
mTORC1 Activity in Psoriatic Lesions Is Mediated by Aberrant Regulation through the Tuberous Sclerosis Complex

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Chemicals
2.2. Immunohistochemistry
2.3. Cell Culture and Western Blotting
2.4. Immunofluorescence Staining
2.5. Statistical Analysis
3. Results
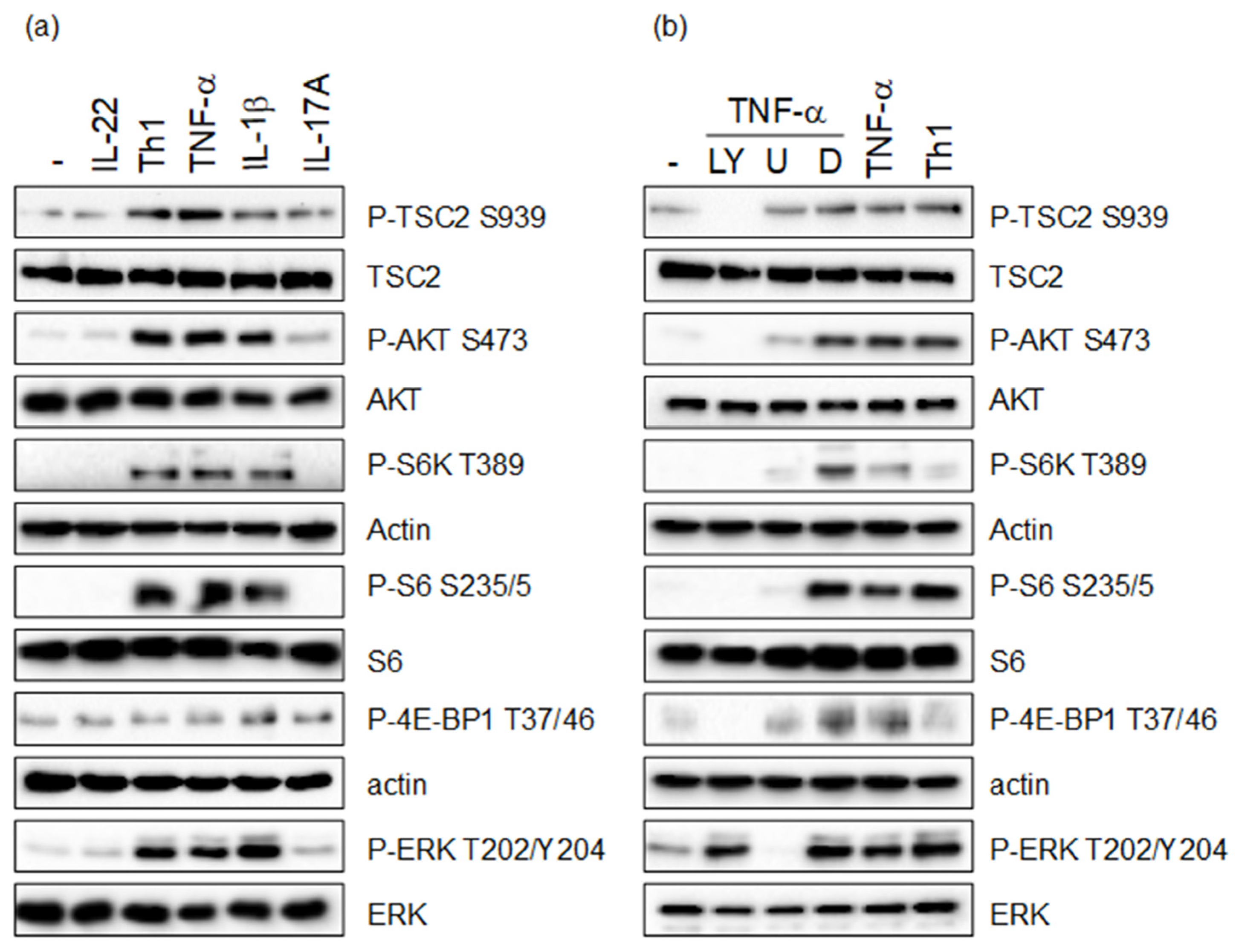
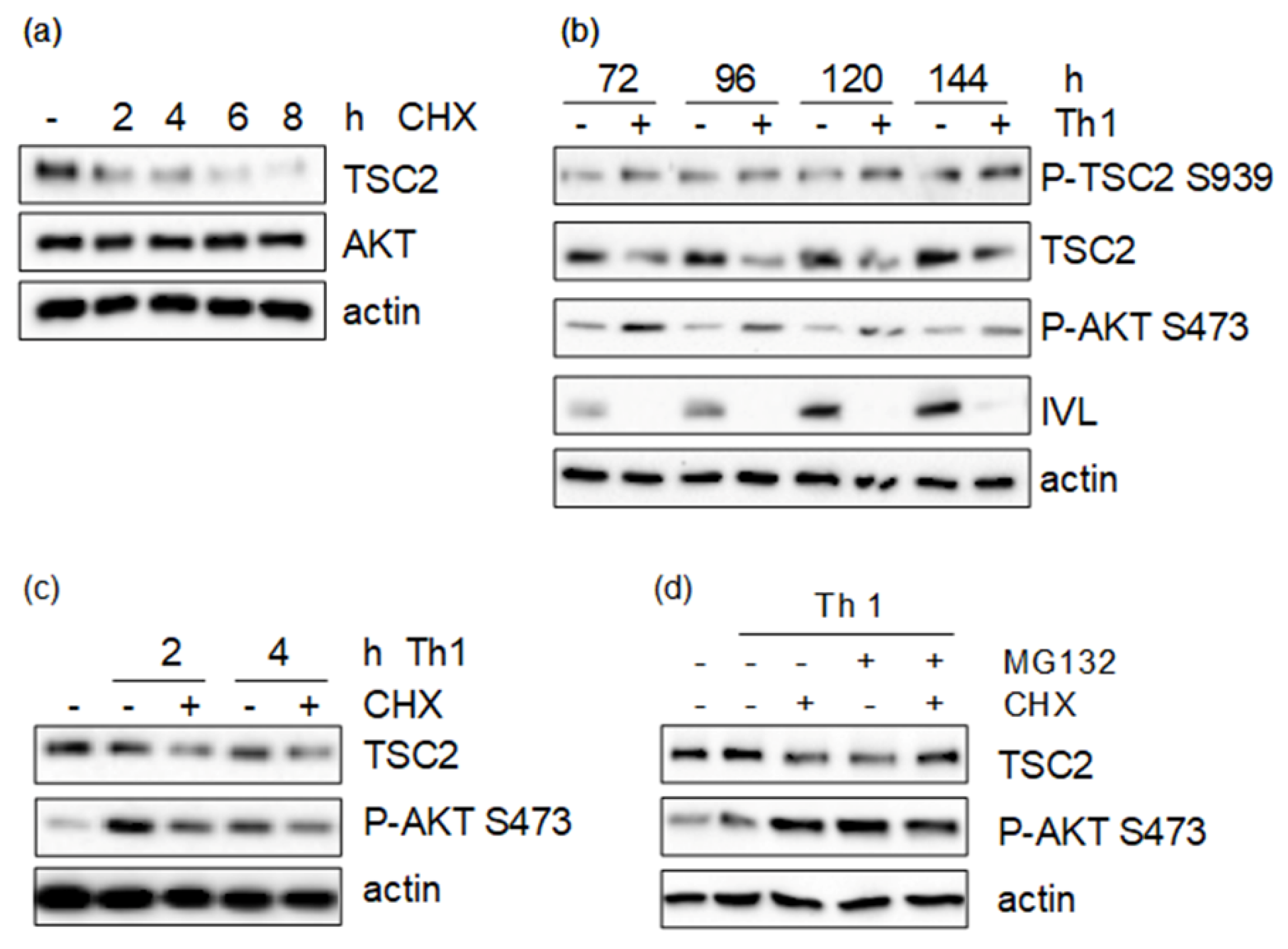
3.1. Th1 Cytokines Induce TSC2 Phosphorylation via the PI3-K/AKT and MAPK Pathway
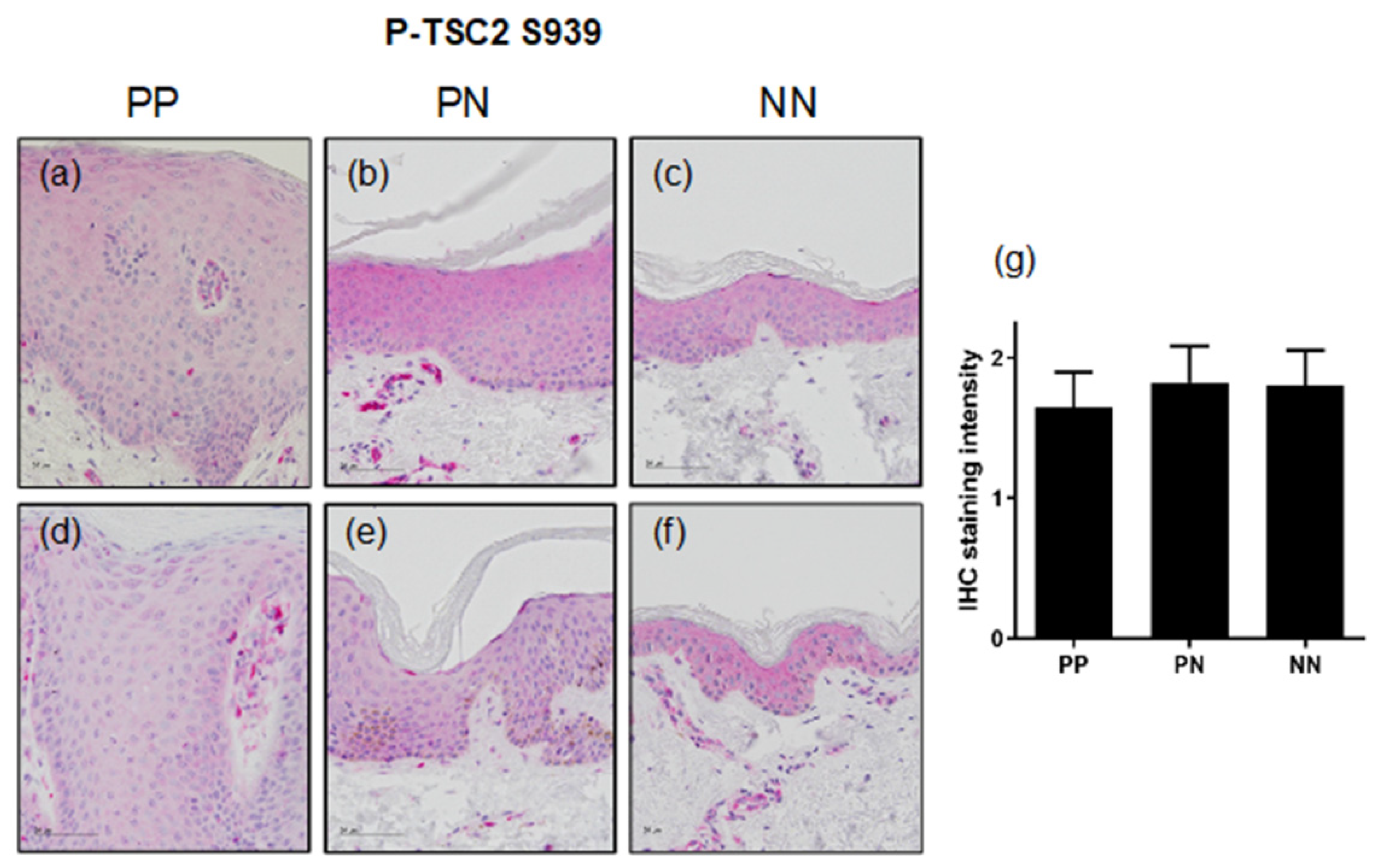
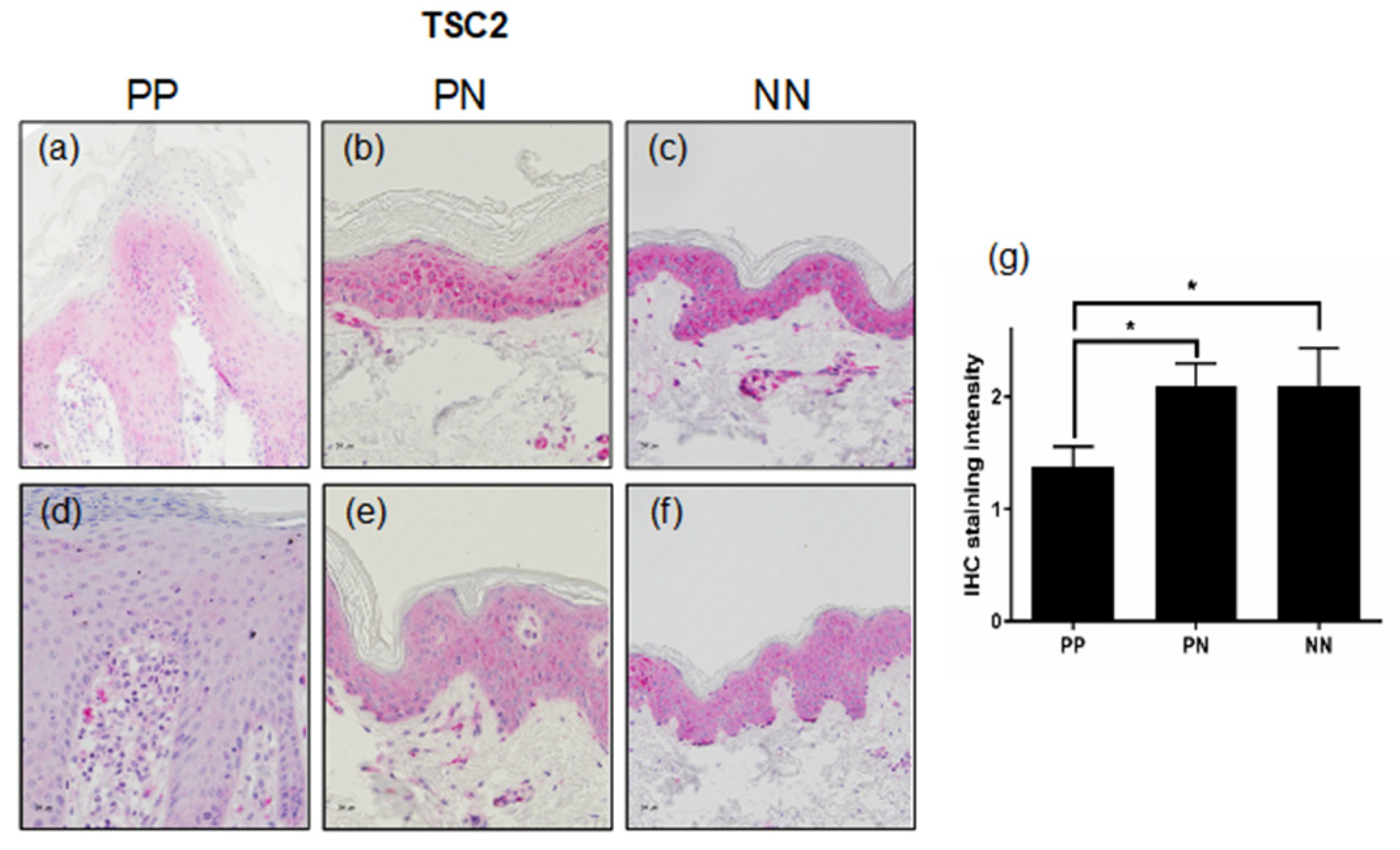
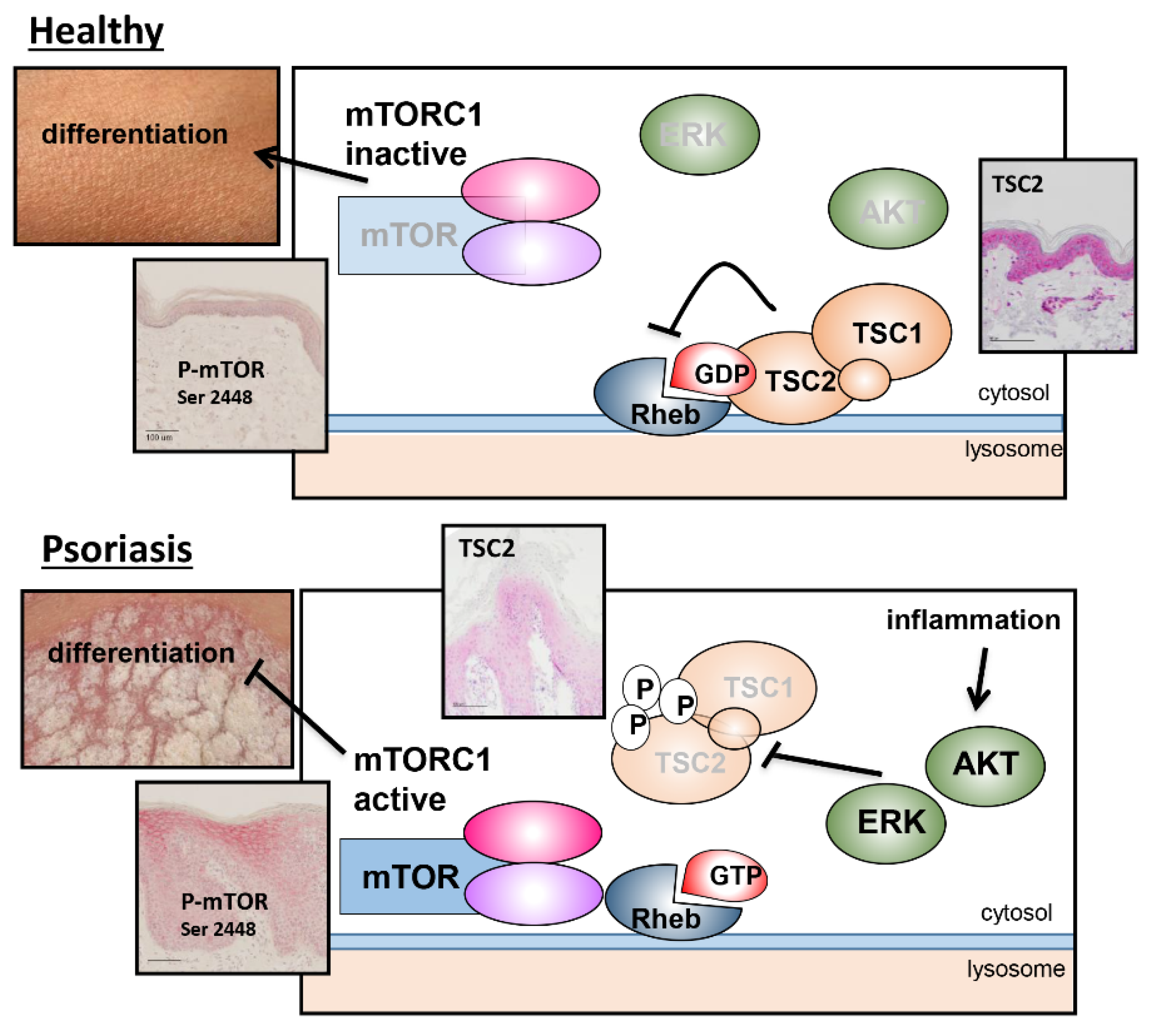
3.2. TSC2 Is Downregulated in Lesional Psoriatic Skin
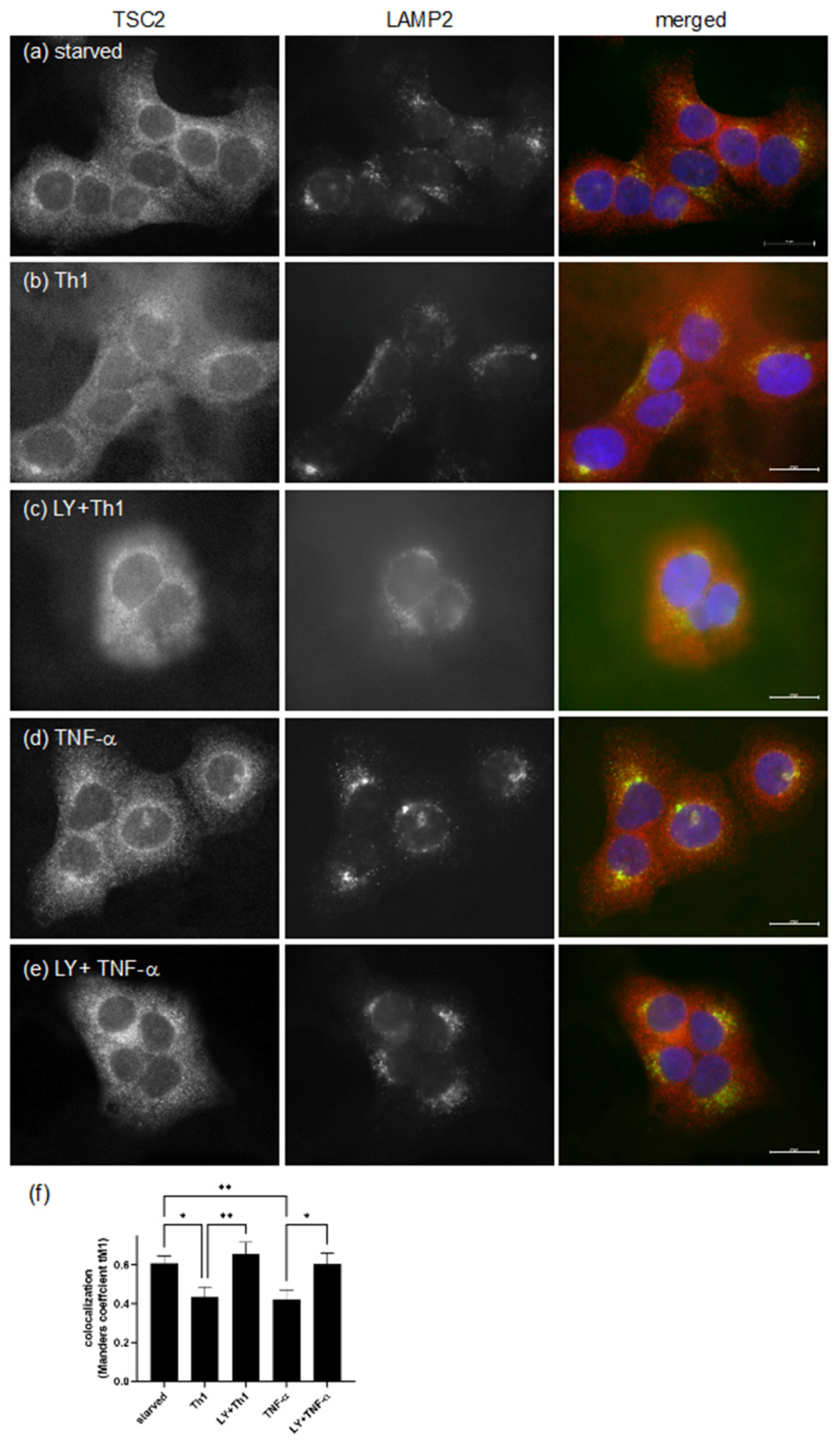
3.3. Pro-Inflammatory Cytokines Regulate TSC Function by Spatial Re-Distribution and Degradation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boehncke, W.H. Systemic Inflammation and Cardiovascular Comorbidity in Psoriasis Patients: Causes and Consequences. Front. Immunol. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed]
- Boehncke, W.H.; Schon, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Lowes, M.A.; Bowcock, A.M.; Krueger, J.G. Pathogenesis and therapy of psoriasis. Nature 2007, 445, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Schakel, K.; Schon, M.P.; Ghoreschi, K. [Pathogenesis of psoriasis]. Hautarzt 2016, 67, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.A.; Russell, C.B.; Martin, D.A.; Towne, J.E.; Krueger, J.G. