K.-S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-Binding UVRAG Targets the Class C Vps Complex to Coordinate Autophagosome Maturation and Endocytic Trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef]
- Cheng, M.L.; Ho, H.Y.; Lin, H.Y.; Lai, Y.C.; Chiu, D.T.Y. Effective NET Formation in Neutrophils from Individuals with G6PD Taiwan-Hakka Is Associated with Enhanced NADP+ Biosynthesis. Free Radic. Res. 2013, 47, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Berghe, T.V.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil Extracellular Trap Cell Death Requires Both Autophagy and Superoxide Generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Shrestha, S.; Youn, Y.J.; Kim, J.K.; Kim, S.Y.; Kim, H.J.; Park, S.H.; Ahn, W.G.; Kim, S.; Lee, M.G.; et al. Autophagy Primes Neutrophils for Neutrophil Extracellular Trap Formation during Sepsis. Am. J. Respir. Crit. Care Med. 2017, 196, 577–589. [Google Scholar] [CrossRef]
- Germic, N.; Stojkov, D.; Oberson, K.; Yousefi, S.; Simon, H.U. Neither Eosinophils nor Neutrophils Require ATG5-Dependent Autophagy for Extracellular DNA Trap Formation. Immunology 2017, 152, 517–525. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Multiple Nuclear Factors Interact with the Immunoglobulin Enhancer Sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-KB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Respect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-ΚB P65 and Strategies for Therapeutic Manipulation. J. Inflamm. Res. 2018, 11, 407. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Castro-Alcaraz, S.; Miskolci, V.; Kalasapudi, B.; Davidson, D.; Vancurova, I. NF-ΚB Regulation in Human Neutrophils by Nuclear IκBα: Correlation to Apoptosis. J. Immunol. 2002, 169, 3947–3953. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Seo, J.Y.; Roh, K.H.; Lim, J.W.; Kim, K.H. Suppression of NF-ΚB Activation and Cytokine Production by N-Acetylcysteine in Pancreatic Acinar Cells. Free Radic. Biol. Med. 2000, 29, 674–683. [Google Scholar] [CrossRef]
- Lapponi, M.J.; Carestia, A.; Landoni, V.I.; Rivadeneyra, L.; Etulain, J.; Negrotto, S.; Pozner, R.G.; Schattner, M. Regulation of Neutrophil Extracellular Trap Formation by Anti-Inflammatory Drugs. J. Pharmacol. Exp. Ther. 2013, 345, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Langereis, J.D.; Raaijmakers, H.A.J.A.; Ulfman, L.H.; Koenderman, L. Abrogation of NF-ΚB Signaling in Human Neutrophils Induces Neutrophil Survival through Sustained P38-MAPK Activation. J. Leukoc. Biol. 2010, 88, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-C.; Lin, H.-C.; Liu, C.-Y.; Wang, C.-H.; Hwang, T.; Huang, T.-T.; Lin, C.-H.; Kuo, H.-P. Neutrophil Elastase Induces IL-8 Synthesis by Lung Epithelial Cells via the Mitogen-Activated Protein Kinase Pathway. J. Biomed. Sci. 2004, 11, 49–58. [Google Scholar] [CrossRef]
- Sun, B.; Dwivedi, N.; Bechtel, T.J.; Paulsen, J.L.; Muth, A.; Bawadekar, M.; Li, G.; Thompson, P.R.; Shelef, M.A.; Schiffer, C.A.; et al. Citrullination of NF-KB P65 Enhances Its Nuclear Localization and TLR-Induced Expression of IL-1β and TNFα HHS Public Access. Sci. Immunol. 2017, 2, eaal3062. [Google Scholar] [CrossRef]
- Keshari, R.S.; Jyoti, A.; Dubey, M.; Kothari, N.; Kohli, M.; Bogra, J.; Barthwal, M.K.; Dikshit, M. Cytokines Induced Neutrophil Extracellular Traps Formation: Implication for the Inflammatory Disease Condition. PLoS ONE 2012, 7, e48111. [Google Scholar] [CrossRef]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.-C.; Goode, J.; Miething, C.; Goktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-ΚB Is a Negative Regulator of IL-1β Secretion as Revealed by Genetic and Pharmacological Inhibition of IKKβ. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D Plays a Vital Role in the Generation of Neutrophil Extracellular Traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Meher, A.K.; Spinosa, M.; Davis, J.P.; Pope, N.; Laubach, V.E.; Su, G.; Serbulea, V.; Leitinger, N.; Ailawadi, G.; Gilbert, R.; et al. Novel Role of IL (Interleukin)-1β in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Tsourouktsoglou, T.D.; Branzk, N.; Wang, Q.; Reincke, S.; Herbst, S.; Gutierrez, M.; Papayannopoulos, V. Reactive Oxygen Species Localization Programs Inflammation to Clear Microbes of Different Size. Immunity 2017, 46, 421–432. [Google Scholar] [CrossRef]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid Killing of Human Neutrophils by the Potent Activator Phorbol 12-Myristate 13-Acetate (PMA) Accompanied by Changes Different from Typical Apoptosis or Necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef]
- Luehong, N.; Khaowmek, J.; Wongsawan, K.; Chuammitri, P. Preferential Pattern of Mouse Neutrophil Cell Death in Response to Various Stimulants. Vitr. Cell. Dev. Biol.-Anim. 2017, 53, 513–524. [Google Scholar] [CrossRef]
- Fadeel, B.; Åhlin, A.; Henter, J.-I.; Orrenius, S.; Hampton, M.B. Involvement of Caspases in Neutrophil Apoptosis: Regulation by Reactive Oxygen Species. Blood 1998, 92, 4808–4818. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Desai, J.; Kumar, S.V.; Mulay, S.R.; Konrad, L.; Romoli, S.; Schauer, C.; Herrmann, M.; Bilyy, R.; Müller, S.; Popper, B.; et al. PMA and Crystal-Induced Neutrophil Extracellular Trap Formation Involves RIPK1-RIPK3-MLKL Signaling. Eur. J. Immunol. 2016, 46, 223–229. [Google Scholar] [CrossRef]
- Amini, P.; Stojkov, D.; Wang, X.; Wicki, S.; Kaufmann, T.; Wong, W.W.L.; Simon, H.U.; Yousefi, S. NET Formation Can Occur Independently of RIPK3 and MLKL Signaling. Eur. J. Immunol. 2016, 46, 178–184. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damascena, H.L.; Silveira, W.A.A.; Castro, M.S.; Fontes, W. Neutrophil Activated by the Famous and Potent PMA (Phorbol Myristate Acetate). Cells 2022, 11, 2889. https://doi.org/10.3390/cells11182889
Damascena HL, Silveira WAA, Castro MS, Fontes W. Neutrophil Activated by the Famous and Potent PMA (Phorbol Myristate Acetate). Cells. 2022; 11(18):2889. https://doi.org/10.3390/cells11182889
Chicago/Turabian StyleDamascena, Hylane Luiz, Wendy Ann Assis Silveira, Mariana S. Castro, and Wagner Fontes. 2022. "Neutrophil Activated by the Famous and Potent PMA (Phorbol Myristate Acetate)" Cells 11, no. 18: 2889. https://doi.org/10.3390/cells11182889