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol. 2013, 34, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Buerger, C.; Richter, B.; Woth, K.; Salgo, R.; Malisiewicz, B.; Diehl, S.; Hardt, K.; Boehncke, S.; Boehncke, W.H. Interleukin-1beta interferes with epidermal homeostasis through induction of insulin resistance: Implications for psoriasis pathogenesis. J. Investig. Dermatol. 2012, 132, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Buerger, C.; Malisiewicz, B.; Eiser, A.; Hardt, K.; Boehncke, W.H. Mammalian target of rapamycin and its downstream signalling components are activated in psoriatic skin. Br. J. Dermatol. 2013, 169, 156–159. [Google Scholar] [CrossRef]
- Buerger, C.; Shirsath, N.; Lang, V.; Berard, A.; Diehl, S.; Kaufmann, R.; Boehncke, W.H.; Wolf, P. Inflammation dependent mTORC1 signaling interferes with the switch from keratinocyte proliferation to differentiation. PLoS ONE 2017, 12, e0180853. [Google Scholar] [CrossRef]
- Buerger, C. Epidermal mTORC1 Signaling Contributes to the Pathogenesis of Psoriasis and Could Serve as a Therapeutic Target. Front. Immunol. 2018, 9, 2786. [Google Scholar] [CrossRef]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Uysal, S.P.; Sahin, M. Tuberous sclerosis: A review of the past, present, and future. Turk. J. Med. Sci. 2020, 50, 1665–1676. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef]
- Castro, A.F.; Rebhun, J.F.; Clark, G.J.; Quilliam, L.A. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J. Biol. Chem. 2003, 278, 32493–32496. [Google Scholar] [CrossRef]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [Green Version]
- Demetriades, C.; Doumpas, N.; Teleman, A.A. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef]
- Monteleon, C.L.; Agnihotri, T.; Dahal, A.; Liu, M.; Rebecca, V.W.; Beatty, G.L.; Amaravadi, R.K.; Ridky, T.W. Lysosomes Support the Degradation, Signaling, and Mitochondrial Metabolism Necessary for Human Epidermal Differentiation. J. Investig. Dermatol. 2018, 138, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Akinduro, O.; Sully, K.; Patel, A.; Robinson, D.J.; Chikh, A.; McPhail, G.; Braun, K.M.; Philpott, M.P.; Harwood, C.A.; Byrne, C.; et al. Constitutive Autophagy and Nucleophagy during Epidermal Differentiation. J. Investig. Dermatol. 2016, 136, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Bochenska, K.; Moskot, M.; Malinowska, M.; Jakobkiewicz-Banecka, J.; Szczerkowska-Dobosz, A.; Purzycka-Bohdan, D.; Plenkowska, J.; Slominski, B.; Gabig-Ciminska, M. Lysosome Alterations in the Human Epithelial Cell Line HaCaT and Skin Specimens: Relevance to Psoriasis. Int. J. Mol. Sci. 2019, 20, 2255. [Google Scholar] [CrossRef]
- Klapan, K.; Frangez, Z.; Markov, N.; Yousefi, S.; Simon, D.; Simon, H.U. Evidence for Lysosomal Dysfunction within the Epidermis in Psoriasis and Atopic Dermatitis. J. Investig. Dermatol. 2021, 141, 2838–2848.e4. [Google Scholar] [CrossRef]
- Chong-Kopera, H.; Inoki, K.; Li, Y.; Zhu, T.; Garcia-Gonzalo, F.R.; Rosa, J.L.; Guan, K.L. TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J. Biol. Chem. 2006, 281, 8313–8316. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Witt, R.M.; Santos, T.M.; Polizzano, C.; Sabatini, B.L.; Ramesh, V. Pam (Protein associated with Myc) functions as an E3 ubiquitin ligase and regulates TSC/mTOR signaling. Cell Signal. 2008, 20, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Ding, H.; Lu, Z.; Li, Y.; Pan, Y.; Ning, T.; Ke, Y.; Liu, W.; Yi, Y.; Zhang, C.; et al. E3 ubiquitin ligase E6AP-mediated TSC2 turnover in the presence and absence of HPV16 E6. Genes Cells 2008, 13, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yi, Y.; Zhang, C.; Zhou, B.; Liao, L.; Liu, W.; Hu, J.; Xu, Q.; Chen, J.; Lu, J. The Expression of TRIM6 Activates the mTORC1 Pathway by Regulating the Ubiquitination of TSC1-TSC2 to Promote Renal Fibrosis. Front. Cell Dev. Biol. 2020, 8, 616747. [Google Scholar] [CrossRef] [PubMed]
- Plas, D.R.; Thompson, C.B. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J. Biol. Chem. 2003, 278, 12361–12366. [Google Scholar] [CrossRef]
- Li, Y.; Inoki, K.; Yeung, R.; Guan, K.L. Regulation of TSC2 by 14-3-3 binding. J. Biol. Chem. 2002, 277, 44593–44596. [Google Scholar] [CrossRef]
- Shumway, S.D.; Li, Y.; Xiong, Y. 14-3-3beta binds to and negatively regulates the tuberous sclerosis complex 2 (TSC2) tumor suppressor gene product, tuberin. J. Biol. Chem. 2003, 278, 2089–2092. [Google Scholar] [CrossRef]
- Madigan, J.P.; Hou, F.; Ye, L.; Hu, J.; Dong, A.; Tempel, W.; Yohe, M.E.; Randazzo, P.A.; Jenkins, L.M.M.; Gottesman, M.M.; et al. The tuberous sclerosis complex subunit TBC1D7 is stabilized by Akt phosphorylation-mediated 14-3-3 binding. J. Biol. Chem. 2018, 293, 16142–16159. [Google Scholar] [CrossRef]
- Man, X.; Zhang, X.; Tang, J.; Chen, Y.; Li, H.; Xu, B.; Pan, L. Downregulation of 14-3-3beta and 14-3-3zeta in lesions of psoriasis vulgaris. Clin. Exp. Dermatol. 2013, 38, 390–395. [Google Scholar] [CrossRef]
- Raaby, L.; Otkjaer, K.; Salvskov-Iversen, M.L.; Johansen, C.; Iversen, L. A Characterization of the expression of 14-3-3 isoforms in psoriasis, basal cell carcinoma, atopic dermatitis and contact dermatitis. Dermatol. Rep. 2010, 2, e14. [Google Scholar] [CrossRef]
- Gen, S.; Matsumoto, Y.; Kobayashi, K.I.; Suzuki, T.; Inoue, J.; Yamamoto, Y. Stability of tuberous sclerosis complex 2 is controlled by methylation at R1457 and R1459. Sci. Rep. 2020, 10, 21160. [Google Scholar] [CrossRef]
- Garcia-Aguilar, A.; Guillen, C.; Nellist, M.; Bartolome, A.; Benito, M. TSC2 N-terminal lysine acetylation status affects to its stability modulating mTORC1 signaling and autophagy. Biochim. Biophys. Acta 2016, 1863, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Gudjonsson, J.E.; Liang, L.; Stuart, P.E.; Li, Y.; Chen, W.; Weichenthal, M.; Ellinghaus, E.; Franke, A.; Cookson, W.; et al. Gene expression in skin and lymphoblastoid cells: Refined statistical method reveals extensive overlap in cis-eQTL signals. Am. J. Hum. Genet. 2010, 87, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R.; Xing, X.; Stuart, P.E.; Chen, C.S.; Aphale, A.; Nair, R.P.; Voorhees, J.J.; Elder, J.T.; Johnston, A.; Gudjonsson, J.E. Heterogeneity of inflammatory and cytokine networks in chronic plaque psoriasis. PLoS ONE 2012, 7, e34594. [Google Scholar] [CrossRef]
- Hickerson, R.P.; Leake, D.; Pho, L.N.; Leachman, S.A.; Kaspar, R.L. Rapamycin selectively inhibits expression of an inducible keratin (K6a) in human keratinocytes and improves symptoms in pachyonychia congenita patients. J. Dermatol. Sci. 2009, 56, 82–88. [Google Scholar] [CrossRef]
- Naeem, A.S.; Tommasi, C.; Cole, C.; Brown, S.J.; Zhu, Y.; Way, B.; Willis Owen, S.A.; Moffatt, M.; Cookson, W.O.; Harper, J.I.; et al. A mechanistic target of rapamycin complex 1/2 (mTORC1)/V-Akt murine thymoma viral oncogene homolog 1 (AKT1)/cathepsin H axis controls filaggrin expression and processing in skin, a novel mechanism for skin barrier disruption in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2017, 139, 1228–1241. [Google Scholar] [CrossRef]
- Lai, M.; Zou, W.; Han, Z.; Zhou, L.; Qiu, Z.; Chen, J.; Zhang, S.; Lai, P.; Li, K.; Zhang, Y.; et al. Tsc1 regulates tight junction independent of mTORC1. Proc. Natl. Acad. Sci. USA 2021, 118, e2020891118. [Google Scholar] [CrossRef]
- Wei, K.C.; Lai, P.C. Combination of everolimus and tacrolimus: A potentially effective regimen for recalcitrant psoriasis. Dermatol. Ther. 2015, 28, 25–27. [Google Scholar] [CrossRef]
- Reitamo, S.; Spuls, P.; Sassolas, B.; Lahfa, M.; Claudy, A.; Griffiths, C.E. Efficacy of sirolimus (rapamycin) administered concomitantly with a subtherapeutic dose of cyclosporin in the treatment of severe psoriasis: A randomized controlled trial. Br. J. Dermatol. 2001, 145, 438–445. [Google Scholar] [CrossRef]
- Frigerio, E.; Colombo, M.D.; Franchi, C.; Altomare, A.; Garutti, C.; Altomare, G.F. Severe psoriasis treated with a new macrolide: Everolimus. Br. J. Dermatol. 2007, 156, 372–374. [Google Scholar] [CrossRef]
- Ormerod, A.D.; Shah, S.A.; Copeland, P.; Omar, G.; Winfield, A. Treatment of psoriasis with topical sirolimus: Preclinical development and a randomized, double-blind trial. Br. J. Dermatol. 2005, 152, 758–764. [Google Scholar] [CrossRef]
- Burger, C.; Shirsath, N.; Lang, V.; Diehl, S.; Kaufmann, R.; Weigert, A.; Han, Y.Y.; Ringel, C.; Wolf, P. Blocking mTOR Signalling with Rapamycin Ameliorates Imiquimod-induced Psoriasis in Mice. Acta Derm. Venereol. 2017, 97, 1087–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreri, A.; Lang, V.; Kaufmann, R.; Buerger, C. mTORC1 Activity in Psoriatic Lesions Is Mediated by Aberrant Regulation through the Tuberous Sclerosis Complex. Cells 2022, 11, 2847. https://doi.org/10.3390/cells11182847
Ferreri A, Lang V, Kaufmann R, Buerger C. mTORC1 Activity in Psoriatic Lesions Is Mediated by Aberrant Regulation through the Tuberous Sclerosis Complex. Cells. 2022; 11(18):2847. https://doi.org/10.3390/cells11182847
Chicago/Turabian StyleFerreri, Antonio, Victoria Lang, Roland Kaufmann, and Claudia Buerger. 2022. "mTORC1 Activity in Psoriatic Lesions Is Mediated by Aberrant Regulation through the Tuberous Sclerosis Complex" Cells 11, no. 18: 2847. https://doi.org/10.3390/cells11182847